





Abstract
SHP (short heterodimer partner) is an unusual orphan receptor that lacks a conventional DNA-binding domain. Previous results have shown that it interacts with several other nuclear hormone receptors, including the retinoid and thyroid hormone receptors, and inhibits their ligand-dependent transcriptional activation. Here we show that SHP also interacts with estrogen receptors and inhibits their function. In mammalian and yeast two-hybrid systems as well as glutathione-S-transferase pull-down assays, SHP interacts specifically with estrogen receptor-α (ERα) in an agonist-dependent manner. The same assay systems using various deletion mutants of SHP map the interaction domain with ERα to the same SHP sequences required for interaction with the nonsteroid hormone receptors such as retinoid X receptor and thyroid hormone receptor. In transient cotransfection assays, SHP inhibits estradiol -dependent activation by ERα by about 5-fold. In contrast, SHP interacts with ERβ independent of ligand and reduces its ability to activate transcription by only 50%. These data suggest that SHP functions to regulate estrogen signaling through a direct interaction with ERα.
INTRODUCTION
The nuclear hormone receptor superfamily is a group of transcription factors that regulate target genes in response to small hydrophobic compounds such as steroids, thyroid hormone, and retinoids. The members of this superfamily play a variety of important roles in development and differentiation by regulating transcription of specific target genes (1–4). In general, these proteins bind DNA with high affinity as dimers. The majority of the type II or nonsteroid receptors, such as the thyroid hormone receptor (TR), retinoic acid receptor (RAR), or the orphan receptors, require heterodimerization with the retinoid X receptor (RXR) for high-affinity binding to their response elements (2). In contrast, the steroid receptors are generally thought to bind to their response elements exclusively as homodimers (e.g. Ref. 1), although the recently described ERβ isoform (5, 6) suggests the possibility of additional interactions.
Estrogen receptor (ER) regulates gene expression in female reproductive tissues in response to estrogen and is an important target in breast cancer therapy, as more than half of breast cancers express high levels of ER and depend on estrogen for growth. Although the ERα and ERβ isoforms recognize the same response elements, ERβ binds estrogen with a lower affinity than ERα and shows weaker transactivation activity (7). In addition, ERβ may activate a different spectrum of genes in response to certain ligands (8). Both the overlapping expression patterns of ERα and β in various tissues and the possibility of formation of ERα-ERβ heterodimers have complicated the understanding of gene regulation by estrogen. Furthermore, recent studies suggest that ER function may be modulated by cyclin D1 (9, 10) and that epidermal growth factor, transforming growth factor-α, and dopamine (11) elicit estrogen-independent, ER-dependent gene transcription of estrogen-responsive genes. Thus, ERs may have functional interactions with a range of other signaling molecules.
SHP (short heterodimer partner) was initially isolated by yeast two-hybrid screening (12) based on its interaction with mCAR (13), the murine homolog of the orphan receptor MB67 (14). Isolation of full-length cDNA clones revealed that SHP, like the orphan DAX-1 (15), lacks a conventional DNA-binding domain (12). Both direct biochemical analysis and results from the yeast two-hybrid system demonstrated that SHP can interact with the thyroid hormone receptor (TR), RAR, RXR, and other members of the receptor superfamily. In addition, SHP inhibits in vitro DNA binding by RAR/RXR heterodimers. In mammalian cell cotransfections, SHP also inhibits transactivation by RAR, mCAR, and other receptor superfamily members with which it interacts (12). These results suggest that the major physiological role of SHP is to repress signaling by other receptor superfamily members. Additional support for this hypothesis was provided by the recent demonstration that SHP itself can act as a direct transcriptional repressor (16). The receptor interaction and repression domains of SHP were mapped to the central region and carboxyl terminus, respectively, by both mammalian and yeast two-hybrid assays (16).
We have examined the potential role of SHP in estrogen signaling. Here we report that SHP interacts with ERα, both in vitro and in yeast and mammalian two-hybrid systems in a ligand-dependent fashion. As expected from its functional effects with other receptors, SHP inhibits transactivation by ERα induced by estrogen. Thus we propose that SHP is a novel inhibitor of ERα activity and may play a role in modulating cellular responses to estrogen.
