





Abstract
Primate herpes simplex viruses are species-specific and relatively harmless to their natural hosts. However, cross-species transmission is often associated with severe disease, as exemplified by the virulence of macacine herpesvirus 1 (B virus) in humans. We performed a genome-wide scan for signals of adaptation of simplexviruses to their hominin hosts. Among core genes, we found evidence of episodic positive selection in three glycoproteins, with several selected sites located in antigenic determinants. Positively selected noncore genes were found to be involved in different immune-escape mechanisms. The herpes simplex virus (HSV)-1/HSV-2 encoded product (ICP47) of one of these genes is known to down-modulate major histocompatibility complex class I expression. This feature is not shared with B virus, which instead up-regulates Human Leukocyte Antigen (HLA)-G, an immunomodulatory molecule. By in vitro expression of different ICP47 mutants, we functionally characterized the selection signals. Results indicated that the selected sites do not represent the sole determinants of binding to the transporter associated with antigen processing (TAP). Conversely, the amino acid status at these sites was sufficient to determine HLA-G up-regulation. In fact, both HSV-1 and HSV-2 ICP47 induced HLA-G when mutated to recapitulate residues in B virus, whereas the mutated version of B virus ICP47 failed to determine HLA-G expression. These differences might contribute to the severity of B virus infection in humans. Importantly, they indicate that the evolution of ICP47 in HSV-1/HSV-2 led to the loss of an immunosuppressive effect. Thus, related simplexviruses finely tune the balance between immunosuppressive and immunostimulatory pathways to promote successful co-existence with their primate hosts.
Introduction
Herpes simplex viruses (genus Simplexvirus, family Herpesviridae, order Herpesvirales) are double stranded (ds) DNA viruses that infect mammals, including humans and other primates. They have long genomes of approximately 155 kbp, organized into two regions of unique sequence (long [UL] and short [US]) flanked by direct or inverted repeats. The two unique sequences contain the great majority of the protein-coding regions, including core genes, which are shared among herpesviruses, and noncore genes, that are specific for members of the Alphaherpesvirinae subfamily and/or of the Simplexvirus genus only (McGeoch et al. 2006).
In analogy to other herpesviruses, the evolutionary history of simplexviruses was mainly characterized by coevolution and codivergence with their hosts (McGeoch et al. 2006). A known exception is represented by human herpes simplex virus 2 (HSV-2), which most likely originated from the cross-species transmission of an ancestor of chimpanzee herpesvirus 1 (PanHV-3) to an ancestor of modern humans, around 1.6 million years ago (Severini et al. 2013; Wertheim et al. 2014; Underdown et al. 2017). Thus, whereas most primates are infected by a single simplexvirus, humans host two: HSV-2 and human herpes simplex virus 1 (HSV-1). These viruses are present at high prevalence in human populations. Estimates vary with geography and reach 67% (HSV-1) and 11% (HSV-2) of the world population (Looker, Magaret, Turner, et al. 2015; Looker, Magaret, May, et al. 2015). Even if HSV-1 is primarily responsible for oro-facial lesions and HSV-2 for genital herpes (Arvin et al. 2007), both viruses can establish latency in trigeminal and lumbosacral ganglia, resulting in life-long infection (Arvin et al. 2007). Whereas a relatively low proportion of infected individuals show clinical manifestations during primary infection or reactivation (Tognarelli et al. 2019), simplexviruses can occasionally determine severe diseases such as infectious blindness, acute encephalitis, and neonatal invasive infection (Whitley 2004; Farooq and Shukla 2012).
In nonhuman primates (NHP), simplexvirus infections show symptoms and seroprevalence generally comparable to those of HSV-1 and HSV-2, and these viruses are species-specific in natural settings (Eberle and Jones-Engel 2017). This feature, together with the near commensal relationship with their hosts, is in line with long-standing virus-host coevolution. Indeed, the consequences arising from disruption of the delicate balance established during millions of years of coexistence are evident when cross-species transmissions occur. For instance, macacine herpesvirus 1 (McHV-1, also known as B virus) is almost asymptomatic in macaques, but infection of humans or African monkeys results in a severe, often fatal form of encephalomyelitis (Loomis et al. 1981; Wilson et al. 1990; Tischer and Osterrieder 2010; Eberle and Jones-Engel 2017). Likewise, the transmission of HSV-1 from humans to marmosets or other New World monkeys is almost invariably fatal (Tischer and Osterrieder 2010; Azab et al. 2018). These examples clearly testify how the viral and host genomes interact to determine the outcome of infection and highlight the potential zoonotic threat posed by simplexviruses.
Most likely, simplexvirus adaptation to their hosts involves the fine tuning of host responses, in a delicate balance of immune evasion and damage containment. For instance, downregulation of major histocompatibility complex (MHC) class I surface expression is a common mechanism adopted by herpesviruses, including HSV-1 and HSV-2 (but not by B virus) (Hill et al. 1995; Vasireddi and Hilliard 2012). Reduced MHC class I surface expression prevents recognition of infected cells by CD8+ T-lymphocytes but also leads to susceptibility to NK cell cytotoxicity, which limits viral spread (Hill et al. 1995; Huard and Früh 2000).
