





Abstract
Ligand-dependent interactions between nuclear receptors and members of a family of nuclear receptor coactivators are associated with transcriptional activation. Here we used fluorescence resonance energy transfer (FRET) as an approach for detecting and quantitating such interactions. Using the ligand binding domain (LBD) of peroxisome proliferator-activated receptor (PPARγ) as a model, known agonists (thiazolidinediones and Δ12, 14-PGJ2) induced a specific interaction resulting in FRET between the fluorescently labeled LBD and fluorescently labeled coactivators [CREB-binding protein (CBP) or steroid receptor coactivator-1 (SRC-1)]. Specific energy transfer was dose dependent; individual ligands displayed distinct potency and maximal FRET profiles that were identical when results obtained using CBP vs. SRC-1 were compared. In addition, half-maximally effective agonist concentrations (EC50s) correlated well with reported results using cell-based assays. A site-directed AF2 mutant of PPARγ (E471A) that abrogated ligand-stimulated transcription in transfected cells also failed to induce ligand-mediated FRET between PPARγ LBD and CBP or SRC-1. Using estrogen receptor (ERα) as an alternative system, known agonists induced an interaction between ERα LBD and SRC-1, whereas ER antagonists disrupted agonist-induced interaction of ERα with SRC-1. In the presence of saturating agonist concentrations, unlabeled CBP or SRC-1 was used to compete with fluorescently labeled coactivators with saturation kinetics. Relative affinities for the individual receptor-coactivator pairs were determined as follows: PPARγ-CBP = ERα-SRC-1 > PPARγ-SRC-1 ≫ ERα-CBP. Conclusions: 1) FRET-based coactivator association is a novel approach for characterizing nuclear receptor agonists or antagonists; individual ligands display potencies that are predictive of in vivo effects and distinct profiles of maximal activity that are suggestive of alternative receptor conformations. 2) PPARγ interacts with both CBP and SRC-1; transcriptional activation and coactivator association are AF2 dependent. 3) Nuclear receptor LBDs have distinct affinities for individual coactivators; thus, PPARγ has a greater apparent affinity for CBP than for SRC-1, whereas ERα interacts preferentially with SRC-1 but very weakly with CBP.
INTRODUCTION
Nuclear receptors are a large family of ligand-activated transcription factors that bind as homo- or heterodimers to their cognate DNA elements in gene promoters (1–5). The nuclear receptor proteins contain a central DNA binding domain (DBD) and a COOH-terminal ligand binding domain (LBD) (1). The DBD is composed of two highly conserved zinc fingers that target the receptor to specific promoter/enhancer DNA sequences (hormone response elements). The LBD is about 200–300 amino acids in length and is less well conserved. There are at least four functions encoded in the LBD: dimerization, ligand binding, binding of coactivators or corepressors, and transactivation. Transactivation can be viewed as a molecular switch between a transcriptionally inactive state and an activated state of the receptor. The COOH-terminal portion of the LBD contains an activation function domain (AF2) that is required for this switch (6–8).
Ligand-induced transactivation is mediated through interactions with members of a growing family of coactivator proteins including CREB-binding protein (CBP/p300) (6, 9), steroid receptor coactivator-1 (SRC-1)/nuclear receptor coactivator-1 (NcoA-1) (6, 10, 11), transcriptional intermediary factor 2 (TIF2)/glucocorticoid receptor interacting protein 1 (GRIP-1)/NcoA-2 (12, 13), and p300/CBP interacting protein (p/CIP) (14). Upon agonist binding, a conformational change in the LBD creates a coactivator binding surface; transcriptional activation occurs after recruitment of coactivator(s) to the receptor. This interaction is viewed as necessary and sufficient for receptor-mediated transcriptional activation. The binding of antagonist ligands to nuclear receptors results in a conformational state that does not allow coactivators to interact (and may also promote interactions with corepressor molecules). The coactivator CBP can form a bridge between nuclear receptors and basal transcriptional machinery (6). In addition, CBP and SRC-1 also contain intrinsic histone acetyltransferase activity that results in local chromatin rearrangement that is crucial for transcriptional activation (14–16). A ligand- and AF2-dependent interaction between selected nuclear receptors — retinoid X receptor (RXR), retinoic acid receptor (RAR), glucocorticoid receptor (GR), vitamin D receptor, thyroid receptor (T3R), and estrogen receptor α (ERα) — and CBP or SRC-1 has been demonstrated using in vitro glutathione-S-transferase (GST) pull-down experiments and with far-Western assessment of protein-protein interactions (6). Thus, AF2 mutants of T3R, ER, or RAR that impair the transcriptional function are also deficient with respect to coactivator interactions (6, 17). The important biological role of nuclear receptor coactivators was elegantly shown by Xu et al. (18) who characterized defects in steroid hormone action in SRC-1 null mice.
Using homogeneous time-resolved fluorescence (HTRF) technology (19), we developed a novel approach for characterizing the ligand-dependent protein-protein interaction between nuclear receptors and coactivators. This approach was employed to characterize differential effects of specific ligands for peroxisome proliferator-activated receptor-γ (PPARγ) and ERα on receptor function, to determine the requirement of the PPARγ AF2 domain for coactivator association, and to assess the affinity and relative specificity of these receptors for CBP vs. SRC-1.