RESULTS
SHP Interacts with ER
We have shown previously that SHP interacts with RXR and several of its heterodimer partners, such as RAR, TR, and mCAR, using two-hybrid, biochemical, and functional assays (12, 16). Unexpectedly, the same approaches demonstrate that SHP also interacts with human estrogen receptor α (ERα). In yeast, ERα interacts with full-length SHP (Table 1). The pattern of interaction of ER with a series of SHP deletion mutants (Fig. 1) previously used to identify SHP sequences required for interaction with other receptors (16) is quite similar to that described for RXR, although interaction of ER with theΔ E1 and W160X mutants is more ligand dependent (Table 1). These results identify SHP residues 72–148 as the minimal ER interaction domain. This same region is sufficient for interaction with RXR, TR, RAR, and mCAR (16).
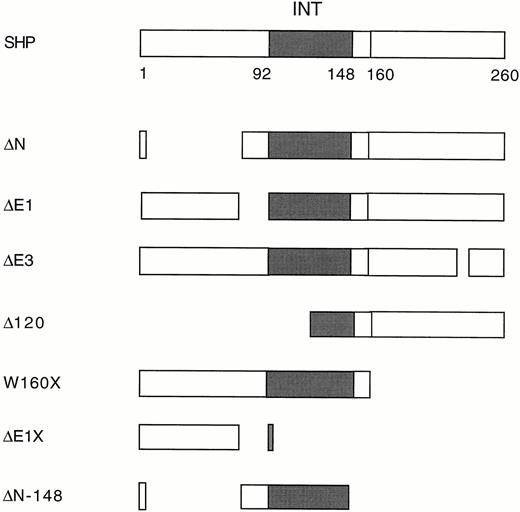
Deletion Mutants of SHP The ER-interacting domain is indicated by a solid box. The deletion constructs were previously described (16 ).
Interaction of Full-Length SHP and Mutants with ER in Yeast
| Lex− | Lex-SHP | |||||
|---|---|---|---|---|---|---|
| WT | ΔE1 | ΔN-148 | ΔE1X | W160X | ||
| B42− | − | − | − | − | − | − |
| −ERα | −/− | +/++ | −/+ | +/++ | −/− | −/++ |
| −RXRα | −/− | ++/++ | +/++ | ++/++ | −/− | ++/++ |
| Lex− | Lex-SHP | |||||
|---|---|---|---|---|---|---|
| WT | ΔE1 | ΔN-148 | ΔE1X | W160X | ||
| B42− | − | − | − | − | − | − |
| −ERα | −/− | +/++ | −/+ | +/++ | −/− | −/++ |
| −RXRα | −/− | ++/++ | +/++ | ++/++ | −/− | ++/++ |
++, Strongly blue colonies after 1 day of incubation and strong interaction; +, light blue colonies after 1 day incubation and weak interaction; −, white colonies and no interaction.
For RXR and ER, 100 μl of a 10−6m stock of the appropriate ligand was added to the plate before plating, as previously described (30). Results are indicated in the absence and presence of ligand (−/+). The LBD of human ERα and human RXRα were used for construction of B42 fusions.
Interaction of Full-Length SHP and Mutants with ER in Yeast
| Lex− | Lex-SHP | |||||
|---|---|---|---|---|---|---|
| WT | ΔE1 | ΔN-148 | ΔE1X | W160X | ||
| B42− | − | − | − | − | − | − |
| −ERα | −/− | +/++ | −/+ | +/++ | −/− | −/++ |
| −RXRα | −/− | ++/++ | +/++ | ++/++ | −/− | ++/++ |
| Lex− | Lex-SHP | |||||
|---|---|---|---|---|---|---|
| WT | ΔE1 | ΔN-148 | ΔE1X | W160X | ||
| B42− | − | − | − | − | − | − |
| −ERα | −/− | +/++ | −/+ | +/++ | −/− | −/++ |
| −RXRα | −/− | ++/++ | +/++ | ++/++ | −/− | ++/++ |
++, Strongly blue colonies after 1 day of incubation and strong interaction; +, light blue colonies after 1 day incubation and weak interaction; −, white colonies and no interaction.
For RXR and ER, 100 μl of a 10−6m stock of the appropriate ligand was added to the plate before plating, as previously described (30). Results are indicated in the absence and presence of ligand (−/+). The LBD of human ERα and human RXRα were used for construction of B42 fusions.
