





Abstract
The development of nuclear hormone receptor antagonists that directly inhibit the association of the receptor with its essential coactivators would allow useful manipulation of nuclear hormone receptor signaling. We previously identified 3-(dibutylamino)-1-(4-hexylphenyl)-propan-1-one (DHPPA), an aromatic β-amino ketone that inhibits coactivator recruitment to thyroid hormone receptor β (TRβ), in a high-throughput screen. Initial evidence suggested that the aromatic β-enone 1-(4-hexylphenyl)-prop-2-en-1-one (HPPE), which alkylates a specific cysteine residue on the TRβ surface, is liberated from DHPPA. Nevertheless, aspects of the mechanism and specificity of action of DHPPA remained unclear. Here, we report an x-ray structure of TRβ with the inhibitor HPPE at 2.3-Å resolution. Unreacted HPPE is located at the interface that normally mediates binding between TRβ and its coactivator. Several lines of evidence, including experiments with TRβ mutants and mass spectroscopic analysis, showed that HPPE specifically alkylates cysteine residue 298 of TRβ, which is located near the activation function-2 pocket. We propose that this covalent adduct formation proceeds through a two-step mechanism: 1) β-elimination to form HPPE; and 2) a covalent bond slowly forms between HPPE and TRβ. DHPPA represents a novel class of potent TRβ antagonist, and its crystal structure suggests new ways to design antagonists that target the assembly of nuclear hormone receptor gene-regulatory complexes and block transcription.
THE NUCLEAR HORMONE receptor (NR) family of transcription factors is a target for pharmaceutical development, and many NR antagonists are in current use (1, 2). For example, estrogen receptor (ER) antagonists such as tamoxifen and faslodex inhibit growth and recurrence of estrogen-dependent breast cancer (3). Likewise, androgen receptor (AR) antagonists such as hydroxyflutamide and bicalutamide are used to treat androgen-dependent prostate cancers (4, 5). Other available NR inhibitors include spironolactone, which reduces mortality after heart attack (6), and RU486, which is used as emergency birth control (7).
New NR inhibitors would most likely be useful for treating certain diseases. Thyroid hormone (TH) receptor (TR) antagonists could provide rapid-acting therapies for hyperthyroidism (excess TH production), particularly for use during thyroid storm, a life-threatening thyrotoxic crisis. Antagonists selective for the TRα isoform, which regulates heart rate, could be used to treat cardiac arrhythmias (8, 9).
NRs are composed of three modular domains (10, 11): the C-terminal ligand-binding domain (LBD), the ligand-dependent transactivation function (AF-2), and the N-terminal transactivation function (AF-1) domain. Hormone binds the LBD and activates AF-2, which, in turn, recruits coactivators (12, 13). All NR antagonists that are currently available for clinical use competitively inhibit hormone binding (5). Most NR antagonists are believed to work either by precluding formation of an active LBD conformation or by inducing an aberrant LBD conformation that does not permit AF-2 activity. A number of alternate mechanisms have been suggested, including inhibition of NR via enhanced corepressor recruitment to a surface that partially overlaps AF-2, increased NR turnover, blockade of NR dimer formation, and inhibition of the AF-1 domain (13).
The molecular basis for NR antagonism is well understood (8, 9, 14). NR LBDs comprise a sandwich of three distinct layers formed by 1–12 α-helices (H1–H12) and four short β-strands with activating ligands enclosed in the hydrophobic core of the domain (15). Agonists enhance the packing of the C-terminal helix (H12) over the lower part of the LBD (H3 and H5), completing the AF-2 surface (15). Many antagonists resemble cognate hormone but contain bulky extensions that displace H12. Others occupy the hormone-binding pocket but fail to form a hydrogen bond network, which is required for H12 packing (8). This knowledge has been already exploited to create new antagonists for TRs and many other NRs (8).
However, despite these successes, new NR antagonists are still needed. Many ligand-dependent NRs can influence transcription in the absence of hormone. For example, ERs and AR acquire the capacity to activate transcription in the presence of antagonists during progression of breast or prostate cancer (17, 18). Furthermore, many NRs are not ligand dependent. The LBD of the orphan nuclear receptor 1 lacks a conventional hormone-binding pocket, and ligands of the hepatocyte nuclear factor-4α (14- to 18-chain fatty acids) bind tightly and are better described as prosthetic groups (19). We and others have suggested that directly blocking the coregulator-binding site would afford antagonists that have been referred to as surface-interacting drugs (SIDs). SIDs inhibit key NR protein-protein interaction surfaces and could block NR activity, irrespective of hormone responsiveness of the target cell or the presence or absence of the ligand (20–22). Additionally, SIDs that target unique regions of the NR surface should exhibit better specificity than conventional antagonists, which show troubling cross-reaction with hormone-binding pockets of closely related NRs.
The NR AF-2 surface is an attractive target for candidate SIDs, because it is deeply articulated and has significant hydrophobic character (15, 20, 23). Combined x-ray structural analysis and scanning-surface mutagenesis approaches indicated that TR AF-2 is a small concave surface and that only six hydrophobic residues (V284, K288, I302, K306, L454, and E457) are crucial for its function (24). This surface binds short coactivator domains (NR boxes) that conform to the consensus NR-interaction motif, leucine-x-x-leucine-leucine, and form short α-helices with one face being predominantly hydrophobic (24, 25).
In previous work, we identified two molecules that inhibit interactions between TRβ and the steroid receptor coactivator 2 (SRC2) with IC50 values of approximately 2 μm (21). We also showed that one of these compounds, 3-(dibutylamino)-1-(4-hexylphenyl)-propan-1-one (DHPPA, Fig. 1) is more than 10-fold selective for TRβ over the closely related TRα isoform. DHPPA 2 does not displace T3 from TR in vitro but inhibits TRβ activity in vivo when the receptor is saturated with TH. It appears to have no gross effects upon protein structure or stability. Other groups have reported compounds that act on the ER (26–29).