RESULTS
HTRF-Based Measurement of Nuclear Receptor-Coactivator Interaction
HTRF technology relies upon europium cryptate, a lathanide (20). In the presence of appropriate chelates, lathanide ions display two remarkable characteristics: first, a pronounced Stoke’s shift with excitation and emission maxima often differing by as much as 300 nm (e.g. for the europium chelate,λ maxex = 307 nm,λ maxem = 620 nm); second, a long fluorescence lifetime, as long as 1 msec, or about 5 to 6 orders of magnitude longer than that of organic fluorophores, thereby allowing measurements to be carried out in a time-resolved mode (20, 21). These two properties result in extremely low backgrounds allowing assays of high sensitivity where quantities as small as tens of attomoles have been detected (22, 23). HTRF technology also incorporates a second fluorophore, XL665 (λmaxex = 620 nm,λ maxem = 665 nm), which mediates the fluorescence resonance energy transfer (FRET). An additional benefit of using lathanide in HTRF FRET measurements is that it has a large R0 value (R0, the distance at which 50% of the energy is transferred). The R0 for the Eu(K)-XL665 pair is about 90 Å. As a consequence, larger complexes can be detected, and the use of appropriately labeled antibody partners becomes much more facile. For the HTRF-based measurement of receptor-coactivator interaction, excitation of Eu(K) at 337 nm results in emission at 620 nm; XL665 can absorb the 620-nm emission if both molecules are close to one another (near or within R0), and subsequently emit at 665 nm. Therefore, FRET between the two fluorophores is a measure of the proximity of the molecules. The extent of the specific FRET is measured as a ratio of the emission intensity at 665 nm vs. that at 620 nm. This ratiometric readout provides additional advantages, in that 620 nm signal serves an internal standard, to minimize interference from variations due sample impurities (color, turbidity), probe concentrations, excitation intensity, and emission sensitivity. As a result, variation between multiple determinations is typically less than 2%.
To mimic the functional interaction between nuclear receptors and coactivators in an HTRF-based format, we used PPARγ-CBP, PPARγ-SRC-1, and ERα-SRC-1 as model systems (Fig. 1). The LBDs of either PPARγ or ERα, each of which is also predicted to contain the ligand-dependent coactivator-binding domain, were expressed in Escherichia coli (PPARγ) or yeast (ERα) and purified as GST-fusion proteins (GST-PPARγLBD and GST-ERαLBD) where the NH2 terminus of the LBDs was fused to the COOH terminus of GST. The nuclear receptor-binding domains of human CBP, CBP1−453 (amino acids 1–453), and human SRC-1, SRC568−780 (amino acids 568–780) were also expressed in Escherichia coli and biotinylated after purification. As shown in Fig. 1, GST-PPARγLBD or GST-ERαLBD was indirectly linked to Eu(K) through an anti-GST antibody, which was covalently liked to Eu(K). CBP1−453 or SRC568−780 was indirectly linked to XL665 through a streptavidin (SA)-biotin adapter. Thus, agonist-induced interaction between the nuclear receptor and its coactivator would bring the two fluorogenic partners together and result in the nuclear receptor ligand-dependent FRET. The stoichiometry of biotin labeling that resulted in maximal ligand-induced FRET was determined for each nuclear receptor-coactivator pair and was generally 2–2.5 biotins to one coactivator molecule.
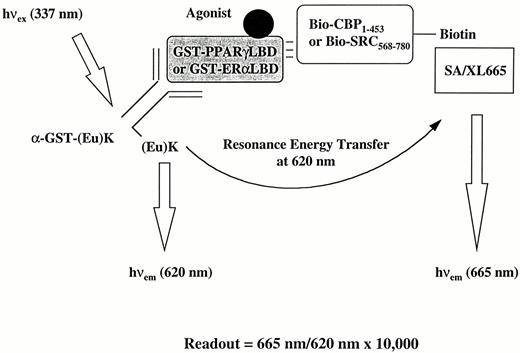
Strategy for Assessment of Nuclear Receptor-Coactivator Association Using the HTRF Approach GST-PPARγLBD or GST-ERαLBD proteins were indirectly linked to Eu cryptate, (Eu)K, through (Eu)K-labeled anti-GST antibody,α -GST-(Eu)K. Purified recombinant CBP1–453 or SRC568–780 proteins were biotinylated and indirectly linked to XL665 through XL665-labeled streptavidin (SA/XL665). The specific 665-nm signal of XL665 is produced only when there is a ligand-induced change in receptor conformation that results in binding to the coactivator. The extent of the FRET is measured as a ratio of 665 nm/620 nm × 10,000.
Known PPARγ Agonists Induce Dose-Dependent FRET Resulting from Interactions between PPARγ LBD and CBP or SRC-1
The antidiabetic thiazolidinediones (TZDs) have been shown to be activating ligands for PPARγ (24, 25). TZDs bind PPARγ with high affinity in vitro and can mediate transactivation of PPAR response element-luciferase reporter constructs in mammalian cells. We first established that TZDs can induce the interaction of PPARγLBD with GST-CBP1−453 using a standard pull-down approach (Fig. 2A); this demonstrated that PPARγLBD and nuclear receptor-binding domain of CBP were sufficient to mediate the agonist-dependent interaction. Figure 2, B and C, shows that, in an HTRF approach, TZDs induced a specific interaction between PPARγ-LBD and CBP1−453 or SRC568−780 in a dose-dependent manner. The EC50s obtained from this HTRF-based approach correlated very well with published IC50s from receptor-ligand binding-experiments and EC50s obtained via transactivation assays. Thus, the rank order of potency for these different compounds, TZDA > TZDB > TZDC = TZDD > TZDE, was preserved relative to published results (24–26). In addition, these effects correlate well with previously reported glucose-lowering potencies in diabetic animals (25). The differential potencies of these PPARγ ligands were also nearly identical when data derived using CBP vs. SRC-1 were compared. The naturally occurring, putative endogenous ligand of PPARγ, 15-deoxyΔ12,14 PGJ2 (27, 28) also induced the PPARγ-CBP interaction, whereas a related compound, PGJ A2, which is reportedly inactive as a PPARγ agonist, failed to mediate FRET between labeled PPARγ LBD and labeled CBP (Fig. 2D).