The possibility of a direct interaction of SHP with ER was tested biochemically. As indicated in Fig. 2A, [35S]methionine-labeled SHP showed 17β-estradiol-dependent interaction with a glutathione-S-transferase (GST)-human ERα ligand-binding domain (LBD) fusion protein (lanes 2–4). This SHP-ER interaction was as strong as or stronger than interaction of SHP with full-length RXR (lanes 3–6). An analogous, although somewhat weaker, ligand-dependent interaction was also observed between a GST full-length SHP fusion and[ 35S]methionine-labeled full-length hERα (data not shown). The interaction of rat ERβ (rERβ) and SHP was also examined using GST-rERβ LBD and [35S]methionine-labeled SHP. A specific, but ligand-independent and relatively weak interaction was observed (data not shown). In contrast to these results with ER isoforms, a GST-human androgen receptor LBD (GST-hAR) fusion protein did not interact with SHP in either the presence or absence of its ligand (Fig. 2A, lanes 7 and 8), and GST-SHP also showed no interaction with radiolabeled full-length hAR (data not shown).
![In vitro Interaction of GST-Receptor Fusion Protein with SHP Full-length murine SHP and various mutants (Fig. 1) were radiolabeled with [35S]methionine by in vitro translation with pT7lacHisMycSHP. A, Specific interaction of SHP with GST-human ERα. GST-ERα LBD, -AR LBD, and -RXRα full-length proteins were expressed in E. coli BL21 strain and tested for interaction with SHP in the absence and presence of their cognate ligands (25 ). For ligands, 1 μm of 17β-estradiol, 9-cis-retinoic acid, and R1881 were used for ER, RXR, and AR, respectively, and indicated as +. SHP did not bind to GST protein. About 20% of SHP used in each lane was shown as INPUT (lane 1). B, Effect of 4-OHT on interaction of SHP with ER. GST-human ERα LBD fusion protein was incubated in the absence or presence of 10 nm (indicated as 1) or 1 μm (indicated as 100) 17β-estradiol (E2). To test whether SHP can interact with ER in the presence of a partial agonist, 1μ m 4-OHT was incubated with or without 10 nm estradiol. About 20% of radiolabeled SHP used in each lane was shown as INPUT (lane 1). c, Mapping of ER-interacting domain of SHP. SHP products were tested for interaction with a GST fusion to LBD of human ERα in the absence and presence of 1 μm 17β-estradiol. Tests of SHP proteins with GST alone did not show any specific interaction.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/mend/12/10/10.1210_mend.12.10.0184/2/m_mg1080184002.jpeg?Expires=1750528028&Signature=QgBNlgUrXUbugZMxlXwHKt~PpD6NzCag5qwuKBLQPb~U7s9gc5nj63zoDPIRLobW0swDY1JL3x6E8rYoeYAlV7t6ajRqItMvHOk49K-lZZht3-D8dNghJvZUXIB5Dwz9PNH8nZ6jXbyMr6pSatIrxv~8CPvAsL9FH8feM1qHaQo1XmtpihK17JkkArK95B~XoKRtKUaaFV5a42yjGMh124Uo0b2V~GLETRYzC-swL5Mo7qizsIhZHwkWheHdMEnQDwqwl1sGfzBt4pAwBop5Eesf-5DRg15NPj1P1St65azJq1W~bZvgeDvm9uo8YH7fxW8evb-~ZBZPJH4b~M044w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
In vitro Interaction of GST-Receptor Fusion Protein with SHP Full-length murine SHP and various mutants (Fig. 1) were radiolabeled with [35S]methionine by in vitro translation with pT7lacHisMycSHP. A, Specific interaction of SHP with GST-human ERα. GST-ERα LBD, -AR LBD, and -RXRα full-length proteins were expressed in E. coli BL21 strain and tested for interaction with SHP in the absence and presence of their cognate ligands (25 ). For ligands, 1 μm of 17β-estradiol, 9-cis-retinoic acid, and R1881 were used for ER, RXR, and AR, respectively, and indicated as +. SHP did not bind to GST protein. About 20% of SHP used in each lane was shown as INPUT (lane 1). B, Effect of 4-OHT on interaction of SHP with ER. GST-human ERα LBD fusion protein was incubated in the absence or presence of 10 nm (indicated as 1) or 1 μm (indicated as 100) 17β-estradiol (E2). To test whether SHP can interact with ER in the presence of a partial agonist, 1μ m 4-OHT was incubated with or without 10 nm estradiol. About 20% of radiolabeled SHP used in each lane was shown as INPUT (lane 1). c, Mapping of ER-interacting domain of SHP. SHP products were tested for interaction with a GST fusion to LBD of human ERα in the absence and presence of 1 μm 17β-estradiol. Tests of SHP proteins with GST alone did not show any specific interaction.
