





Abstract
MicroRNAs (miRNAs) are involved in posttranscriptional gene regulation by repressing the expression of their target genes through inhibition of translation and/or cleavage of mRNAs. In plants, the target genes of miRNAs usually belong to large gene families, and miRNA regulation is gained for individual genes throughout gene family evolution. To explore the selective effect of miRNA regulation on their target genes, we investigated the pattern of polymorphism and interspecific divergence in 11 multigene families that include target genes of ancient miRNAs in Arabidopsis thaliana. We found that the levels of polymorphism and divergence in target genes are significantly reduced, whereas those for nontarget genes are very similar to the genomic averages. This pattern is particularly clear at synonymous sites; we found that the reduction of synonymous variation is caused by selection for optimal codons, which increase the efficiency and accuracy of translation. Based on these results, we conclude that tuning via miRNA regulation has a strong impact on the evolution of target genes through which highly sophisticated regulation systems have been established.miRNA system, gene duplication, codon usage bias.
INTRODUCTION
MicroRNAs (miRNAs) are noncoding small RNAs that regulate genes posttranscriptionally and they are in both the animal and the plant kingdoms (Lagos-Quintana et al. 2001, Lau et al. 2001, Lee and Ambros 2001, Reinhart et al. 2002). miRNAs are 19–25 nt endogenous small RNAs, which are generated from RNA precursors with imperfect foldback structures (Carrington and Ambros 2003, Bartel 2004, He and Hannon 2004). Regulation via miRNAs is activated by the binding of miRNAs to the mRNAs of target genes that have miRNA-binding sites. These binding sites are defined by near-perfect complementation to the mature miRNA sequences. Following binding, the expression of the target genes is repressed by inhibition of translation and/or cleavage of mRNAs, both of which are mediated by the ARGONAUTE protein family (Carrington and Ambros 2003, Bartel 2004, He and Hannon 2004). miRNA-mediated gene regulation is classified into two types—that is, the fully repressing “switch” type and moderately repressing “tuner” type. The latter is considered to be a major role of miRNAs (Bartel and Chen 2004, Flynt and Lai 2008).
A single miRNA can simultaneously regulate a number of target genes, which usually belong to a large gene family. This relationship emphasizes the crucial role of gene duplication in the evolution of target gene families (Rhoades et al. 2002, Li et al. 2008, Takuno and Innan 2008). In fact, target and nontarget genes often coexist in a single gene family in various species (Rhoades et al. 2002, Li et al. 2008, Takuno and Innan 2008) while maintaining similar protein sequences and functions. One expects that the gain of miRNA regulation should change the pattern of gene expression relative to gene family members that are not miRNA regulated. We hypothesized that this change in regulation would, in turn, affect the pattern and level of selective pressures on target genes. Motivated by this prediction, we explore the effect of the selective pressure on the pattern of molecular evolution and polymorphism in miRNA target genes.
We used Arabidopsis thaliana as a model species because the miRNA target gene interactions have been comprehensively identified (e.g., Rhoades et al. 2002, Jones-Rhoades and Bartel 2004, Schwab et al. 2005). With a high-quality genome sequence and reliable annotation, it is straightforward to perform evolutionary analyses of multigene families from which we can identify when gain events occurred on the phylogeny. For example, figure 1 illustrates an neighbor joining tree of the auxin response factor (ARF) multigene family that consists of 27 gene copies in A. thaliana. In this gene family, two copies are regulated by miR160 and three are regulated by miR167. It is easy to place the gain events of the miRNA interactions on the tree (represented by stars) because the two copies under regulation of miR160 are most closely related to each other as are the three copies regulated by miR167. These patterns indicate that each interaction has originated from a single event and that subsequent gene duplication created multiple target copies. Thus, in such gene families, there coexist both miRNA target genes and nontarget genes.
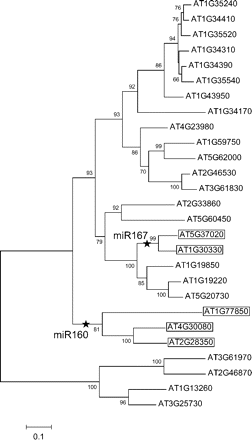
Neighbor joining tree of a multigene family under the regulation of miRNAs in Arabidopsis thaliana. As an example, the phylogeny of ARF gene family is shown. Putative gain events of miRNA regulation are shown on the gene tree by stars. The IDs of target genes are boxed. The numbers at nods indicate the bootstrap values (only ≥ 50% are shown).