These observations imply that simplexviruses must have adapted to their hosts and that the signatures of such adaptation may be detected using molecular evolution approaches. We thus performed a genome-wide scan of positive selection to identify variants in simplexvirus coding genes that arose during adaptation to the hominin lineage. As a proof of concept, we tested the functional effect of selected variants in US12. This gene encodes the Infected Cell Protein 47 (ICP47) protein, the molecule responsible for MHC class I down-modulation by HSV-1/HSV-2 (Früh et al. 1995).
Results
Selective Patterns of Catarrhini-Infecting Simplexvirus Coding Genes
We first explored the selective patterns of primate simplexvirus coding genes. We thus analyzed six complete genomes of simplexviruses that infect different primates, from hominins (HSV-1, HSV-2, and chimpanzee herpesvirus (ChHV)) to Old World African and Asian monkeys (Cercopithecine alphaherpesvirus 2, CeHV-2; Papiine alphaherpesvirus 2, PaHV-2; McHV-1) (fig. 1A and supplementary table S1, Supplementary Material online).
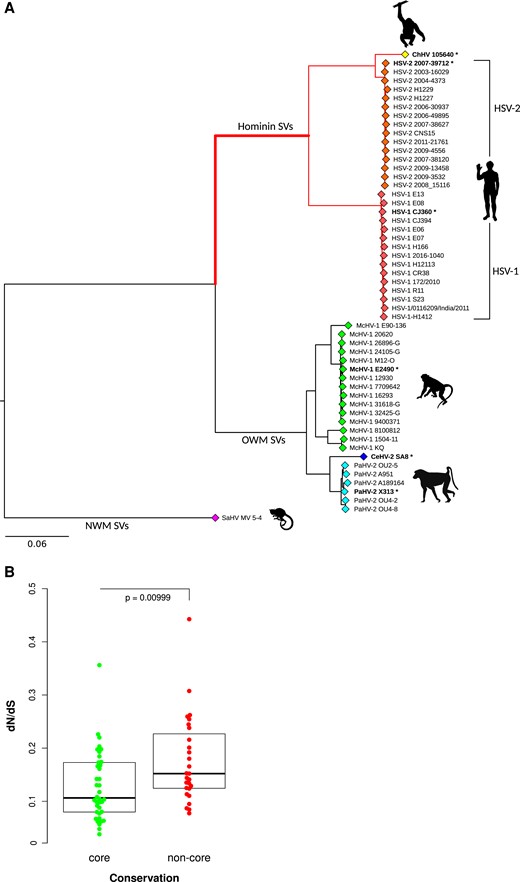
Selective patterns of primate simplexviruses. (A) A maximum-likelihood tree of the longest nonrecombining region of UL30 (encoding the DNA polymerase catalytic subunit) is drawn to exemplify the phylogenetic relationships among primate simplexviruses. (Strain information and GeneBank IDs are reported in supplementary table S1, Supplementary Material online.) The Saimiriine alphaherpesvirus 1 (GeneBank ID: NC_014567) was used as the outgroup, and the tree was constructed using PhyML (see Materials and Methods). Asterisks denote viruses that were included in the analysis of selective patterns of catarrhini-infecting simplexviruses. The hominin simplexvirus branch, which was specifically tested for episodic positive selection, is bolded. SV: simplexvirus (B) Comparison of dN/dS between core and noncore genes. The P value was calculated by the Wilcoxon Rank-Sum test.
Because high sequence diversity can affect evolutionary inference, viruses that infect New World primates were excluded from these analyses. Analysis of selective patterns was performed for all coding genes with reliable one-to-one orthologs. Gene sequences were rigorously filtered to ensure high quality alignments (see Materials and Methods). Genes for which few orthologous sequences were retrieved or with extended overlapping ORFs were discarded (supplementary table S2, Supplementary Material online). The average nonsynonymous substitution/synonymous substitution rate (dN/dS, also referred to as ω) was calculated for the resulting 65 genes and was found to be lower than 1 (indicating purifying selection) in all cases (supplementary table S3, Supplementary Material online). This is expected as negative selection is a major force driving the evolution of viral and non-viral coding sequences (Lin et al. 2019). However, the strength of purifying selection may differ among genes. We thus compared dN/dS values among core genes (conserved among Herpesviridae, n = 38) and noncore genes (specific to simplexviruses, n = 27). Results indicated that these latter show significantly less evolutionary constraint (Wilcoxon rank sum test, P = 0.0099) (fig. 1B and supplementary table S3, Supplementary Material online).
Adaptive Evolution in the Hominin-Infecting Simplexvirus Lineage
In order to assess whether adaptation to hominins drove the evolution of specific simplexvirus coding genes, we applied a branch-site test (Zhang et al. 2005) to an extended phylogeny of 53 viruses that infect hominins, Old World African monkeys, and Old world Asian monkeys (fig. 1A and supplementary table S1, Supplementary Material online). In the branch-site test, the branches of the tree are divided a priori into foreground and background lineages, and models that allow or disallow positive selection on the foreground lineage(s) are compared. The branch-site test can thus detect lineage-specific selected genes (episodic positive selection), and it also provides information on which codons were targeted by selection. Herein, we set the branch leading to the hominin-infecting simplexviruses as foreground (fig. 1A).
After accounting for recombination (see Materials and Methods), we found evidence of adaptive evolution for 11 genes (16.9%). To assess whether the results were affected by the tree topology, analyses for the 11 positively selected genes were repeated using a tree derived from the longest nonrecombining region of UL30 (encoding the DNA polymerase catalytic subunit). All results were confirmed using this tree (not shown).