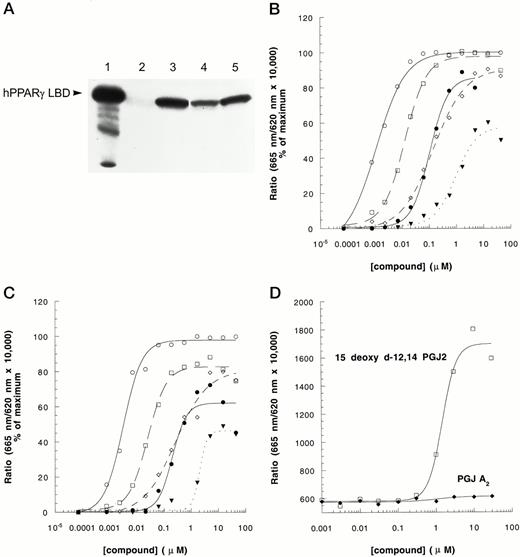
Induction of PPARγ-CBP and PPARγ-SRC-1 Interaction by Known PPARγ Agonists A, TZDs induced interaction between hPPARγLBD and GST-CBP1–453 in pull-down experiments. Purified hPPARγLBD (0.2 μg) was incubated with glutathione-bound GST-hCBP1–453 (1–2 μg) in the absence or presence of 1μ m of each TZD compound (lane 1, input; lane 2, dimethylsulfoxide; lane 3, TZDB; lane 4, TZDC; lane 5, TZDD). Bound hPPARγLBD was eluted and analyzed by Western blotting. Using the HTRF approach, effects of TZDs on PPARγ-CBP interaction (B), PPARγ-SRC-1 interaction (C), and effects of PGs on PPARγ-CBP interaction (D) were analyzed in the presence of 10 nm SA/XL665, 10 nm biotin-CBP1–453 or 10 nm biotin-SRC568–780, 1 nm GST-PPARγLBD, and 2 nm α-GST-(Eu)K as described in Materials and Methods. For panels B and C, the symbols are: ○, TZDA; □, TZDB; ⋄, TZDC; •, TZDD; ▾, TZDE. The experiment was repeated three times with similar results.
The PPARγ AF2 Domain Is Required for Transcriptional Activation and Coactivator Association
It has been shown for other nuclear receptors that AF2 function is required for both receptor-mediated transcriptional activation in cells and in vitro coactivator interaction. To ascertain whether the PPARγ AF2 domain is similarly required for transactivation and interactions with coactivators, a PPARγ mutant (E471A) was made, in which glutamic acid at position 471, located in a conserved AF2 core sequence, was substituted by alanine via site-directed mutagenesis. This mutant, as well as the wild-type receptor, was expressed in COS1 cells as fusion protein constructs containing the DNA-binding domain of the glucocorticoid receptor and the ligand-binding domains of human PPARγ: GR/PPARγLBD and GR/PPARγLBDE471A. As observed with other tested nuclear receptors, this mutant completely lost its ability to transactivate the reporter gene (Fig. 3A). Western blot analysis showed that the mutant chimeric receptor protein was expressed at a level similar to the wild-type chimer (data not shown). GST-PPARγLBD-E471A protein was subsequently expressed in Escherichia coli and purified. Using the HTRF assay, it was shown to have lost its ability to interact with both CBP and SRC-1 (Fig. 3B), indicating that ligand-mediated PPARγ interactions with both CBP and SRC-1 depend on the AF2 domain of the receptor.
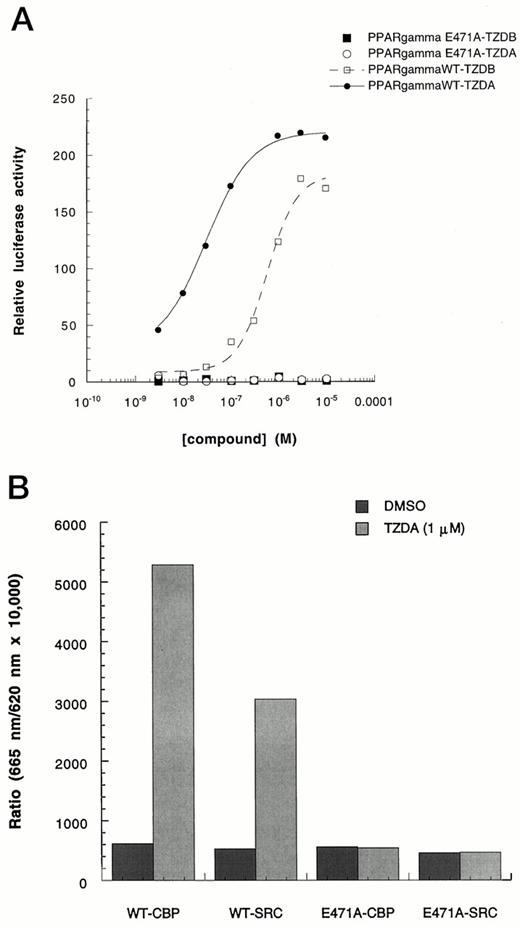
Requirement of PPARγ AF-2 Function for Transactivation and Coactivator Interaction A, Ligand-activated PPARγ/GR transactivation was abolished by an AF2 domain mutation. COS-1 cells were cotransfected with pSG5-hPPARγ/GR or pSG5-hPPARγE471A/GR, pMMTV/luc reporter, and pSV-β-galactosidase plasmids. Five hours after transfection, cells were treated with TZDA and TZDB for 36 h. Luciferase activity was then determined and normalized with β-galactosidase activity. Each point represents the mean of three determinations. B, Purified recombinant LBD protein corresponding to wild-type vs. PPARγE471A, 1 nm each, were compared using HTRF. Only wild-type PPARγ LBD was able to undergo ligand (TZDA)-induced association with either CBP or SRC-1. The experiment was repeated three times with similar results. Similar results were also obtained using 5 nm or 10 nm GST-PPARγLBD and GST-PPARγLBDE471A.