As 4-hydroxy-tamoxifen (4-OHT) acts as a partial ERα agonist in liver, where SHP is highly expressed, the effect of 4-OHT on the in vitro interaction between SHP and ERα was examined. As shown in Fig. 2B, SHP did not bind to ER in the presence of 1μ m 4-OHT (lane 6). Moreover, addition of 1μ m of 4-OHT abolished the hormone-dependent interaction observed in the presence of 10 nm estradiol (lane 5). These results indicate that SHP specifically recognizes the estrogen-bound form of ERα.
Various deletion mutants of SHP were used to confirm the mapping of the ER interaction domain of SHP (Fig. 2C). Consistent with the results observed in yeast, the pattern of in vitro interactions was almost identical to that reported for RXR (16). SHP-ER interaction was retained with several SHP mutants, including W160X, which lack the I-box or the 9th heptad, as well as internal deletions of either this region (ΔE3), or of the more highly conserved signature motif located near the N terminus of the LBD (ΔE1). The SHP N-terminal deletionΔ 120 did not show specific interaction in vitro with GST-ER, relative to GST alone. The smallest protein retaining specific in vitro interaction was the ΔN-148 mutant, as observed in yeast (Fig. 2C and Table 1).
To further confirm these results, the interaction of SHP with ERα was tested using the mammalian two-hybrid system, with GAL4-hERα LBD and VP16-SHP fusion constructs. Cotransfection of GAL4-ER with VP16 alone in HepG2 cells showed approximately 5-fold activation by 17β-estradiol relative to cotransfection of both empty vectors, GAL4 and VP16. However, cotransfection of VP16-SHP fusion with GAL4-ER LBD increased transactivation to more than 35-fold, or 7-fold greater than that of the GAL4-ERα LBD with VP16 alone (Fig. 3). Very similar results were obtained in transfections using U2-OS cells (data not shown).
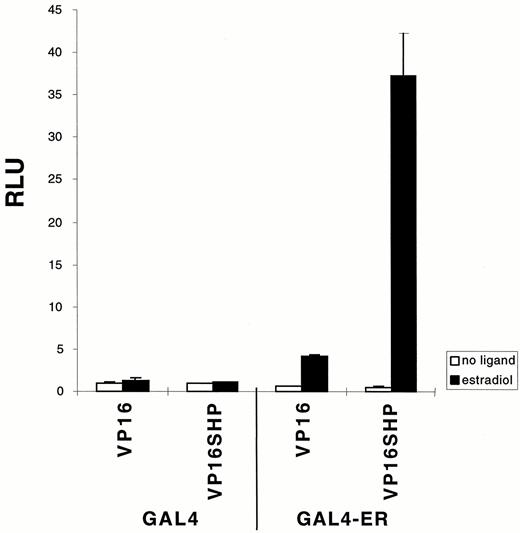
Specific Interaction of ER with SHP in HepG2 Cells by a Mammalian Two-Hybrid Assay GAL4-human ERα LBD (0.1 μg/well) and VP16-SHP (0.5 μg/well) were cotransfected by diethylaminoethyl-dextran method (28 ) with TKGH (0.5 μg/well) and luciferase reporter plasmid containing three copies of GAL4 DNA-binding element (0.5 μg/well) into HepG2 cells grown in 12-well plates. Fresh media containing either 1 μm 17β-estradiol or ethanol were added about 16 h after DMSO shock. Results are expressed relative to luciferase expression with empty vectors, GAL4 alone plus VP16, in the absence of ligand. The data were reproducible, and a similar pattern was obtained with transfection of U2-OS cells.