Positive selection in hominin simplexviruses similarly targeted core and noncore genes (selected fraction = 15.8% and 18.5%), irrespective of the higher selective constraint observed in core genes during viral evolution in catarrhini (supplementary table S4, Supplementary Material online).
We next analyzed positively selected sites. To be conservative, these were detected by the intersection of two approaches (see Materials and Methods). Among the core genes, we found evidence of episodic positive selection for three glycoproteins: gB (UL27), gH (UL22), and gM (UL10) (fig. 2 and supplementary fig. S1, Supplementary Material online). UL27 encodes the viral envelope glycoprotein B (gB), which is a major target antigen in herpesviruses (Malito et al. 2018). Both selected sites in gB are located in the ectodomain and one of them, A334, is part of an epitope recognized by the SS55 neutralizing antibody (Cairns et al. 2014) (fig. 2). Interestingly, an R-to-Q substitution at residue 335 confers resistance to the SS55 mAb (Cairns et al. 2014). As for gH, two positively selected sites, Y85 and E170, flanked amino acids that, if mutagenized, confer resistance to the potent LP11 neutralizing antibody (fig. 2) (Chowdary et al. 2010). Because the LP11 antibody competes with gB for binding to the gH-gL complex, the gB binding site was proposed to be in close proximity to (or maybe overlapping) with the LP11 epitope surface (Chowdary et al. 2010). E170 is part of this surface, together with other sites we found under positive selection (fig. 2). Overall, these observations suggest that the selective pressure acting on these two glycoproteins is exerted by the host immune system.
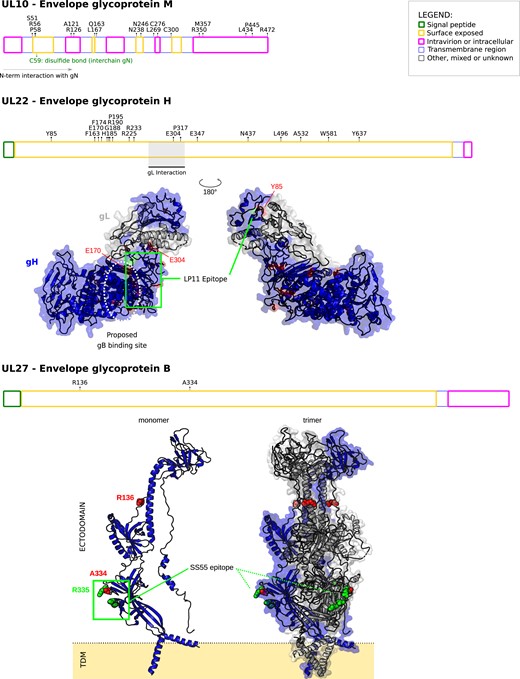
Positive selection in simplexvirus glycoproteins. Positively selected sites were mapped onto HSV-1 glycoproteins together with the location of functional domain/sites (gray). Topological features are color-coded according to the legend. For gH, positively selected sites were mapped onto the 3D structure of the gH-gL complex (PDB ID: 3m1c) (Chowdary et al. 2010). The location of the LP11 epitope and of the gB binding sites (dotted oval) are reported. The two views are rotated 180° around the vertical axis. For gB, positively selected sites were mapped onto the 3D structures of the gB monomer (PDB ID: 6bm8) (Cooper et al. 2018) and the trimeric gB complex. This latter was obtained by a structural imposition of the monomer, using the 2gum structure as scaffold (Heldwein et al. 2006). The location of the SS55 epitope is reported. Positions refer to the reference HSV-1 strain 17 (NC_001806).
We also found many positively selected sites in gM (UL10); the N-terminus of gM is predicted to interact with the glycoprotein N (gN), to form a stable complex, which modulates the viral fusion machinery (El Kasmi and Lippé 2015). Three of the positively selected sites (S51, R56, P58) we found are located in the surface exposed region of gM, just upstream the cystein (C59) residue which is responsible for an interchain disulphide bond that stabilize the gM-gN complex (Striebinger et al. 2016), strongly suggesting that these residues could contribute to gM-gN interaction. Several other positively selected sites were located along the whole sequence of gM (fig. 2).
Among noncore genes showing evidence of positive selection, four (UL46, US8, US1, and US12) are involved in different immune-escape mechanisms. US8 codes for glycoprotein E (gE) that, in complex with gI, forms an Fc receptor for immunoglobulin G (IgG) (Dubin et al. 1991; Sprague et al. 2006). The gE-gI complex binds the Fc region of IgG leading to an antibody bipolar bridging on infected cells, preventing IgG-mediated immune response. In US8, six positively selected sites were found in the protein domain involved in Fc interaction; among them, E227 and G313 lies at Fc interaction surface boundaries (fig. 3) (Sprague et al. 2006).
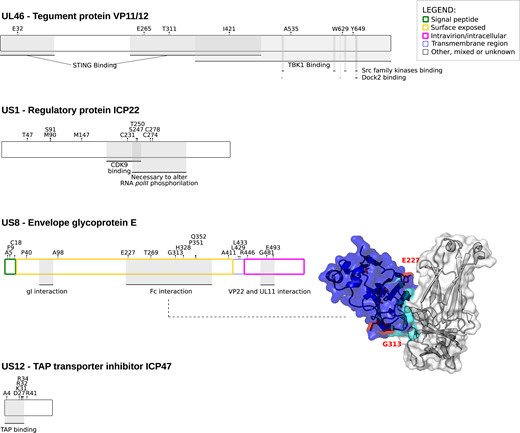
Positive selection in simplexvirus proteins involved in host immune system-escape. Proteins and positively selected sites are reported as in figure 2. For gE, the 3D structure of the complex with Fc (PDB ID:2gj7) is reported (Sprague et al. 2006). gE is represented in blue, with the Fc interaction surface in cyan. Positively selected sites are in red. Positions refer to the reference HSV-1 strain 17 (NC_001806).