Functional Characterization of ER Ligands Using the HTRF Approach
To explore the possibility that the HTRF coactivator association strategy can be readily applied to other nuclear receptors, an analogous system was examined in which GST-ERαLBD was used (Fig. 1). As shown in Fig. 4, the ERαLBD-SRC-1 interaction was also ligand dose-dependent. The EC50s for 17β-estradiol (E2) and diethylstilbestrol were 0.25 nm and 0.6 nm, respectively, which correlates well with their reported potencies using ligand-receptor binding and transactivation assays (29, 30). 4-OH-tamoxifen and clomiphene have been shown to antagonize the activity of ER by binding to ER LBD and preventing agonist-mediated recruitment of coactivators (31, 32). Figure 4B shows that the E2-induced ERα-SRC interaction was effectively blocked by the addition of 4-OH-tamoxifen or clomiphene. The IC50s obtained using this approach were also similar to reported results obtained from binding determinations or functional antagonism in transactivation experiments (31, 32).
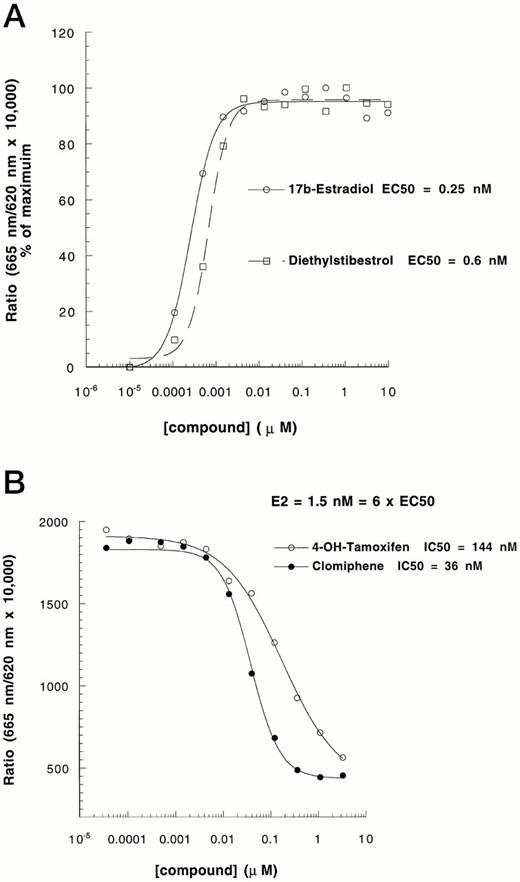
Effects of Known ER Agonists and Antagonists on ERβ-Coactivator Interaction A, The ability of known ER agonists (E2 and diethylstilbestrol) to induce an interaction between ERα and SRC-1 was tested following methods described in the legend to Fig. 2. B, The effects of known ER antagonists (4-OH-tamoxifen and clomiphene) on E2-induced ERα-SRC-1 interaction were analyzed in the presence of 1.5 nm E2 (6 × EC50). The experiment was repeated three times with similar results.
CBP and SRC-1 Interact with PPARγ or ERα with Different Affinities
Figure 5A shows that under the same experimental conditions, ligand binding induced the interaction of ERα only with SRC-1, whereas PPARγ interacted with both CBP and SRC-1. This finding may be the result of an intrinsic difference in affinity between each of these nuclear receptors and the two coactivators. The affinity between PPARγ or ERα and between CBP or SRC-1 was analyzed using saturating concentrations of potent agonists, TZDA for PPARγ and E2 for ERα. As shown in Fig. 5, B–D, unbiotinylated CBP or SRC-1 effectively competed for the TZDA-induced FRET with saturation kinetics. The PPARγ-CBP interaction showed a higher apparent affinity than PPARγ-SRC-1 interaction in both experimental paradigms, either using biotinylated-CBP or biotinylated-SRC-1. In contrast, unbiotinylated CBP had little effect on the ERα biotinylated-SRC-1 interaction, although unbiotinylated SRC-1 could compete efficiently with biotinylated-SRC-1. Further evidence of ERα specificity for SRC-1 vs. CBP was obtained from data depicted in Fig. 5E. In this experiment, unlabeled full-length ERα was used to compete for interactions of PPARγ with biotinylated-SRC-1 or -CBP. In the presence of E2, unlabeled ERα effectively competed with PPARγ for biotinylated-SRC-1 by disrupting TZDA-induced FRET. In contrast, increasing ERα concentrations did not affect the PPARγ-BioCBP interaction. These data show that under our experimental conditions, there was a distinct profile of preferred receptor-coactivator interactions, which follows a rank order of PPARγ-CBP1−453 = ERα-SRC-1568−780 > PPARγ-SRC-1568−780 ≫ ERα-CBP1−453.