![Structures of Inhibitors of the Interaction of TRβ LBD and SRC2 A, The β-amino-ketone DHPPA [3-(dibutylamino)-1-(4-hexylphenyl)propan-1-one] was used in the soaking experiments. B, The α,β-unsaturated ketone HPPE [1-(4-hexylphenyl)prop-en-1-one] was the compound seen in the structure. C, The inactive compound HPPA [1-(4-hexylphenyl)propan-1-one] was used as the control.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/mend/21/12/10.1210_me.2007-0174/3/m_zmg0120741560001.jpeg?Expires=1750391027&Signature=kvCGEbCKXAB1FMQTYcUZpoDZCOF5Nt2oOv3TY2jwEXZJlCEvLVHGt9YmSYy-H-oF6S4eFW6I0Kh~g~BDpvWUehhUeZZIBZb77NfT57-jxZlWJG3YcL0XUnjoMgGeB1-P0GDppaOA-7PoH-VHWvgmQP2IIQ3E4bwxH-VMDbNuILhHzW3tndVoU-ZlLlCtHu-Fa9hEE14lMIIxZUINskBrT26LV5DVpKrVXrM9KTXabLbGIsUgPOsZz-9u-8x8KRczds~yaj9O1IK7ymLv7lIYf7zKvUBBVxQ4B0NxB4URyvCmWl6YSqk3BomyNduGKFKiyCS54KsX9G4Jz2JyawixMQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Structures of Inhibitors of the Interaction of TRβ LBD and SRC2 A, The β-amino-ketone DHPPA [3-(dibutylamino)-1-(4-hexylphenyl)propan-1-one] was used in the soaking experiments. B, The α,β-unsaturated ketone HPPE [1-(4-hexylphenyl)prop-en-1-one] was the compound seen in the structure. C, The inactive compound HPPA [1-(4-hexylphenyl)propan-1-one] was used as the control.
Several lines of evidence suggested that DHPPA was a prodrug, the active species of which, β-enone 1-(4-hexylphenyl)-prop-2-en-1-one (HPPE) acted irreversibly. DHPPA is a member of a class of compounds called Mannich bases, which undergo slow β-elimination in solution at physiological pH to form α,β-unsaturated ketones that alkylate nearby electron-rich groups, with a strong preference for nucleophilic sulfur such as that of cysteine side chains (30). We found that HPPE potently inhibits TRβ AF-2 activity in biochemical and cell culture models. Moreover, DHPPA inhibition of TRβ interactions with coactivators is time dependent and requires stoichiometric amounts of compound, hallmarks of irreversible inhibition. Finally, incubation of TRβ LBD with DHPPA increases TRβ molecular weight in a manner consistent with adduct formation between TRβ and a single HPPE molecule. We also noted that several cysteine residues are exposed on the TRβ surface, including three close to AF-2 (Cys309, Cys298, and Cys294). Mutation of Cys309 weakens the actions of DHPPA, although we did not confirm a covalent bond between this residue and HPPE.
In this study, we investigate the mechanism of DHPPA action by using x-ray crystallography and directed mutagenesis of the TRβ coactivator-binding surface. The results suggest that DHPPA inhibits TRβ action by rapidly liberating the reactive α,β-unsaturated ketone HPPE at the TRβ AF-2 surface and that this intermediate, in turn, reacts in a slower step with the nearby Cys298 residue to occlude AF-2. This mechanism exploits the intrinsic activity of the SRC-binding site to generate intermediates that modify nucleophilic groups that are accessible and activated by their environment. This is a mechanism of action reminiscent of enzyme-suicide inhibition. These characteristics may be a useful paradigm for development of new selective NR antagonists.
RESULTS
Reaction of the TRβ LBD with DHPPA
To understand the mechanism of reaction between DHPPA and TRβ, we carried out several studies to determine the reactivity of individual LBD-exposed cysteine residues and the degree to which unbound HPPE is generated. If HPPE is generated locally and reacts with an immediately adjacent sulfhydryl, then DHPPA activity should be resistant to exogenous thiols in the buffer, and the majority of the conjugated HPPE should be directed to the TRβ. Indeed, when coactivator-binding experiments were carried out in the presence of increasing amounts of β-mercaptoethanol (BME, 10 nm to 10 mm), no external product resulting from the conjugation of HPPE with BME was detected by mass spectrometry (see supplemental data Fig. 1 published on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org). Additionally, the DHPPA inhibitor remained fully active with no shifts in potency or efficacy until the concentration of BME exceeded 10 mm (a ∼20,000-fold excess relative to protein). These findings suggest that the HPPE is generated within the binding site and remains bound to that site until it reacts with one of the local cysteines.
We also assessed the sensitivity of TRβ to nonspecific alkylators that attack surface-exposed sulfhydryls to determine whether a particularly reactive cysteine residue was present or if the protein was particularly sensitive to electrophiles. To assess these issues, we conducted coactivator-binding experiments in the presence of increasing concentrations (10 nm to 10 mm) of the sulfhydryl-reactive reagents iodoacetamide and N-ethylmaleimide (see supplemental data Figs. 2 and 3). Neither of these reagents had strong influence on the behavior of TRβ. In fact, no difference in coactivator binding was detected for either reagent until its concentration exceeded 100 μm (iodoacetamide) and 1 mm (N-ethylmaleimide), which, relative to the amounts of protein present, were about 1,000-fold and 10,000-fold excess, respectively. The potency of iodoacetamide was also approximately 100-fold lower than that of DHPPA or HPPE, and the potency of N-ethylmaleimide was 1000-fold lower. These findings imply that some characteristic of HPPE provides a specific alkylation event that blocks function.