UL46 encodes an abundant tegument protein that mediates viral evasion from foreign DNA-sensing pathways (Deschamps and Kalamvoki 2017). In particular, the UL46 protein of HSV-1 interacts with both TMEM173/STING and TBK1 through separate domains and blocks the DNA-sensing pathway. We detected positively selected sites both in the STING and in the TBK1 binding regions (fig. 3).
US1 encodes the ICP22 protein (fig. 3), a general transcription regulator that also down-modulates the expression of CD80 in dendritic cells (Matundan and Ghiasi 2019). Finally, US12 encodes the ICP47 protein, which down-regulates the expression of MHC class I molecules on the cell surface (Früh et al. 1995; Hill et al. 1995). In particular, the ICP47 proteins of HSV-1 and HSV-2 act as inhibitors of the transporter associated with antigen processing (TAP), which translocates antigenic peptides into the endoplasmic reticulum lumen for loading onto MHC class I (HLA-ABC) molecules (Früh et al. 1995; Hill et al. 1995; Tomazin et al. 1998). The TAP-binding region resides in the N-terminal portion of the ICP47 protein, where all the positively selected sites are located (Galocha et al. 1997; Matschulla et al. 2017) (figs. 3 and 4A and B).
Positively Selected Sites in US12 Modulate the Surface Expression of MHC Class I Molecules
A previous study indicated that B virus does not down-modulate HLA-ABC expression at the cell surface. However, a distinctive feature of B virus, not shared by human simplexviruses, is the ability to up-regulate the membrane-bound form of HLA-G, an immunomodulatory nonclassical MHC class I molecule (Vasireddi and Hilliard 2012). We thus reasoned that these differences among closely related viruses might be mediated by the positively selected sites in ICP47, as the protein encoded by HSV-1 and HSV-2 is known to inhibit TAP and cause HLA-ABC retention in the endoplasmic reticulum. In particular, the 55 N-terminal residues of ICP47, where all positively selected sites are located (fig. 4A), are sufficient to interact with and inhibit TAP (Galocha et al. 1997; Matschulla et al. 2017). The N-terminal region of ICP47 is poorly conserved across simplexviruses, and considerable divergence is also observed between HSV-1 and HSV-2, which however both bind and inhibit human TAP (Tomazin et al. 1998). Despite poor conservation, the two human viruses share the same amino acid at all but one positively selected sites (fig. 4B), supporting their role in conferring the ability to inhibit TAP. We thus investigated whether the positively selected sites in ICP47 modulate the different ability of B virus and HSV-1/HSV-2 to regulate HLA-ABC expression. We also investigated whether ICP47 of B virus and the selected sites can modulate HLA-G up-regulation.
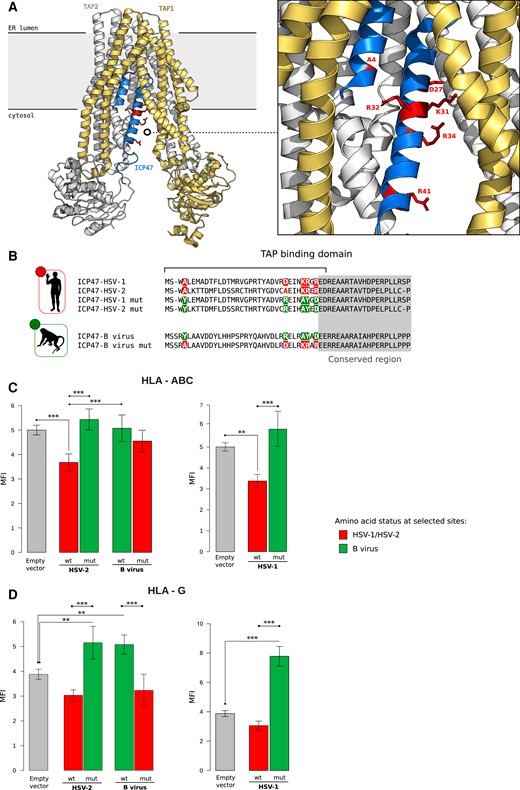
Functional characterization of positive selected sites of US12 (ICP47). (A) Ribbon representation of the 3D structure of ICP47 bound to the TAP transporter, which in turn is composed by two subunits, TAP1 and TAP2 (PDB ID: 5u1d) (Oldham et al. 2016). Positively selected sites are represented as red sticks in the enlargement. (B) Schematic view recapitulating the amino state of positively selected sites tested in our analyses. The positively selected sites are colored. Green represents the amino acid status observed in HSV-1/HSV-2, whereas red denotes the status in B virus. The same colors are also used for the barplots in (C) and (D). HLA-ABC (C) and HLA-G (D) expression at the cell surface. Jurkat cells were transfected with the ICP47 constructs and the amounts of total HLA-ABC or HLA-G antigen was quantified by cytofluorimetry after 48 hours. MFI bar plots represent the mean and standard deviation of four replicates. P values were calculated using ANOVA with Tukey’s post hoc tests (*P < 0.05; **P < 0.01; ***P < 0.001).