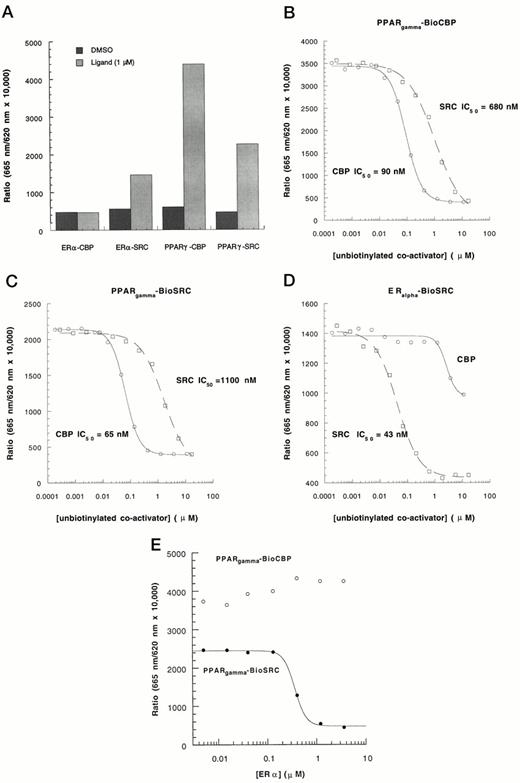
Analysis of the Interaction Affinities between Nuclear Receptor Coactivators by HTRF A, PPARγ interacts with both CBP and SRC-1 while ERα does not interact with CBP. Experimental conditions were the same as described in the legends to Figs. 2 and 4. The ligands (1 μm each) were E2 for ERα and TZDA for PPARγ. Unbiotinylated CBP and SRC-1 were used to compete for interactions of PPARγ with biotinylated-CBP (B), PPARγ with biotinylated-SRC-1 (C), and ERα with biotinylated-SRC-1 (D). E, Unlabeled full-length ERα was used to compete for interactions of PPARγ with biotinylated-CBP (○) and biotinylated-SRC-1 (•). The concentrations of other components are the same as described in legends to Figs. 2 and 4. The experiment was repeated three times with similar results.
DISCUSSION
Ligand binding promotes the association of nuclear receptors with a diverse group of universally expressed nuclear proteins, including CBP/p300, SRC-1/NcoA-1, and TIF2/GRIP-1NcoA-2, which are believed to function as coactivators by forming a bridge with basal transcriptional machinery and conferring a local increase in histone acetyltransferase activity. Thus, ligand-induced transcriptional activation of target genes by nuclear receptors involves the formation of a large protein complex on the receptor homo- (e.g. ER) or heterodimeric (e.g. PPAR/RXR) receptors bound to their cognate DNA response elements. A key role for coactivators is evident from studies that show that ligand-induced and AF2-dependent interactions between nuclear receptors and their coactivators (such as SRC-1 or CBP) are both necessary and sufficient for activation of transcription (6, 17). This in vivo functional interaction can be mimicked in the absence of a receptor heterodimer partner or a DNA response element by using isolated receptors in vitro in a GST pull-down approach, or via far-Western analysis, and using the yeast two-hybrid system (6, 17, 33).
The HTRF nuclear receptor strategy described here provides a novel quantitative tool for studying functional interactions of nuclear receptors with coactivator molecules (and possibly corepressors as well). The components of this system, purified recombinant nuclear receptors and nuclear receptor binding domains of coactivators, are simple compared with transcription complexes that exist within the nucleus of cells. We achieved further simplification by using only the LBDs of the PPARγ and ERα. However, we have not observed significant differences when comparing results obtained using full-length PPARγ2 rather than PPARγLBD (data not shown). Compared with other approaches for measuring nuclear receptor-coactivator interactions noted above, the HTRF-based strategy provides the first in vitro homogeneous method allowing for quantitation of such events under equilibrium conditions.
Importantly, the HTRF-based assessment of nuclear receptor activation was shown to be specific and predictive of in vivo receptor function, as evidenced by the following three sets of experimental results. First, EC50s of TZDs and ER agonists obtained using the HTRF approach correlated very well with other data available from ligand-binding and cell-based transactivation assays as well as with in vivo potencies in the case of TZD PPARγ agonists. In addition, the putative natural ligand for PPARγ, 15-deoxyΔ12,14 PGJ2, was fully active with an EC50 that reflects its reported activity using other approaches (27, 34), whereas PGJ A2, which is reported to be functionally inactive, was unable to induce FRET. Second, AF2 function, which is required for ligand-dependent function of nuclear receptors in cells, was shown to be necessary for transcriptional activation of a chimeric GR-PPARγLBD protein in transfected cells and for interaction of PPARγ with both tested coactivators in the cell-free HTRF context (Fig. 3). Third, two known ER antagonists, 4-OH-tamoxifen and clomiphene, failed to induce the association of ERα with SRC-1 and could efficiently block the E2-induced ERα-SRC-1 interaction (Fig. 4B). These effects occurred with half-maximal activities that could be predicted from their reported potencies as ligands and antagonists in cell-based assays (31, 32). It is interesting to note that different agonists of PPARγ resulted in different degrees of maximal FRET induction. As shown in Fig. 2, B and C, testing of TZDE repeatedly yielded 50–60% maximum FRET induced by TZDA, using either CBP or SRC-1; for other PPARγ ligands, distinct profiles of maximal FRET that differed from TZDA or TZDF were also observed using SRC-1 (Fig. 2C). Since FRET is a measure of the proximity of the receptor LBD and coactivator, saturating concentrations of individual ligands may have resulted in some variation in net proximity achieved. Alternatively, subtle changes in the receptor-coactivator affinity may underlie these differences. In either case, alternative conformational states adopted by PPARγ upon binding to structurally distinct ligands can be implicated. It is clear that functionally distinct classes of ER ligands (agonists vs. partial agonists vs. antagonists) exert alternative receptor conformations as measured by protease protection (35). Furthermore, we have observed that certain PPARγ ligands that appear to function as partial agonists in cell-based assays also exhibit reduced maximal FRET in the HTRF assay (J. Berger and G. Zhou, unpublished). Further understanding of potential variation in ligand-induced PPARγ conformation might provide a molecular basis for variation in biological responses to in vivo treatment with different agonists.