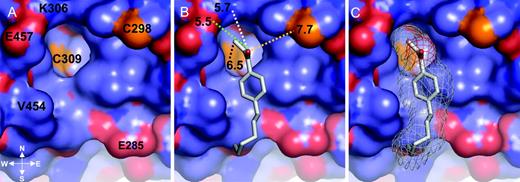
Close-up of HPPE Bound into the TRβ LBD AF-2 Pocket A, Solid representation of TRβ LBD is depicted. The negatively charged residues are red, the positively charged residues are blue, the hydrophobic residues are lilac, and the cysteine residues are yellow. B, The TRβ AF-2 surface and the compound HPPE are shown as a gray stick model placed inside the AF-2 pocket. The distance between HPPE and K306 was 5.7 Å (white dotted line); that to E457, 5.7 Å (green dotted line); that to the unsaturated carbon and C309, 6.5 Å (black dotted line); and that to C298, 7.7 Å (yellow dotted line). C, The TRβ AF-2 surface and the compound HPPE (shown as a gray stick model). A pale gray mesh is included to show the volume of the AF-2 pocket that the compound occupies. The figure was generated using PyMOL (16 ).
![Mass Spectrometric Identification of HPPE Modification of Cys298 The fragmentation spectrum collected for a peptide spanning K289 to K306 and containing a covalent HPPE adduct at C298 is shown. The peptide ([M+H]+: 2349.26) was observed as the triple-charged form ([M+3H]3+: 784.3). The inset shows the peptide sequence and the observed y and b ion fragments. The modified cysteine is indicated in red.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/mend/21/12/10.1210_me.2007-0174/3/m_zmg0120741560003.jpeg?Expires=1750391027&Signature=cz4L3RZ2N6Fz4~41Up~uhW4-yjoZ4d~8Q8KJVN70SEIBBfzA2jd7-rpyRvPfL8gdsSug9wH9VwvZ0YjgPhjZi1Os1rKWi3O9tIBqDajICGOOK5MKJLXJvH6NAjfrd6tWQoVPDbZ3mfr6VLc85QPNqE4nMpqRyXXc3mvyDn-IWBBxgQGP7KQQSgmoLM90pSpOKuHUtzoEUrkbMZ1e9qiNUpTHmw-28Nv-vkOHfIHsqGIqXiehPidlG~f9C0GkW-XrEbQQu20~dKC-nMoUVTkas2QE4KiSeeIVa4ScNsNK6sI89kbQIfI9J4tGHa7~IFTtaxXb2uwbWf3tbrkabscBqw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Mass Spectrometric Identification of HPPE Modification of Cys298 The fragmentation spectrum collected for a peptide spanning K289 to K306 and containing a covalent HPPE adduct at C298 is shown. The peptide ([M+H]+: 2349.26) was observed as the triple-charged form ([M+3H]3+: 784.3). The inset shows the peptide sequence and the observed y and b ion fragments. The modified cysteine is indicated in red.
In addition to these experiments, we reexamined previously published high-resolution TR crystal structures resulting from crystals that had been soaked with or grown in the presence of such vast excesses of nonspecific alkylators [(Protein Data Bank (PDB) identification nos. 2H6WX, 2H77A, 2H79A, and others). Surface-exposed cysteine residues in these structures were found to have reacted with buffer components, and there was an even distribution of alkylation events among exposed cysteines of Cys294, Cys298, Cys388, and Cys434 and their TRα equivalents. Together, these findings imply that the inactivation of TRβ by DHPPA is the result of a specific, targeted alkylation event that results from interaction of the particular electrophile with the protein surface, probably through positioning of the electrophilic pharmacophore element and not due to a particularly reactive cysteine residue.
Structure of the TRβ LBD-Ketone Complex
To understand how DHPPA inhibits coactivator binding to TRβ, we set out to image the compound at the TR surface. Attempts to cocrystallize TRβ LBD in complex with the TH Triac (3,3′,5-triiodothyroacetic acid) and DHPPA were not successful. We therefore first obtained crystals of a TRβ mutant (TRβ D355R) in complex with Triac and then soaked crystals for varying times with DHPPA solutions, as described in Materials and Methods. This TRβ mutant formed stable dimers in solution but otherwise displayed normal transcriptional activity (Ref. 31 ; and Jouravel, N., M. Togashi, E. Sablin, J. D. Baxter, P. Webb, and R. J. Fletterick, manuscript in preparation) and was chosen here because it forms long-lived crystals that are relatively stable (data not shown).
Long-term treatment of preformed TRβ D355R:Triac crystals with DHPPA solution resulted in significant dimensional changes in the lattice. Crystals failed to diffract beyond 10 Å when soaked for 2 h. Crystals that were soaked with DHPPA for as long as 1 h diffracted to 2.3 Å, thereby permitting structural analysis and assignment of the compound to the TR surface. The secondary and tertiary structures of the TRβ D355R mutant were identical to those of wild-type TRs. Two molecules of TRβ D355R formed a dimer (monomer A and B) with a root mean square between monomers of 0.47 Å; Triac was buried inside the ligand-binding pocket. Given extensive similarities between this and previously elucidated TR structures (32–34), the details of TRβ LBD organization will not be further described.
We detected a single compound at the TR AF-2 surface. Consistent with our predicted mechanism, this was HPPE, the product of DHPPA β-elimination, and not the parental DHPPA compound that was used in the soaks (Fig. 1). HPPE has three pharmacophore components: a hydrophobic alkyl chain, a hydrophobic benzyl ring, and a hydrophilic unsaturated ketone substituent that make up the reactive site. All of these features were clearly visible. No evidence indicated the presence of the amine group that is characteristic of DHPPA. We were not able to detect DHPPA or HPPE binding to any other region of the TR LBD surface. Our analysis of the relative electron densities of the compound and receptor suggested that there was 1:1 stoichiometry.