To this aim, we designed constructs carrying the TAP binding domains of HSV-1, HSV-2, or the corresponding region of B virus ICP47. Two additional constructs carried the HSV-1 or HSV-2 ICP47 N-terminal domain mutagenized at the positively selected sites to recapitulate the amino acid state observed in the macaque virus. In turn, mutations reproducing the amino acids observed in the human viruses were introduced in the B virus N-terminal domain (fig. 4B).
These constructs were transiently transfected in Jurkat cells and the amount of translated protein was assessed by Western-blot. Comparable expression levels were observed for the HSV-2 and B virus ICP47 fragments (supplementary fig. S2, Supplementary Material online). Conversely, possibly as a result of protein degradation, amounts were lower for the HSV-1 ICP47 constructs (supplementary fig. S2, Supplementary Material online). Thus, the wild-type and mutant HSV-1 proteins were compared to each other and to the control (empty vector), but not to the other constructs.
Analysis of the surface expression of HLA-ABC molecules was evaluated by cytofluorimetry after cell transfection. Significant differences in the HSV-2 and B virus proteins’ ability to modulate HLA-ABC expression was evident (Analysis of variance, ANOVA; F = 14.399, P = 7.25 × 10−5). Tukey post hoc tests indicated that the wild-type TAP binding domain of HSV-2 significantly reduced HLA-ABC expression compared to the control and to cells transfected with the B virus constructs (fig. 4C). Mutation of the positively selected sites in the HSV-2 TAP binding domain of ICP47 totally abolished this effect (fig. 4C), suggesting that the selected sites play an important role in TAP binding. The same result was observed for the wild-type and mutated ICP47 fragments of HSV-1, although the interpretation is complicated by the fact that the wild-type molecule is expressed at higher levels than the mutated one (supplementary fig. S2, Supplementary Material online). In line with previous results (Vasireddi and Hilliard 2012), the ICP47 domain of B virus did not affect HLA-ABC expression. However, mutation of the selected sites to recapitulate the amino acids observed in the HSV-1/HSV-2 molecules was not sufficient to restore TAP inhibition (fig. 4C). Overall, these results indicate that the positively selected sites are not the sole determinants of TAP binding.
We next assessed the effect of the different ICP47 constructs on HLA-G expression. Again, a significant effect for the HSV-2 and B virus constructs (ANOVA, F = 115.697, P = 1.25e−06) was observed. The short N-terminal domain of B virus ICP47 was sufficient to significantly increase HLA-G expression compared the control (fig. 4D). Mutation of the positively selected sites to those observed in HSV-1 and HSV-2 fully abrogated the increased expression of HLA-G. Interestingly, whereas the expression of the N-terminal fragment of HSV-2 ICP47 did not affect HLA-G expression, introduction of mutations that recapitulate amino acids observed in B virus conferred the ability to induce HLA-G (fig. 4D). The same pattern was observed for the HSV-1 N-terminal fragments, despite the lower abundance of the mutated molecule (supplementary fig. S2, Supplementary Material online). These results indicate that the modulatory effect of B virus on HLA-G expression is at least partially mediated by the N-terminal domain of ICP47 and that the positively selected sites are major determinants of HLA-G regulation. Clearly, the effect on HLA-G expression must be TAP-independent.
Discussion
Primate simplexviruses are often regarded as an epitome of virus-host coevolution and codivergence (McGeoch et al. 2006; Eberle and Jones-Engel 2017). These viruses establish life-long infections and usually cause little harm to their hosts, whereas periodic viral reactivation allows transmission in the population. Indeed, virulence and host range are often interconnected traits in viruses (Rothenburg and Brennan 2020), which are expected to evolve to maximize their transmission potential in the host and to tune their virulence accordingly.
Whereas several herpesviruses are unable to infect species other than their natural host, the occasional cross-species transmission of primate simplexviruses has been documented several times, indicating that few barriers exist in terms of infection potential (Azab et al. 2018). However, most spill-overs result in a very severe disease in the new host, especially when the phylogenetic distance from the original host is considerable (Azab et al. 2018). For instance, HSV-1 infection is almost invariably fatal in New World monkeys, whereas limited data on gorillas and Old World monkeys suggest that the symptoms are milder (Tischer and Osterrieder 2010; Gilardi et al. 2014). The best known example of the severe effects of cross-species transmission is that of B virus. Although the virus is rarely acquired, even in people who are in frequent contact with macaques, mortality due to central nervous system involvement is extremely high when infection occurs (Tischer and Osterrieder 2010; Azab et al. 2018; Eberle and Jones-Engel 2018).