An important question concerns the extent to which different receptors (or different conformations of the same receptor in response to binding of alternative ligands) can differentially interact with subsets of the spectrum of coactivators. In other words, does signaling specificity reside at the level of direct interactions between receptors and coactivator? There appear to be clear differences in the ability of individual receptors to interact with corepressors; thus, unlike TR or RAR, PPARγ does not readily associate with nuclear receptor corepressor (N-CoR) (36). In addition, ligand-induced association of coactivators (SRC-1 and p140) with RXR is differentially regulated by the heterodimeric partner; this is permitted in the case of PPARγ/RXR but not in the case of RAR/RXR heterodimers (36). However, RAR was shown to actually block binding of ligands to RAR/RXR heterodimers. Therefore, the ability of any given receptor LBD to exhibit differential interactions with multiple coactivators has not been adequately explored.
Several investigators have reported that PPARγ exhibits a ligand-induced association with SRC-1 (11, 33, 36). Our data indicate that PPARγ can also be induced to strongly associate with CBP, suggesting an important role for both SRC-1 and CBP in PPARγ-mediated regulation of gene transcription. In addition, the results we obtained with either SRC-1 or CBP using several compounds from the two important classes of PPARγ ligands appeared to be nearly identical. CBP has been shown to be a universal coactivator or integrator required for many transcription factors, including nuclear receptors, AP-1 (6, 37), signal transducer and activator of transcription-1 (STAT-1) (38), cAMP-regulated enhancer binding protein (CREB) (39), and nuclear factorκ B (NF-κB). Since cellular CBP concentrations may be limiting, recruitment of CBP to PPARγ in response to agonist stimulation might antagonize the activity of other CBP-requiring transcription factors. This is a potential explanation for recently reported observations concerning the ability of 15-deoxyΔ12,14 PGJ2 and synthetic PPARγ ligands to inhibit macrophage activation and cytokine production (40, 41).
In contrast to results obtained with PPARγ, known ER agonists promoted the interaction of ERα with SRC-1 but not with CBP. Further characterization of the relative affinities of PPARγ and ERα for either coactivator revealed that PPARγ interacted with CBP with higher affinity than with SRC-1. Moreover, CBP1−453, which did not interact efficiently with ERα, was unable to compete for the ERα-SRC-1 interaction. The overall rank order of affinities was: PPARγ-CBP = ERα-SRC-1 > PPARγ-SRC-1 ≫ ERα-CBP. Poor interaction between a peptide motif derived from the nuclear receptor-binding motif of CBP with ERα has been documented using a yeast two-hybrid system (17). However, the pattern of potential coactivators that may interact directly with ERα has not been otherwise assessed. The fact that ERα does not interact with CBP with high affinity does not exclude a role for CBP in ERα function. CBP, via a discrete domain, constitutively interacts with the CBP-binding domain of SRC-1 (6). Thus, the high-affinity interaction of ERα with SRC-1 could result in CBP recruitment to the transcriptional complex. In this regard, it is interesting to note that RAR has been shown to bind to the NH2-terminal domain of CBP; however, this binding is not required for its function since a CBP NH2-terminal deletion mutant retained the ability to augment RAR activation (42).
It is clear that the differential capacity of certain nuclear receptor classes (such as TR and RAR) to interact with corepressors has important physiological consequences for in vivo repression of gene transcription. Recently reported data suggest that the degree to which receptors can associate with specific coactivators in response to ligand binding can serve to modulate the subsequent biological response. Thus, in HeLa cells in which 4-OH-tamoxifen functions as an ER antagonist, increasing the SRC-1 expression level was able to confer agonist activity upon this compound (31). The present studies provide a new and precisely quantitative approach for characterizing interactions between selected nuclear receptors and coregulators. Using this approach, we have characterized the molecular pharmacology of ligand-induced interactions between PPARγ or ERα and CBP or SRC-1. Clearly, interaction of these receptors with coactivators that occurs within a complex nuclear environment in vivo may differ from the observations reported here. However, our results suggest that differential affinity profiles may exist for any given combination of receptor and coactivator. Such differences may contribute to cell type-specific variation in biological responses that arise because of relative changes in the pattern of coactivator expression.
MATERIALS AND METHODS
Reagents
SA/XL665 and (Eu)K were from CIS Biointernational (Cedex, France) and Packard Instrument Company (Meriden, CT). The goat anti-GST antibody and glutathione Sepharose were from Pharmacia (Piscataway, NJ). SULFO-SMCC and SPDP were from Pierce (Rockford, IL). Dry milk was from Bio-Rad (Richmond, CA). The full-length recombinant human ER protein was purchased from ABR (Affinity Bioreagents, Inc., Golden, CO). The following six TZD compounds were kindly provided by Dr. Gerard Kieczykowski, Dr. Philip Eskola, Dr. Conrad Santini, Mr. Joseph F. Leone, and Mr. Peter A. Cicala (Merck Research Laboratories, Rahway, NJ): TZDA (AD5075) = 5-[4-[2-(5-methyl-2-phenyl-4-oxazoly)-2-hydroxyethoxy]benzyl]-2,4-thiazolidinedione; TZDB (BRL49653) = 5-(4-[2-[methyl-(2-pyridyl)amino]ethoxy]benzyl)thiazolidine-2,4-dione; TZDC (pioglitazone) = 5-[[4-[(5-ethyl-2-pyridyl)ethoxy]phenyl]-methyl]-2,4-thiazolidinedione; TZDD (troglitazone) = 5-[4-(6-hydroxy-2, 5, 7, 8-tetramethylchroman-2-yl-methoxy)Benzyl]-2,4-thiazolidinedione; and TZDE (englitazone) = 5-[(2-benzylbenzopyranyl)-6-methyl]-2,4-thiazolidinedione.