As observed in the crystal structure, HPPE was unreacted with TR but positioned close to several cysteine residues (Fig. 2). As described above, TRβ AF-2 is a small concave surface that contains the following six hydrophobic residues that are essential for coactivator recruitment: V284, K288, I302, K306, L454, and E457. The HPPE alkyl chains bind to a hydrophobic patch formed by L454 and V284; the aromatic ring binds to the concave AF-2 surface, making contacts with L454, V284, and I302. The ketone carbonyl oxygen engages in a water-mediated electrostatic interaction with the K306 amino group, although the distance between these residues (5.7 Å) and that from E457 (∼5.5 Å) eliminates the possibility of a true hydrogen bond forming. Overall, these interactions guide the reactive hydrophilic portion of the HPPE molecule close to four cysteine residues. The unsaturated enone part of the molecule lies about 6.5 Å from Cys309, which is located in the base of the AF-2 cleft. The side chain of Cys308 is buried within the core of the domain with side chain atoms and main chain atoms forming a separation wall between it and the enone. The reactive group of HPPE also lies within 10 Å of Cys298 and Cys294, which are represented on the upper right and right corners of the AF-2 site.
Together, our results suggest that HPPE is liberated from DHPPA in the TRβ crystal and stably binds within the AF-2 cleft. Previous chemical analyses showed that HPPE binds irreversibly to the TRβ LBD with strong time dependency (21). The fact that our crystal contains an unreacted HPPE molecule suggests that the structure corresponds to an intermediate stage of this two-step reaction.
Identification of Covalent Attachment Sites of HPPE on TRβ LBD
To understand how HPPE inhibits TR AF-2 activity, we set out to identify the most likely target(s) of HPPE modification using mass spectrometry of treated protein. Trypsin-digested control and HPPE-treated protein samples were used to define the precise site(s) on TR that was modified by treatment with HPPE. We analyzed the resulting peptides by nanoscale liquid chromatography coupled online to a tandem mass spectrometer. Fragmentation spectra were acquired automatically and interpreted manually and via the use of the MASCOT protein database-searching program (Matrix Science, Boston, MA). Alkylation by HPPE was observed in a peptide spanning residues 289–306 (KLPMFCELPCEDQIILLK) (Fig. 3). Fragmentation spectra were obtained for both the [M+2H]2+ and [M+3H]3+ precursor ion forms, and in both cases, Cys298 was conclusively shown to be the site of modification. Comparison of the integrated ion intensities from the normal and modified forms of this peptide suggested stoichiometric modification at this site; however, possible differences in the relative ability of the two species to ionize precluded a quantitative appraisal of the extent of modification.
Despite observing peaks corresponding to fragment peptides spanning most of the HPPE-binding site, we did not observe peptides that included Cys308 and/or Cys309 in either the control or experimental samples. Thus, although Cys298 is readily modified by HPPE, we cannot rule out some degree of reaction with Cys309 or its neighbor. Nevertheless, mass spectrometry did confirm that Cys294 was unreacted.
Two other sites were modified at very low stoichiometry (<0.1), Lys211 and Cys388. Lys211 is a highly accessible residue located at the beginning of the N-terminal fragment of the LBD TRβ structure, before helix 1. It is fully solvent accessible, and it is not located in any secondary structure element. Cys388 is also surface exposed and located at the C terminus of helix 9, on the opposite side of the receptor from AF-2. Together, our results suggest that Cys298, which is adjacent to the AF-2 surface, is the major target for HPPE modification.
Mutation of Cys 298 Creates a TR that Binds Coactivators but Is Resistant to DHPPA
To assess the role of Cys298 in DHPPA inhibition of TR activity, we determined the effect of mutation of Cys298 on cofactor binding and DHPPA reactivity. Significant amounts of T3-liganded wild-type TRβ were retained on an affinity column in which the NR-interaction domain of the coactivator SRC2 was attached to the solid support (Fig. 4). TRβ alone (i.e. uncoupled from its ligand) did not bind to the column (data not shown). Incubation with increasing amounts of DHPPA or HPPE inhibited TRβ binding to its coactivator SRC2 in a dose-dependent manner, whereas an unreactive control compound 1-(4-hexylphenyl)propan-1-one (HPPA) did not. In parallel, a TRβ mutant in which serine was substituted for Cys298 (C298S) bound strongly to the column, confirming that this residue is not needed for TRβ to bind the coactivator and is insensitive to DHPPA and HPPE action. Thus, Cys298 is not required for cofactor binding but is necessary for DHPPA activity.
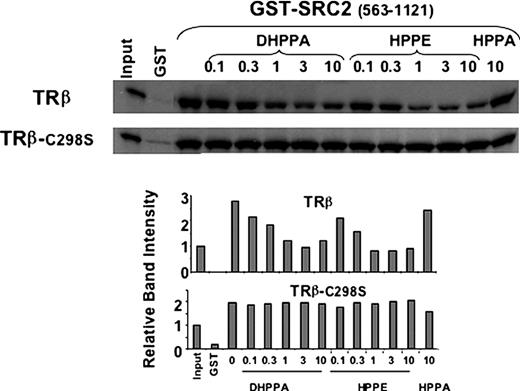
Mutation at Cys298 Abolishes the Sensitivity of TRβ to DHPPA Inhibition SDS-PAGE gel showing quantities of in vitro-translated T3-liganded TRβ or the mutant TRβ C298S retained in pull-down assays using bacterially expressed GST-SRC2 (amino acids 563-1121) at 3 μg per assay. Binding is shown in assays performed with increasing concentrations (micromolar) of DHPPA, HPPE, or the control inactive compound HPPA.
To explore the roles of the cysteine residues located near the AF-2 pocket in DHPPA action, we examined the effects of mutations that targeted key residues surrounding the AF-2 surface but permitted some cofactor binding. Cys298 mutations C298S and C298R suppressed the ability of DHPPA to inhibit cofactor binding, whereas a mutation at Cys294 (C294K) did not (Fig. 5, upper panel). A mutation of Cys309 (C309A) that permitted coactivator binding also failed to reverse the ability of DHPPA to inhibit SRC2 binding (Fig. 5, lower panel).