These observations clearly indicate that simplexviruses have been adapting to their hosts to balance virulence and transmission. Such a balance is most likely the result of multiple interactions between virus- and host-encoded factors, and the interplay between the host immune response and the viral evasion strategies is expected to determine the outcome of infection. We thus searched for signals of adaptation of simplexviruses to their hominin hosts. The approach we used differs from previous analyses of positive selection in simplexviruses (Szpara et al. 2014; Mozzi, Forni, et al. 2020; Mozzi, Biolatti, et al. 2020), as we specifically searched for selective events that occurred on the branch of hominin-infecting viruses. In particular, we applied a branch-site test, which is well-suited to identify episodic positive selection—that is, selection events that occurred on a specific branch of a phylogeny. This test was shown to have good power for divergence levels comparable to those of the simplexvirus phylogeny we analysed, and it is robust to substitution saturation (Anisimova and Yang 2007; Yang and dos Reis 2011; Gharib and Robinson-Rechavi 2013). Although the branch-site test does not allow for positive selection on background branches, violation of this assumption does not increase the rate of false positives, but affects power (Anisimova and Yang 2007; Gharib and Robinson-Rechavi 2013). Thus, because it is possible, and even likely, that branches other the one we set as foreground experienced positive selection at some genes, we may have missed a proportion of selection signals. Also, our data should not be taken to imply that selection on the branch we analyzed was stronger or more widespread than for other lineages. We tested this branch because we were specifically interested in identifying selection events that contributed to adaptation to the hominin hosts.
Among core genes, we found evidence of episodic positive selection in three glycoproteins, namely gB, gM, and gH, all of which contribute to virus cell entry via membrane fusion (El Kasmi and Lippé 2015; Arii and Kawaguchi 2018). For gB and gH, we found that some of the positively selected sites map to antigenic determinants, suggesting that the host adaptive immune response represents the underlying selective pressure. Moreover, these glycoproteins participate in other processes that contribute to the alteration of the host immune responses. In fact, gB affects the trafficking of MHC class II molecules and diverts them to the exosome pathway (Temme et al. 2010), whereas gH interacts with both αvβ3-integrin and TLR2, which sense the virus and activate the innate immune response (Gianni et al. 2012; Leoni et al. 2012). Both gH and gM were also reported to counteract tetherin, a cellular restriction factor for several enveloped viruses (Blondeau et al. 2013; Liu et al. 2015). In line with the view that hosts and viruses are engaged in genetic conflicts, tetherin was shown to have evolved under positive selection in primates (Gupta et al. 2009; McNatt et al. 2009; Lim et al. 2010). Indeed, this is a general finding for a number of genes involved in defense mechanisms, which display unusually rapid rates of evolution in response to the selective pressure imposed by pathogens (Sironi et al. 2015). Clearly, several infectious agents can insist on the same defense pathway, implying that pathogens are faced with a fast-evolving array of host defense mechanisms. For instance, STING, a stimulator of interferon responsive genes, which is positively selected in primates (Mozzi et al. 2015), is targeted by several viruses. We found three positively selected sites in the STING-binding domain of UL46, suggesting virus adaptation to modulate interaction with the host molecule. Another cellular system commonly antagonized by viruses is the antigen processing and presentation pathway, many components of which show rapid evolutionary rates (Forni et al. 2014). In particular, different herpesviruses employ distinct strategies to interfere with the antigen presentation pathway, thus protecting themselves from the host immune response (van de Weijer et al. 2015; Verweij et al. 2015). In addition to the above-mentioned effect of gB on MHC class II sorting, simplexviruses express the ICP22 protein, which is positively selected and down-modulates CD80 (Matundan and Ghiasi 2019), as well as the ICP34.5 protein (the product of RL1), a neurovirulence factor that blocks MHC II expression on the surface of infected cells (Trgovcich et al. 2002). Due to the small number of confirmed orthologs of RL1, we could no test whether positive selection acted on this gene.
We instead analyzed the selection pattern of US12, which encodes ICP47. All but one of the positively selected sites we detected were located within the N-terminal domain. For the HSV-1 ICP47 protein, this region is sufficient to bind TAP and freeze it in an inactive conformation (Galocha et al. 1997; Matschulla et al. 2017). Because peptide loading is necessary to allow folding of HLA class I molecules in their active configuration, this in turn results in the retention of HLA-ABC molecules in the endoplasmic reticulum. HLA-ABC down-regulation prevents the recognition of infected cells by CD8+ T-lymphocytes, which explains why TAP inhibition is a common viral strategy of immune subversion (Hill et al. 1995). The TAP binding activity of ICP47 was demonstrated for both the HSV-1 and HSV-2 proteins, although sequence similarity is limited in the N-terminal portion. Conversely, infection with B virus does not result in the down-modulation of HLA-ABC expression (Vasireddi and Hilliard 2012). We thus reasoned that the selected sites might underlie the different ability of simplex viruses to inhibit TAP. However, our data indicate that, although the amino acid status at these sites is clearly important, as their mutation in HSV-1/HSV-2 ICP47 restored HLA-ABC expression to the same level as non-transfected cells, they do not represent the sole determinants of TAP binding. In fact, when the amino acids observed in the human viruses were introduced in the N-terminus of B virus ICP47, no HLA-ABC down-modulation was observed. Conversely, the amino acid status at the positively selected sites is sufficient to determine HLA-G up-regulation. In fact, the N-terminal domains of both HSV-1 and HSV-2 ICP47 induced HLA-G when mutated to recapitulate residues in B virus. Conversely, the mutated version of B virus ICP47 failed to determine HLA-G expression. Overall, these results imply that the ability of B virus to induce HLA-G resides in the N-terminal domain of ICP47 and that it does not depend on TAP. This is consistent with the notion that HLA-G can be loaded with peptides by both TAP-dependent and TAP-independent pathways (Lee et al. 1995). The mechanism underlying the up-regulation of HLA-G by B virus ICP47 remains unexplored, and further experiments will thus be required to determine how the positively selected sites exert their effect. As a corollary, our data indicate that the short region of ICP47 we analyzed herein could be used as an inducer of HLA-G expression, which is regarded as a potential biotherapy in allogenic transplantation (Deschaseaux et al. 2011).