Overall, we conclude that SHP specifically interacts with ER in a ligand-dependent manner, as demonstrated by both yeast and mammalian two-hybrid systems, and by in vitro assays.
SHP Inhibits Transactivation by ER
SHP inhibits transactivation by RAR, mCAR, and other nuclear receptors (12). Thus, the effect of SHP on transactivation by human ERα was tested in transient cotransfections using a luciferase reporter containing three copies of the vitellogenin estrogen response element (ERE). As shown in Fig. 4, A and B, this reporter was transactivated by approximately 30-fold by ERα in U2-OS cells. However, this transactivation was decreased in a dose-dependent manner by SHP coexpression, with only approximately 20% of the response remaining with a 10-fold molar excess of SHP expression vector (Fig. 4B). This level of inhibition is similar to that previously observed with mCAR and RXR, and stronger than that with RAR (12). As expected, a similar inhibitory effect of SHP on ERα was also observed in HepG2 cells (data not shown). Consistent with the relatively weak, ligand-independent interaction of SHP with ERβ observed in vitro, SHP coexpression decreased ERβ transactivation by approximately 50% in either the presence or absence of ligand (Fig. 4A).
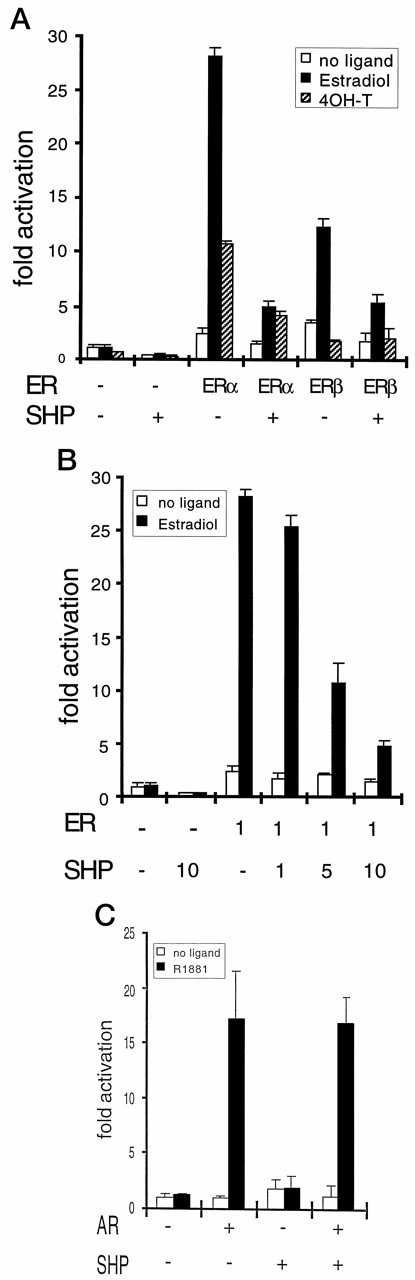
Specific Inhibition of ERα by SHP A, Inhibition of transactivation by ER with SHP. Plasmids containing human ERα and rat ERβ1 in pCDNA3 vector (25 ng/well) and SHP in CDM8 vector (250 ng/well) were cotransfected with TKβ-gal (125 ng/well) and ERETKluciferase (250 ng/well), a luciferase reporter plasmid containing three copies of vitellogenin ER response element, into U2-OS cells grown in 24-well plates. Results are expressed relative to luciferase activity with pCDNA3 alone without 17β-estradiol. The data were reproducible, and a similar pattern was obtained with transfection of HepG2 cells. B, Dose-dependent inhibition of SHP. Plasmids containing human ERα in pCDNA3 vector (25 ng/well) were cotransfected with 1-, 5-, or 10-fold concentration of SHP in CDM8 vector (25, 125, or 250 ng/well, respectively) in addition to TKβ-gal (125 ng/well) and ERETKluciferase (250 ng/well) into U2-OS cells grown in 24-well plates. Results are expressed relative to luciferase activity with pCDNA3 alone without 17β-estradiol. The data were reproducible and a similar pattern was obtained with transfection of HepG2 cells. c, No effect of SHP on transactivation by AR. Plasmids containing human AR with SV 40 promoter (0.05 μg/well) and SHP in CDM8 vector (0.5 μg/well) were cotransfected with TKβ-gal (0.25 μg/well) and luciferase reporter plasmid MMTV promoter (0.5 μg/well) into U2-OS cells grown in 24-well plates. Results are expressed relative to luciferase activity with empty vector alone without a ligand of AR, R1881. In all experiments, 1μ m of appropriate cognate ligands were added about 16 h after transfection, and cells were incubated for one more day before harvest for assays.