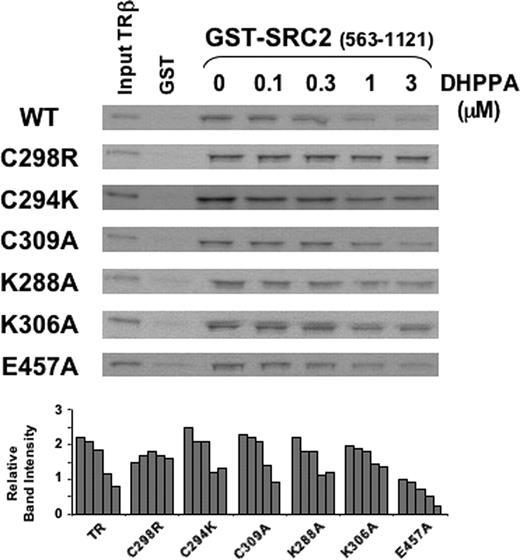
Mutation at Cys298 But Not at Cys309 Uniquely Abolishes DHPPA Action Two SDS-PAGE gels showing quantities of in vitro-translated T3-liganded wild-type (WT) TRβ or mutant TRs retained in pull-down assays using bacterially expressed GST-SRC2 (amino acids 563-1121) at 3 μg per assay in the presence of increasing concentrations (micromolar) of DHPPA.
Both the elimination of DHPPA to form HPPE and the reaction of DHPPA with nucleophilic groups at the TR surface should be affected by mutations that alter the electrostatic environment at the reaction site. Not surprisingly, mutations in charged residues that surround the AF-2 cleft modestly reduced the efficacy of DHPPA. Removal of the positive charge at lysine residue 306 reduced the efficacy of DHPPA (Fig. 5, lower panel), even though we did not detect modification of this residue in mass spectroscopic analysis. In contrast, removal of the charge at glutamate residue 457 or lysine 288 on H12 had no effect (Fig. 5, lower panel).
DHPPA Inhibits TRβ Binding to the NR Corepressor
Coactivators and corepressors bind to an overlapping TR surface that comprises most of the H3–H5 region of the AF-2 surface (15, 24) but differ in their requirements for H12. DHPPA and HPPE inhibited TRβ coactivator binding via a mechanism that involves attachment to AF-2 and modification of Cys298; thus, we examined the effects of DHPPA on corepressor binding.
Results from our pull-down assays suggested that DHPPA inhibits NR corepressor (N-CoR) binding via a mechanism that is similar to its effects on coactivator binding. DHPPA inhibited interactions between wild-type TRβ alone (i.e. uncoupled from its ligand) and N-CoR. The C298R mutation did not inhibit corepressor binding but rendered the receptor insensitive to DHPPA, whereas the C309A mutation had no obvious effect on DHPPA action (Fig. 6). In addition, a mutation at residue 451 (451X) that truncated H12 and permitted strong corepressor binding by exposing the complete N-CoR binding surface (24) did not affect the sensitivity of TRβ to DHPPA (Fig. 6). Thus, DHPPA inhibits TRβ interactions with corepressors via a mechanism that requires Cys298 but is independent of H12.
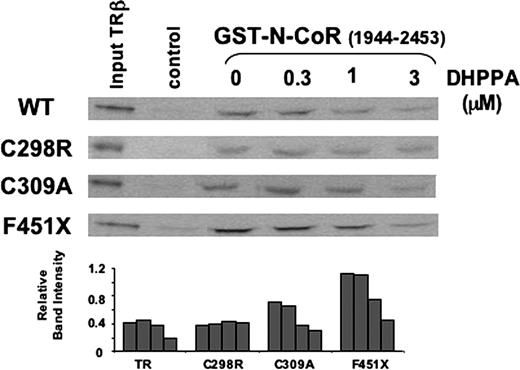
DHPPA Inhibits Corepressor N-CoR Binding to TRβ SDS-PAGE gel showing quantities of in vitro-translated, unliganded wild-type (WT) TRβ or mutant TRs retained on GST-N-CoR (amino acids 1944–2453) in pull-down assays in the presence of increasing concentrations (micromolar) of DHPPA.
DISCUSSION
In this paper, we report the x-ray structure of TRβ in complex with our prototype SID, DHPPA, at 2.3-Å resolution. In addition, results of our mass spectroscopic and mutational analyses reveal that the primary target of DHPPA is a specific cysteine residue (Cys298). These combined structural and functional data also indicate that DHPPA is a prodrug that produces the active compound HPPE by β-elimination, and HPPE specifically targets cysteines in the binding interface. The resulting covalent complex is then unable to recruit coregulators.
Our x-ray structure of TRβ in complex with HPPE confirms that our lead compound interacts specifically with AF-2. We observed a single HPPE molecule that binds to the TRβ AF-2 surface with 1:1 stoichiometry, and there is no evidence that the compound binds elsewhere. The structure also confirms our prediction that DHPPA liberates a reactive intermediate (HPPE) that is in contact with the TRβ surface. Although we incubated TRβ-Triac crystals with the parental β-amino-ketone DHPPA, the α,β-unsaturated ketone HPPE bound to the TRβ AF-2 pocket. Nevertheless, we did not detect electron density that would be consistent with a covalent bond forming between the unsaturated part of HPPE and nearby cysteine residues. Finally, biochemical experiments indicated that little, if any, HPPE escapes into solution. Thus, our structure probably represents a reaction intermediate in which the active form of the compound interacts with TRβ but has not yet completed reaction with the receptor surface.