Plasmids
pGEXKG-PPARγLBD expressing GST fused with LBD (from amino acids 176 of PPARγ1 to 477) of human PPARγ was constructed by subcloning an XhoI-XbaI (XbaI was blunt-ended with T4 DNA polymerase) fragment of pSG5- hPPARγ/GR into pGEXKG (43) digested with XhoI and HindIII (HindIII site was blunt-ended with T4 DNA polymerase).
pGEXhCBP1–453, which expresses human CBP NH2-terminal 1–453 amino acids (hCBP1–453), was cloned by PCR. The primers for hCBP1–453 are: 5′-ACTCGGATCCAAGCCATGGCTGAGAACTTGCTGGACGG-3′ and 5′-CTCAGTCGACTTATTGAATTCCACTAGCTGGAGATCC-3′, and the expected DNA fragment is 1.5 kb. The template used for the PCR was human fetal brain cDNA libary (Stratagene, La Jolla, CA). The PCR-amplified 1.5 kb DNA fragment was digested with NcoI and HindIII and ligated with pGEXKG expression vector digested with NcoI and HindIII to generate pGEXhCBP1−453.
pGEXhSRC568−780 expressing GST fused with a human SRC-1 fragment containing amino acids 568–780 was prepared by subcloning an SRC-1 PCR fragment derived from the human fetal brain cDNA libary. The primers are: 5′-ACTCGGATCCAATTCACCTAGCAGATTAAATATACAACC-3′ and 5′-CACATCT-AGATTACTGTTCTTTCTTTTCGACTTTCACC-3′. The 0.6-kb fragment was then digested with BamHI and XbaI and subcloned into the pGEX-4T-1 vector (Pharmacia).
pSG5-hPPARγ/GR, which contains human PPARγLBD fused to murine GRDBD, was provided by Dr. Azriel Schmidt (Merck Research Laboratories). pMMTV/luc, which contains the murine mammary tumor virus (MMTV) promoter adjacent to the luciferase (luc) gene, was also provided by Dr. Azriel Schmidt.
pESP1ERαLBD expressing GST fused with the LBD (amino acids 302–595) of human ERα was constructed by performing PCR on a human ERα clone using primers HW234 (5′-CGCGGATCCAAGAACAGCCTGGCCTTGTCCCTG-3′) and HW209 (5′-TCAGACTGTGGCAGGGAAACCCTCTGCCTCCCCCGTGATG-3′). The product was subcloned into pTGEM (Promega, Madison, WI) and sequenced by PCR (Pharmacia) and subsequent gel electrophoresis (Stratagene Castaway System). The clone was digested with BamHI (site introduced by HW234) and SpeI (blunt ended using Klenow DNA polymerase) and cloned into pESP-1(Stratagene) cut with BamHI and SmaI.
Preparation of GST Fusion Proteins from E. coli
E. coli strain DH5α (GIBCO BRL, Gaithersburg, MD) served as a host for either pGEXhCBP1–453 or pGEXhSRC568–780 and BL21 (Stratagene) for pGEXhPPARγLBD. DH5α or BL21 was cultured in LB medium (GIBCO BRL) to a density of OD600 0.7–1.0 and induced for overexpression by addition of IPTG (isopropylthio-β-galactoside) to a final concentration of 0.2 mm. The IPTG-induced cultures were grown at room temperature for an additional 2–5 h. The cells were harvested by centrifugation for 10 min at 5000 × g. The cell pellet was used for GST-fusion protein purification according to the recommended procedure from Phamacia Biotech using glutathione Sepharose beads. hCBP1–453 and hSRC568–780 proteins were generated by cleaving the corresponding GST fusion proteins with thrombin.
Purification of GST-ERα LBD from Yeast
The LBD of ERα was expressed in yeast as a GST fusion protein. Yeast strains were grown in fermentors at 28 C. The cell pellet (0.8 g/ml) was resuspended in TEGM [10 mm Tris, pH 7.2, 1 mm EDTA, 10% glycerol, 0.7% β-mercaptoethanol, 1 mm dithiothreitol (DTT), 10 mm sodium molybdate] containing one pellet/2 ml (40 μl/ml) Protease inhibitor cocktail PI (Sigma, St. Louis, MO; 50 μg/ml antipain, 0.7 μg/ml pepstatin, 2 μg/ml benzamidine), and lysed by shaking with glass beads in a bead beater. Unbroken cells were removed by low-speed centrifugation at 3000 × g for 10 min. The supernatant was centrifuged at 100,000 × g for 60 min yielding about 1 ml of cell extract per gram of cells. One milliliter of extract was allowed to bind 1 h at 4 C to 2 ml of glutathione-Sepharose 4B (Pharmacia) in TEGM containing 5 mm DTT, 0.1% Triton X-100 (Sigma), and PI cocktail. The beads were washed extensively with PBS containing PI cocktail and finally eluted with the following buffer: 50 mM Tris, pH 8.0, 5 mm DTT, 0.1% Triton, 0.1% BSA, 0.1 m sodium chloride, PI, and 20 mm glutathione.