4-OHT acted as a partial agonist with ERα but not ERβ in U2-OS cells. Somewhat unexpectedly, SHP also decreased this activation of ERα, although this effect was much weaker than that observed with estradiol- activated ERα. The basis for this inhibition is unknown, but it suggests that the partial agonist activity of 4-OHT in these cells may be due, in part, to a weak, but direct, activation of the ER LBD.
In striking contrast to the results with ERα, SHP showed no effect on transactivation of the mouse mammary tumor promoter (MMTV) promoter by the AR (Fig. 4C). This lack of effect is consistent with the lack of apparent in vitro interaction between SHP and AR (Fig. 2A) and clearly demonstrates that SHP is not a general inhibitor of all members of the nuclear hormone receptor superfamily.
DISCUSSION
The results described here confirm and significantly extend previous indications that SHP can interact with a number of nuclear receptor superfamily members (12, 16). Based on the presumption that the type I steroid receptors act as homodimers (notwithstanding the ability of ERβ to heterodimerize with ERα) the addition of ERα to the list of SHP partners is surprising.
At least superficially, the interactions of SHP with its partners appear similar to those of RXR with its partners, but with opposite consequences. In contrast to RXR, SHP generally inhibits transactivation by the superfamily members with which it interacts. Direct interaction with SHP also blocks, rather than stimulates, DNA binding by RAR/RXR heterodimers and other SHP targets (12). This latter finding led to the suggestion that the inhibitory effects of SHP are a simple consequence of the inhibition of DNA binding (12). However, further studies have demonstrated additional complexities in the interactions of SHP with its partners. Thus, the mapping results described here confirm the previous conclusion that the interaction of SHP with its partners involves sequences quite distinct from the more C-terminal subdomain that is involved in both ER homodimerization (17, 18) and RXR heterodimerization (19). When combined with the additional demonstration that SHP has the capacity to act directly as a transcriptional repressor (16), a somewhat more complex alternative mechanism for its inhibitory effects is suggested. In this model, inhibition could be a consequence of the recruitment of the SHP repressor activity to response elements via a novel SHP-receptor interaction, which would differ significantly from standard receptor homodimer or heterodimer interactions.
This latter model may account for the inhibitory effect of SHP on ERα. Steroid receptors have a potent dimerization function associated with their DNA-binding domains and are able to bind their response elements and transactivate even in the absence of their LBDs. Thus, based on the well-documented modular nature of the ER, interaction of SHP with the ER LBD should not preclude the homodimerization of the DNA- binding domain, and therefore should not block DNA binding. Indeed, we have so far been unable to demonstrate an inhibitory effect of SHP on DNA binding by ERα in vitro using conditions in which binding by RAR/RXR heterodimers is strongly decreased. While this negative result may not reflect the results in vivo, it is most consistent with the model in which the inhibition of ERα transactivation by SHP is a consequence of the action of the SHP repressor domain, rather than an inhibition of DNA binding. This model is also supported by cotransfection results with SHPW160X, which contains the interaction domain but not the repression domain and shows little inhibitory effect on ERα transactivation (data not shown). At least one of its predictions has not been borne out, however, since we have been unable to demonstrate the existence of a multimeric DNA-ER-SHP complex using standard gel shift approaches. The similar failure to demonstrate an analogous DNA-SF1-DAX-1 complex (20) suggests either that the interactions of SHP (or the closely related DAX-1) with their receptor targets may be rather transient, or that other technical issues may preclude the identification of such complexes in vitro.