Functional analysis indicates that the major target for HPPE modification is Cys298, which lies close to the AF-2 cleft. Mass spectroscopic analysis reveals that this residue is modified by HPPE at stoichiometric levels in solution. Moreover, Cys298 mutations uniquely render TRβ insensitive to DHPPA and HPPE action, as revealed by inhibition of TRβ interactions with SRC2 and N-CoR in pull-down assays. Although we mostly detect modification at Cys298, HPPE is not completely specific; there are low levels of modification at Lys211 and Cys388. In addition, we were not able to detect tryptic peptides that overlap Cys309 by mass spectroscopic analysis, so we are not yet able to rule out the possibility that HPPE reacts with this residue at low levels. Nevertheless, Cys309 mutations do not affect DHPPA and HPPE action, suggesting that this residue is not an important target for these SIDs. Supporting a specific molecular mode of action, none of these residues is highly reactive with nonspecific electrophiles.
Together, our findings suggest a likely mechanism for DHPPA action. We postulate that the time dependency of the reaction and the loss of diffraction in the crystal are indicative of a two-step binding mode. The first step is reversible binding of HPPE in the AF-2 pocket, with orientation of the unsaturated part of the ketone toward the nearby nucleophilic side chain of Cys298. After effective positioning, the second step, a slow interconversion of this complex to the covalent adduct, alters the structure sufficiently to prevent formation of diffracting crystals. Because Cys298 is dispensable for coactivator binding to TRβ per se but is absolutely required for HPPE action, the formation of the Cys298/HPPE adduct most likely interferes in a steric manner with coactivator binding to the AF-2 cleft. Prolonged soaking destroys the crystals; therefore, we believe that other residues, probably those identified by mass spectrometry, are sufficiently reactive to be alkylated and damage lattice associations.
Some aspects of the reaction mechanism remain unclear. First, we do not know whether the conversion of DHPPA to HPPE occurs in solution with subsequent binding to TRβ AF-2, or whether it takes place at the AF-2 surface. Mannich base elimination occurs slowly in solution and rapidly at protein surfaces; therefore, we favor the latter possibility. This is bolstered by the fact that we do not detect the expected external products of reaction between buffer components and HPPE. When bound in the mode shown in our structure, the ketone carbonyl oxygen is 5.5 Å from Glu457, and the ketone α-carbon is 5.7 Å from Lys306. This structure raises the possibility that these charged residues facilitate the β-elimination reaction. If so, then the nature of the TRβ active site may play an important role in reactivity of the compound. Second, it is not obvious why Cys298 is the preferred target for modification. There are four possible target cysteine residues near AF-2. Our structure reveals that the reactive group of HPPE is not positioned appropriately to modify Cys294 and that Cys308 is buried in the core of the receptor. Furthermore, Cys309 lies at the base of AF-2 close to HPPE’s unsaturated ketone group. It is not clear why this residue is not important for HPPE action. Perhaps dynamic structural alterations that affect the organization of the AF-2 cleft and are apparent in comparisons of TRα and TRβ structures (21) render the Cys309 side chain inaccessible for HPPE modification in solution.
We expect that our results will facilitate the development of improved second-generation SIDs for TRs. Although DHPPA and HPPE are relatively specific inhibitors of TRβ, they are not potent enough to be useful TR antagonists in the therapeutic setting. Our structural and functional analyses reveal important features of the TR surface that are needed for HPPE binding and action. The TRβ AF-2 pocket contains a narrow hydrophobic passageway that leads to a defined subpocket featuring Cys309 at its bottom, charge beacons provided by Glu457 and Lys306 at its rim, and a flatter subsite surrounded by Cys298 and Lys288. HPPE exploits these topological features to bind TRβ. The compound is captured by the concerted binding of the alkyl chain into the hydrophobic passageway and the aromatic enone moiety into the deep concave hole; the compound targets Cys298, which flanks the flatter hydrophobic subsite. Furthermore, mutational analysis reveals an important but undefined role for Lys306 in DHPPA action; these include possibly orienting the molecule in the appropriate manner for cysteine modification or facilitating elimination of the parental compound, DHPPA. Furthermore, our analysis also suggests ways to improve HPPE binding. Interactions with the flatter AF-2 subsite that is surrounded by Cys298 and Lys288 appear to be suboptimal, and the compound does not bind at all to a large part of the coactivator-binding surface bridged by Glu285. In addition, the fact that DHPPA inhibits TRβ/N-CoR interactions in a manner that is independent of H12 implies that this helix must be dispensable for DHPPA and HPPE action. Thus, chemical modifications that preserve hydrophobic and polar interactions revealed in our structure while simultaneously enhancing suboptimal interactions of HPPE with unoccupied regions of the AF-2 surface and H12 should increase the affinity of DHPPA for TRβ and improve its specificity.
Because it was possible to identify at least one SID that binds specifically to the TRβ AF-2 surface, we expect that we will identify similar inhibitors that bind to the AF-2 of other NRs or perhaps to alternate hydrophobic interaction surfaces such as dimer sites. Given the likely complexity and dynamic nature of such protein-interaction surfaces, we do not think that it will be easy to identify these compounds with standard computer-based molecular modeling approaches. Instead, we suggest that high-throughput screening approaches will identify useful leads and that a combination of x-ray structural analysis and further chemical modification will yield SIDs with high specificity for NRs. Given the mechanism of action of HPPE outlined here, we suggest that NRs with adventitiously placed cysteine residues in close proximity to protein-interaction surfaces would be useful targets.
In summary, few molecules are known to interrupt protein-protein interactions, and of those, the structures of only a few have been characterized. This study presents the first crystal structure of a small molecule that binds to an NR AF-2 pocket, inhibits transcription through its interactions at the AF-2 pocket, and provides important guidelines for future development of improved versions of such compounds. Molecules such as DHPPA and HPPE are the first members of a new class of NR antagonists that are active in the presence of hormone and will provide new options for manipulating the actions of these receptors.