The reason why related viruses use the same protein to differentially modulate host responses remains to be clarified. The loss of TAP-binding activity by B virus ICP47 may represent a strategy to limit NK cell activation (Vasireddi and Hilliard 2012). In fact, reduced HLA-ABC expression on the cell surface results in NK-mediated killing, unless inhibitory ligands are also expressed (Früh et al. 1995; Hill et al. 1995; Huard and Früh 2000). Indeed, NK cells play a central role in limiting HSV-1/HSV-2 infection, as demonstrated by mouse models (Rager-Zisman et al. 1987), as well as by the extremely severe infection outcome in humans with genetic defects resulting in low/absent NK cell counts (Orange 2013). It was instead suggested that B virus, due to its lack of TAP-inhibitory activity, does not trigger NK responses(Vasireddi and Hilliard 2012). In addition, at least in human cells, this virus up-regulates HLA-G (Vasireddi and Hilliard 2012), which is associated with diverse immunosuppressive functions, including inhibition of T cell and NK cell responses (Morandi et al. 2016). On one hand these observations might contribute to the extreme virulence of B virus in humans. On the other, as noted elsewhere(Eberle and Jones-Engel 2018), they do not explain why infection in macaques is poorly pathogenic. Notably, though, rhesus macaques do not express the ortholog of HLA-G, as it is a pseudogene (Boyson et al. 1997). Through alternative splicing, the Mamu-AG gene of these NHP encodes glycoproteins functionally similar to HLA-G (Slukvin et al. 2000), which is also alternatively spliced. Mamu-AG shares several features with human HLA-G, including a role in the establishment of maternal-fetal immune tolerance, but it is phylogenetically more similar to HLA-A (Boyson et al. 1997). It is thus possible that Mamu-AG glycoproteins are not up-regulated by ICP47 and that, therefore, infection in macaques elicits weaker immunomodulatory effects, eventually resulting in mild presentation. Addressing this point will require further analyses and the generation of antibodies against Mamu-AG, which are not commercially available.
In summary, we performed a genome-wide scan of positive selection on the hominin simplexvirus branch. We detected several positively selected sites, many of which most likely evolved in response to immune-mediated selective pressure. As these sites were positively selected, they are expected to affect some viral traits, as phenotypes are the ultimate target of selection. As a proof of concept, we tested the functional effects of positively selected sites in ICP47. Such sites were found to be sufficient to determine the inability of the viral protein to up-regulate HLA-G expression. Thus, the evolution of ICP47 in HSV-1/HSV-2 determined the loss of an immunosuppressive effect, suggesting that the trait under selection was decreased virulence. This possibility parallels findings in human cytomegalovirus, another herpesvirus, whereby different mechanisms promoting viral temperance were described (Dunn et al. 2003; Mozzi, Forni, et al. 2020; Mozzi, Biolatti, et al. 2020). These analyses may also suggest that closely related viruses finely tune the balance between immunosuppressive and immunostimulatory pathways to promote successful co-existence with their primate hosts.
Materials and Methods
Sequences and Alignments
Viral genome sequences were retrieved from the NCBI (http://www.ncbi.nlm.nih.gov/) database. A detailed list of accession numbers is reported in supplementary figure S2, Supplementary Material online. Alignments of whole genome sequences were performed with Progressive MAUVE 2.3.1, using default parameters (Darling et al. 2004, 2010). For each viral genome, we retrieved coding sequences of all annotated ORFs. Orthology was inferred according to MAUVE attribution and to genome annotation.
Gene alignments were generated using GUIDe tree based AligNment ConfidencE 2 (GUIDANCE2) (Sela et al. 2015) with multiple alignment using fast Fourier transform (MAFFT) (Katoh and Standley 2013) as the aligner and setting sequence type as codons. Unreliably aligned codons were filtered using GUIDANCE2 with a cutoff of 0.90 (Privman et al. 2012). The resulting alignments were manually inspected.
Only reliable one-to-one orthologs were included in the subsequent analyses (supplementary table S3, Supplementary Material online).
Selective Patterns in Primate-Infecting Simplexviruses
We generated alignments of 65 genes from 6 simplexviruses infecting different primate species (supplementary table S1, Supplementary Material online). Each alignment was screened for the presence of recombination using the Genetic Algorithm Recombination Detection (GARD) (Kosakovsky Pond et al. 2006), an algorithm implemented in the HYPHY suite (version 2.2.4). No evidence of recombination was detected. The average dN/dS parameter was calculated using the fixed effects likelihood method, which applies a maximum-likelihood approach (Kosakovsky Pond and Frost 2005).
Phylogenetic trees were generated with the phyML program (version 3.1), by applying a general time reversible model plus gamma-distributed rates and four substitution rate categories, a fixed proportion of invariable sites, and a BioNJ starting tree (Guindon et al. 2009).
Differences in dN/dS among catarrhini-infecting simplexvirus genes grouped on the basis of gene conservation in the Herpesvirales order (Davison 2007) were evaluated using the Wilcoxon rank sum test.