Regardless of the mechanism of the inhibitory effect, it could have a significant impact on ERα function. SHP is most abundantly expressed in liver and adrenal gland, with lower expression in other tissues (Refs. 12, 21 and H-S. Choi, unpublished data). In contrast, ERα has generally been considered to function in reproductive tissues such as uterus and breast. However, ERα is also active in liver, where its expression is regulated by GH and various ligands of the nuclear hormone receptors such as T3 and dexamethasone (22, 23). Recently, interest in ERα function in the liver has been significantly heightened by results suggesting that the cardioprotective effects of estrogen are based, at least in part, on its effects on liver gene expression. For example, the demonstration of ERα- mediated transactivation of low density lipoprotein (LDL) receptor promoter in transiently transfected HepG2 cells (24) suggests that direct effects of SHP on ERα transactivation could have quite significant physiological consequences. Further work will be required to identify the most important targets of ERα in the liver and the role of SHP in modulating cellular responses to estrogen.
MATERIALS AND METHODS
Plasmids
Construction of pT7lacHisMyc murine SHP full length and the various deletion mutants of murine SHP were reported previously (16).
The GAL4-ERα and B42-ERα were constructed by insertion of the ERα cDNA digested with EcoRI from pGEX-hERα LBD cDNA into pCMX-GAL4 DBD (DNA binding domain) and pJG4–5 (25) treated with EcoRI, respectively. ERβ was isolated from rat prostate cDNA library by PCR and inserted in pCDNA3 vector (Invitrogen, San Diego, CA). Sequencing confirmed that this isolate is identical to the ERβ clone reported previously (6).
In Vitro Interaction
[35S]methionine-labeled proteins were prepared by in vitro translation using pT7lacHisMyc vectors containing cDNAs coding for full-length SHP and deletion mutants (16) and the TNT-coupled transcriptional translation system with conditions as described by the manufacturer (Promega, Madison, WI). GST fusion proteins containing the LBD of human ERα or human AR, or the full-length human RXRα (GST-ER, -AR, and -RXR) were expressed in the E. coli BL21 strain and purified using glutathione-sepharose affinity chromatography as suggested by the vendor (Pharmacia, Piscataway, NJ). In vitro protein-protein interaction assays were carried out as described (26).
Yeast Two-Hybrid Assay
For the yeast two-hybrid system (25, 27), LexA-murine SHP full length and deletion (16) and B42-human ERα and -human RXRα LBD fusion plasmids were cotransformed into Saccharomyces cerevisiae EGY48 strain containing the β-galactosidase (β-gal) reporter plasmid, 8H18–34. Characterization of β-gal expression on plates was carried out as described (28). Similar results were obtained in more than two similar experiments with independently isolated transformants.
Cell Culture and Transfections
HepG2 and U2-OS cells were propagated in DMEM plus 10% FBS. For transfection of HepG2 cells, cells were grown in 12-well plates with medium supplemented with 10% charcoal-stripped serum for 24 h and transfected as described (29) using the diethylaminoethyl-dextran/chloroquine method followed by dimethyl sulfoxide (DMSO) treatment. Transfections included indicated amounts of plasmids expressing proteins of interest and 0.5 μmg/well of both TKGH (30) and a reporter plasmid containing a luciferase gene and GAL4-binding elements. Approximately 16 h after DMSO treatment, 1 μm 17β-estradiol was added with fresh medium, and cells were incubated for 1 day.
For transfection of U2-OS cells, the calcium phosphate method (29) or superfect transfection reagent (Qiagen, Valencia, CA) was used with cells grown in 24-well plates. A plasmid containing a luciferase gene and vitellogenin ERE was used as a reporter and TKβ-galactosidase as the internal control plasmid. Approximately 16 h after transfection, a final concentration of 1 μm 17β-estradiol, 4-OHT, or R1881 was added with fresh medium, and cells were incubated for one more day. Luciferase was assayed as described by the manufacturer (Promega), and the results were normalized using either GH or β-gal expression from the internal control plasmids. Similar results were obtained in more than two similar experiments.
AR mammalian expression vector and GST-AR plasmid were gifts from Dr. Z. Sun (Boston, MA).
This work was supported by NIH Grants RO1 DK-46546 to D.D.M. and RO1 CA-57374 to M.B. W.S. was supported by a Helen Hay Whitney Foundation fellowship.
Present address: Molekulargenetisches Labor der Universitäts-Frauenklinik im, BMFZ, Gebäude 23.12.04, Univeritäts skr. 1, 40225 Düsseldorf, Germany.

{kind=link}
{kind=link}
{kind=link}
{kind=link}