MATERIALS AND METHODS
Protein Expression and Purification
The human (h)TRβ (D355R) LBD (His6 E209-D461) cDNA sequences were cloned into the BamHI and HindIII restriction sites downstream of the hexahistidine tag of the expression vector pETDuet-1 (Novagen, Madison, WI). The replacement of Asp355 for arginine in the hTRβ LBD construct was performed with the QuikChange XL Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA). The sequence was verified by DNA sequencing (Elim Biopharmaceuticals, Inc., Hayward, CA).
The hTRβ (D355R) LBD was expressed in BL21(DE3) cells added at OD600 = 0.6). When the OD600 reached 4, cells were harvested, resuspended in 20 ml buffer per 1 liter culture medium (20 mm Tris; 300 mm NaCl; 0.025% Tween 20; 0.10 mm phenylmethylsulfonylfluoride; 10 mg of lysozyme, pH 7.5) incubated for 30 min on ice, and then sonicated three times for 3 min on ice. The lysed cells were centrifuged at 100,000 × g for 1 h, and the supernatant was loaded onto Talon resin (20 ml; CLONTECH Laboratories, Inc., Mountain View, CA). Protein was eluted with 500 mm imidazole (3 × 5 ml) plus ligand [3,3′,5-triiodo-l-thyronine (Sigma, St. Louis, MO)]. Protein purity (>90%) was assessed by SDS-PAGE and size-exclusion chromatography (FPLC), and protein concentration was measured by the Bradford protein assay. The protein was dialyzed overnight against assay buffer (3 × 4 liters, 50 mm sodium phosphate; 150 mm NaCl, pH 7.2; 1 mm dithiothreitol; 1 mm EDTA; 0.01% Nonidet P-40; and 10% glycerol).
Crystallization, Structure Determination, and Refinement
A pregrown TRβ LBD crystal was soaked with 3 μl of a 10 mm DHPPA compound solution in dimethylsulfoxide for 1 h. The crystal was obtained by vapor-diffusion methods (hanging-drop technique) in 25% ethylene glycol. The protein solution (1 μl) was mixed with 1 μl of the reservoir solution and concentrated against 300 μl of the reservoir. The crystal was flash cooled using liquid nitrogen and measured using the synchrotron radiation at the 8.3.1 beam line at the Advanced Light Source (University of California, Berkeley), where a complete dataset was collected at 2.3-Å resolution.
The crystal belongs to space group P21 and contains two molecules per asymmetric unit. The diffraction data were integrated and scaled using the computer program ELVES (University of California Berkeley) (35). Molecular-replacement solution for the TRβ LBD structure was obtained using rotation and translation functions from Crystallography and Nuclear Magnetic Resonance Systems (CNS) (36). The first electron maps calculated after the rigid body refinement that followed the molecular replacement displayed clear electron density for the compound and less-defined density indicating flexibility for its alkyl chain. During the improvement of the protein model, the Fourier maps revealed perfectly traceable electron density for the entire compound. A composite-omit map that did not include the compound was calculated during refinement for overcoming phase bias. This map was calculated by omitting 5% of the total model, thereby allowing a better tracing of the main alkyl chain in the compound. Model building was done using QUANTA software (Accelrys Software, San Diego, CA), which was monitored using the R-free factor.
Calculation of the electron density maps and crystallographic refinement was performed with CNS software using the target parameters of Engh and Huber (37). Several cycles of model building, conjugate gradient minimization, and simulated annealing using CNS resulted in structures with good stereochemistry. A Ramachandran plot showed that most of the residues fall into the most favored or allowed regions. The statistics for data collection and refinement are presented in Table 1. The structure has been deposited with the Protein Data Bank (PDB) and assigned the following ID number: PDB ID 2F9E, RCSB ID RCSB035615.
Statistics for Data Collection and Refinement of TR Mutant TR-D355R Crystals Soaked with the Unsaturated Ketone HPPE
| Measure | Statistic |
|---|---|
| No. of molecules per asymmetric unit | 2 |
| Space group | P21 |
| Cell constants a/b/c (Å) | 55.13/92.87/58.35 |
| β | 109.65 |
| Resolution (Å) | 2.3 |
| No. of unique reflections | 24,968 |
| Completeness | |
| Overall (%) | 96.0 |
| Outermost shell (%) | 99.9 |
| R mergea | |
| No. of reflections used per refinement | 24,966 |
| Resolution range (Å) | 500–2.29 |
| R factorb (%) | 21.6 |
| R freec (%) | 25.6 |
| No. of water molecules | 348 |
| Matthews coefficient | 2.34 |
| Solvent content (%) | 47.59 |
| Ramachandran plot | |
| Most favored (%) | 92 |
| Allowed (%) | 7.5 |
| Measure | Statistic |
|---|---|
| No. of molecules per asymmetric unit | 2 |
| Space group | P21 |
| Cell constants a/b/c (Å) | 55.13/92.87/58.35 |
| β | 109.65 |
| Resolution (Å) | 2.3 |
| No. of unique reflections | 24,968 |
| Completeness | |
| Overall (%) | 96.0 |
| Outermost shell (%) | 99.9 |
| R mergea | |
| No. of reflections used per refinement | 24,966 |
| Resolution range (Å) | 500–2.29 |
| R factorb (%) | 21.6 |
| R freec (%) | 25.6 |
| No. of water molecules | 348 |
| Matthews coefficient | 2.34 |
| Solvent content (%) | 47.59 |
| Ramachandran plot | |
| Most favored (%) | 92 |
| Allowed (%) | 7.5 |
R merge = ∑hkl – I / ∑hkl I
R factor = ∑hkl ∥Fo – Fc / ∑hkl Fo
R free set contained 5% of total data