Detection of Positive Selection in the Hominin-Infecting Simplexvirus Lineage
We analyzed a viral phylogeny composed of 53 catarrhini-infecting viral strains of Simplexvirus genus. Specifically, we include 22 fully sequenced strains infecting Old word monkey species (i.e., macaques and baboons), 1 strain infecting chimpanzee, and 30 strains infecting humans (both HSV-1 and HSV-2, n = 15 each). HSV-1 and HSV-2 strains were selected from clinical isolates with no history of passaging in cell culture, sampled in different countries in order to have an heterogeneous pool of viral genomes representative of the diversity among circulating strains (supplementary table S1, Supplementary Material online).
For each coding-gene, phylogenetic trees were reconstructed using phyML. Each alignment was screened for the presence of recombination using GARD (Kosakovsky Pond et al. 2006). When evidence of recombination was detected (P-value < 0.01), the coding alignment was split accordingly; sub-regions were than used as the input for molecular evolution analyses. Only resulting alignments that after GUIDANCE filtering had a length ≥ 250 nt were considered for subsequent analyses.
Episodic positive selection on the Hominin-infecting simplexviruses branch was detected by applying the branch-site likelihood ratio tests from codeml (“test 2”) (Zhang et al. 2005). In this test, a likelihood ratio test is applied to compare a model (MA) that allows positive selection on the foreground lineages with a model (MA1) that does not allow such positive selection. Twice the difference of likelihood for the two models (ΔlnL) is then compared to a χ2 distribution with one degree of freedom (Zhang et al. 2005). The analyses were performed using an F3X4 codon frequency models. An FDR correction was applied to account for multiple tests.
To identify sites evolving under positive selection, we used BEB analysis from MA (with a cutoff of 0.90) and the mixed effects model of evolution (Murrell et al. 2012) (MEME, implemented in the HYPHY suite, cutoff of 0.1), that allows ω to vary from site to site and from branch to branch at a site. To limit false positives, only sites confirmed by both methods were considered as positively selected. All positively selected sites were mapped onto protein alignments and carefully checked for local alignment quality.
Plasmids, Cell Culture, Transfection, and Western-Blot
The coding sequences of ICP47 N-terminus from HSV-1 (55aa, YP_009137148), HSV-2 (55aa, YP_009137225.1), and B-virus (56aa, NP_851932) were synthesized and cloned in pCMV6-Entry vector by Origene custom service. The pCMV6 vectors coding for the corresponding mutagenized sequences were synthesized and cloned as well (fig. 4B).
Jurkat cells were cultured in Roswell Park Memorial Institute (RPMI) complete media without antibiotics and supplemented with 10% fetal bovine serum. Cells were cultured at 37 °C and 5% CO2 in Forma Steri-Cycle CO2 incubator (Thermo). Every 3 days, cells were split to 0.5–1 × 106 cells/mL in a T25 culture flask with fresh media. ∼5 × 105 Jurkat cells were electroporated in a solution of R-buffer (100 μL; Invitrogen) containing 1ug of plasmid (Empty vector, N-term HSV-1, N-term HSV-1-mut N-term HSV-2, N-term HSV-2 mut, N-term B virus, N-term B virus mut) using a Neon® Transfection System (Invitrogen) under the recommended electroporation condition (1,350 V, 10 ms, 3 pulse). The transfected cells were then seeded into 24-well plate. All experiments were run in four replicates and cells electroporated with pCMV6-Entry plasmid were considered as the control.
Post transfection Jurkat cell viability was ≥90% as determined by an automatic cell counter (Digital Bio, NanoEnTek Inc, Korea).
For Western-blot, cells were lysed in Tris-HCl 0.125 M pH 6.8 and 2.5% SDS, loaded on 15% polyacrylamide gel, blotted onto nitrocellulose membranes and probed with primary antibodies: anti-DDK (Origene) and anti-βactin (Santa cruz). Horseradish peroxidase-conjugated secondary antibodies were used and signals were detected using enhanced chemiluminescence (ECL) (GE Healthcare, Chalfont St. Giles, UK).
Immunofluorescent Staining and Flowcytometry Analysis
Jurkat cells were stained with HLA-ABC PE (Clone W6/32, eBioscience) and HLA-G PE-Cy7 (Isotype IgG2 Mouse, Clone 87G, eBioscience), for 15 min at room temperature in the dark. After incubation, cells were washed and resuspended in PBS.
Flow cytometric analyses were performed after 2 days post-transfection using a Beckman Coulter Gallios Flow Cytometer equipped with two lasers operating at 488 and 638 nm, respectively, interfaced with Gallios software and analyzed with Kaluza v 1.2. Two-hundred-thousand events were acquired and gated on HLA-ABC or HLA-G for Jurkat cells.
Data were collected using linear amplifiers for forward and side scatter and logarithmic amplifiers for fluorescence (FL) 1, FL2, FL3, FL4, and FL5. Samples were first run using isotype control or single fluorochrome-stained preparations for color compensation. The mean intensity fluorescence (MFI) was measured on a log scale from 100 to 103, with negative cells <100. Rainbow Calibration Particles (Spherotec, Inc. Lake Forest, IL) were used to standardize flow-cytometry results.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Acknowledgments
This work was supported by the Italian Ministry of Health (“Ricerca Corrente 2021-2022” to M.S.).
Data Availability
The list of NCBI IDs of the viral sequences analyzed is provided in supplementary table S1, Supplementary Material online.
Conflict of Interest
The authors declare that they have no competing interests.
References
Author notes
A.M. and R.C. contributed equally.

{kind=link}
{kind=link}
{kind=link}
{kind=link}