Preparation of Eu(K)-α-GST
The anti-GST antibody (500 μg, 3.33 nmol) at 5 mg/ml in 150 mm NaCl-0.02% azide was buffer exchanged into PBS, pH 6.8, with a BioSpin-30 (Bio-Rad, Richmond, CA) desalting column. To this was added 0.9 μl of a 9.4 mg/ml solution of SPDP in absolute ethanol (8.3 μg, 26.7 nmol, 8.0 eq), and the reaction was allowed to stand at room temperature for 30 min. The thiopyridine moiety was then cleaved by addition of 5 μl of 400 mm DTT in H2O (final concentration, 20 mm). After 15 min at room temperature, removal of excess DTT and buffer exchange into 50 mm Pi, 5 mm EDTA, pH 6.5, was achieved by increasing the total sample volume to 200 μl with PBS and then passaging through BioSpin 30 columns. To this was added activated Eu(K) that had been prepared by reacting 7.8 μl of a 2.5 mg/ml solution of Eu(K) in 10% DMF/PBS (19.6 μg, 13.4 nmol, 4 eq) with 1.3μ l of a 10 mg/ml solution of Sulfo-SMCC in H20 [13.0μ g, 29.8 nmol, 2.9 eq with respect to Eu(K)] at 4 C for 30 min. The reaction was allowed to stand at 4 C overnight.
Purification was achieved by a two-step process. First, the material taken up to a total volume of 500 μl with PBS and buffer exchanged into PBS, pH 6.8, with a Nap-5 column. Subsequently, the protein was further purified by size exclusion chromatography using a BioSep-S2000 column coupled with a Waters 625 HPLC system using the same buffer. The materials were then frozen at −70 C until needed.
HTRF Assay Protocol
SA/XL665 was used as supplied by the manufacturer. Reaction conditions were as follows: 198 μl of reaction mixture [100 mm HEPES, 125 mm KF· 0.125% (wt/vol) 3-[(3-cholamidopropyl) dimethyl-ammonio]-1-propanesulfonate (CHAPS), 0.05% dry milk, 1 nm GST-PPARγLBD or GST-ERβLBD, 2 nm anti-GST-(Eu)K, 10 nm biotin-CBP1–453 or biotin-SRC568–780, 20 nm SA/XL665] were added to each well, followed by addition of 2 μl test sample in appropriate wells. Plates were mixed by hand and covered with TopSeal. The reaction was incubated overnight at 4 C, followed by measurement of fluorescence reading on a Discovery instrument (Packard). One 96-well plate requires about 3 min. The fluorescence signal is stable for at least 3 days at 4 C. Data were expressed as the ratio, multiplied by a factor of 104, of the emission intensity at 665 nm to that at 620 nm. The ratios were directly used for plotting to obtain EC50 values. Alternatively, the ratio obtained from each tested well was normalized by subtracting results obtained from control (dimethylsulfoxide) wells, followed by calculation of percent maximum activation (using maximal ligand-induced FRET as 100%).
Transient Transfections
COS-1 cells were transfected with plasmid DNA constructs using a standard lipofectamine method (GIBCO/BRL). COS-1 cells were seeded in 96-well plates at 1.2 × 104 cells per well in DMEM containing 10% charcoal stripped FCS for 16–20 h before transfection. Cells were cotransfected with pSG5-hPPARγ/GR wild-type or pSG5-hPPARγE471A/GR mutant, pMMTV/luc reporter construct, and pSV-β-galactosidase as internal standard for variation in transfection efficiency and sample toxicity. The DNA-lipid complex was removed after 5 h. The cells were exposed to test compounds in DMEM containing 5% charcoal-stripped FCS. Cell lysates were prepared with Reporter Lysis Buffer (Promega, Madison, WI). Luciferase activity in cell extracts was determined using Luciferase Assay Buffer (Promega) in a ML3000 Luminometer (Dynatech Laboratories, Chantilly, VA).
GST Pull-Down Assay
GST-hCBP1–453 protein (1–2 μg) bound to glutathione Sepharose (10 μl) was incubated with 0.2 μg of purified hPPARγLBD in 100 μl Buffer A [8 mm Tris, pH 7.4, 120 mm KCl, 8% glycerol, 0.5% (wt/vol) CHAPS, 1 mg/ml BSA] for 12–16 h at 4 C. Samples were pelleted by centrifugation at 11,000 × g for 20 sec and washed four times with 300μ l cold Buffer A. The samples were then suspended in 20 μl Laemmli sample buffer, heated for 5 min at 100 C, and electrophoresed in 4–20% SDS-polyacrylamide gels. Electrophoretically separated proteins were electroblotted onto polyvinylidene difluoride (PVDF) membrane. The membrane was blocked in TBST buffer (10 mm Tris, pH 8.0, 150 mm NaCl, 0.2% Tween 20) containing 5% dry milk for 1 h at room temperature. The primary rabbit antihuman PPARγLBD antibody was raised against purified recombinant hPPARγLBD. Primary antibody incubation was performed in TBST buffer containing 5% dry milk at 4 C for 12–16 h. Secondary antibody incubation and chemiluminescent detection with enhanced chemiluminescence (ECL) (Amersham, Arlington Heights, IL) were performed as described in instructions provided by the manufacturer.
Acknowledgments
We thank Drs. Joel Berger and Mark D. Leibowitz for helpful discussions. We appreciate the preparation of TZDA, TZDB, TZDC, TZDD, and TZDE by Dr. Gerard Kieczykowski, Dr. Philip Eskola, Dr. Conrad Santini, Mr. Joseph F. Leone, and Mr. Peter A. Cicala (Merck Research Laboratories, Rahway, NJ). We thank Dr. Azriel Schmidt (Merck Research Laboratories) for plasmids pSG5-hPPARγ/GR and pMMTV/luc. We also thank Dr. Barbara Leiting for providing a protein purification procedure. We gratefully acknowledge the excellent technical assistance of Sheng-Jian Cai and Gretel Salzmann.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}