Statistics for Data Collection and Refinement of TR Mutant TR-D355R Crystals Soaked with the Unsaturated Ketone HPPE
| Measure | Statistic |
|---|---|
| No. of molecules per asymmetric unit | 2 |
| Space group | P21 |
| Cell constants a/b/c (Å) | 55.13/92.87/58.35 |
| β | 109.65 |
| Resolution (Å) | 2.3 |
| No. of unique reflections | 24,968 |
| Completeness | |
| Overall (%) | 96.0 |
| Outermost shell (%) | 99.9 |
| R mergea | |
| No. of reflections used per refinement | 24,966 |
| Resolution range (Å) | 500–2.29 |
| R factorb (%) | 21.6 |
| R freec (%) | 25.6 |
| No. of water molecules | 348 |
| Matthews coefficient | 2.34 |
| Solvent content (%) | 47.59 |
| Ramachandran plot | |
| Most favored (%) | 92 |
| Allowed (%) | 7.5 |
| Measure | Statistic |
|---|---|
| No. of molecules per asymmetric unit | 2 |
| Space group | P21 |
| Cell constants a/b/c (Å) | 55.13/92.87/58.35 |
| β | 109.65 |
| Resolution (Å) | 2.3 |
| No. of unique reflections | 24,968 |
| Completeness | |
| Overall (%) | 96.0 |
| Outermost shell (%) | 99.9 |
| R mergea | |
| No. of reflections used per refinement | 24,966 |
| Resolution range (Å) | 500–2.29 |
| R factorb (%) | 21.6 |
| R freec (%) | 25.6 |
| No. of water molecules | 348 |
| Matthews coefficient | 2.34 |
| Solvent content (%) | 47.59 |
| Ramachandran plot | |
| Most favored (%) | 92 |
| Allowed (%) | 7.5 |
R merge = ∑hkl – I / ∑hkl I
R factor = ∑hkl ∥Fo – Fc / ∑hkl Fo
R free set contained 5% of total data
Pregrown TRβ crystals soaked with the same 10 mm HPPE compound solution in dimethylsulfoxide for longer than 1 h failed to diffract beyond 10-Å resolution at Advanced Light Source.
Tandem Mass Spectrometric Analysis
TRβ sample in buffer (3 × 4 liters, 50 mm sodium phosphate; 150 mm NaCl, pH 7.2; 1 mm dithiothreitol; 1 mm EDTA; 0.01% Nonidet P-40; and 10% glycerol) was denatured by addition of 8 m urea, diluted to 1 m urea, and then digested overnight by addition of trypsin at 1:50 ratio by mass. The resulting peptides were subjected to nanoscale liquid chromatography/mass spectrometry analysis using a QTrap mass spectrometer (Applied Biosystems/Sciex, Foster City, CA) coupled to an LC Packings UltiMate on-line reverse-phase chromatography system (Dionex, Sunnyvale, CA). Peptides were eluted over the course of 2 h by using a gradient of 5–30% acetonitrile at a flow rate of 150 nl/min. Peptide-fragmentation spectra were automatically acquired using the Enhanced Product Ion scan modality. The resulting data were analyzed using MASCOT software (Matrix Science). The software was asked to consider possible HPPE-mediated alkylation of cysteine and lysine residues.
Pull-Down Assays
TRβ labeled with 35[S] methionine was produced in vitro using the TNT-Coupled Reticulocyte Lysate System (Promega Corp., Madison, WI). The glutathione-S-transferase (GST) fusions were expressed in Escherichia coli BL21 purified, and anchored to a solid support (agarose-glutathione beads) according to the manufacturer’s instructions. For binding assays, bead suspensions containing 10 μg GST fusion protein were incubated with 3 μl 35S-labeled wild-type or mutant TRβ in 150 μl IPAB-80 buffer containing 2 μg/ml BSA, 10–6m T3, and various concentrations of DHPPA, HPPE, or controls. After incubation for 2 h at 4 C, beads were washed (three times) with 1 ml IPAB-80 buffer and heated to 100 C for 3 min. Bound proteins were separated by SDS-PAGE (10% polyacrylamide) and visualized by autoradiography and quantified on a Kodak M1 apparatus with Molecular Imaging software.
Acknowledgments
Acknowledgments
We thank Elena Sablin for useful discussions. James Holton and George Meigs for assistance with crystallization data collection and processing at the Advanced Light Source 8.3.1 beamline (University of California Berkeley), and A. J. McArthur in Scientific Editing at St. Jude’s Children’s Research Hospital for comments on the manuscript.
This work was supported in part by The Prostate Cancer Foundation and the American Lebanese Syrian Associated Charities (ALSAC) and by National Institutes of Health Grants DK58080 (to R.J.F. and R.K.G.), DK41482, 61468, and 51281 (to J.D.B.), and the SPORE National Cancer Institute Grant CA8952 and Herbert-Boyer Foundation (to E.E.P.).
Disclosure Statement: J.D.B. has proprietary interests in and serves as a consultant to Karo Bio AB (Huddinge, Sweden), which has commercial interests in this area of research. The other authors have nothing to disclose.
E.E.-P., L.A.A., and N.J. contributed equally to this work.
The compounds DHPPA and HPPE were named generically L1 and L3, respectively (21 ). Their official names are SJ-000000001 and SJ-000000002, respectively. The compound HPPA is officially named SJ-000000055.
Abbreviations:
- AF,
Activation function;
- BME,
β-mercaptoethanol;
- DHPPA,
3-(dibutylamino)-1-(4-hexylphenyl)propan-1-one;
- ER,
estrogen receptor;
- GST,
glutathione S-transferase;
- HPPA,
1-(4-hexylphenyl)propan-1-one;
- HPPE,
1-(4-hexylphenyl)prop-2-en-1-one;
- LBD,
ligand-binding domain;
- N-CoR,
nuclear receptor corepressor;
- NR,
nuclear hormone receptor;
- PDB,
Protein Data Bank;
- SID,
surface interacting drugs;
- SRC2,
steroid receptor coactivator 2;
- TH,
thyroid hormone;
- TR,
thyroid receptor;
- Triac,
3,3′,5-triiodothyroacetic acid.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}