Here, we focus on multigene families for which miRNA targeting is conserved in A. thaliana, Oryza sativa, Populus trichocarpa, and Vitis vinifera (Axtell and Bartel 2005, Griffiths-Jones et al. 2006, Jaillon et al. 2007, Velasco et al. 2007). This conservation across species indicates an ancient origin of miRNA targeting, in some cases predating the origin of land plants (Floyd and Bowman 2004, Axtell and Bartel 2005, Axtell et al. 2007). Therefore, such multigenes will represent the overall evolutionary patterns of the target genes in plants. Computational prediction and experimental validation for the target genes of ancient miRNAs are reliable (e.g., Rhoades et al. 2002, Jones-Rhoades and Bartel 2004, Schwab et al. 2005). We found 11 anciently targeted gene families with well-aligned sequences of target and nontarget genes, consisting of a total of 77 target genes and 294 nontarget genes (table 1). The functions of these multigene families are biased toward transcription factors as noted previously (Rhoades et al. 2002). Three multigene families are targeted by more than one miRNA. One is the ARF family as mentioned above. Another is the MYB family that is targeted by both miR828 and miR858 coexist, both of which are conserved in A. thaliana and V. vinifera (Jaillon et al. 2007, Velasco et al. 2007). Members of the laccase families are regulated by three different miRNAs, of which miR857 has been so far found only in A. thaliana (table 1), whereas the other two are shared by all five plant species. In all cases, we found no gene that is simultaneously regulated by more than one miRNA.
List of the Multigene Families Regulated by Ancient miRNAs Used in this Study
| Multigene Family a | Function a | miRNA | Number of Target Genes | Number of Nontarget Genes |
| SBP | Transcription factor | miR156 | 11 | 5 |
| MYB | Transcription factor | miR159 | 7 | 114 |
| miR828 | 2 | — | ||
| miR858 | 8 | — | ||
| ARF | Transcription factor | miR160 | 3 | 22 |
| miR167 | 2 | — | ||
| NAC | Transcription factor | miR164 | 7 | 72 |
| HAP2 | Transcription factor | miR169 | 7 | 3 |
| SCL | Transcription factor | miR171 | 3 | 19 |
| AP2 | Transcription factor | miR172 | 6 | 12 |
| TCP | Transcription factor | miR319 | 5 | 5 |
| TIR1/F-box | Hormone signaling | miR393 | 4 | 6 |
| GRF | Transcription factor | miR396 | 7 | 2 |
| Laccase | Metabolism | miR397 | 3 | 34 |
| miR408 | 1 | — | ||
| miR857 | 1 | — | ||
| Total | 77 | 294 |
| Multigene Family a | Function a | miRNA | Number of Target Genes | Number of Nontarget Genes |
| SBP | Transcription factor | miR156 | 11 | 5 |
| MYB | Transcription factor | miR159 | 7 | 114 |
| miR828 | 2 | — | ||
| miR858 | 8 | — | ||
| ARF | Transcription factor | miR160 | 3 | 22 |
| miR167 | 2 | — | ||
| NAC | Transcription factor | miR164 | 7 | 72 |
| HAP2 | Transcription factor | miR169 | 7 | 3 |
| SCL | Transcription factor | miR171 | 3 | 19 |
| AP2 | Transcription factor | miR172 | 6 | 12 |
| TCP | Transcription factor | miR319 | 5 | 5 |
| TIR1/F-box | Hormone signaling | miR393 | 4 | 6 |
| GRF | Transcription factor | miR396 | 7 | 2 |
| Laccase | Metabolism | miR397 | 3 | 34 |
| miR408 | 1 | — | ||
| miR857 | 1 | — | ||
| Total | 77 | 294 |
According to Jones-Rhoades et al. (2006), Gustafson et al. (2005), Rajagopalan et al. (2006), and Fahlgren et al. (2007).
List of the Multigene Families Regulated by Ancient miRNAs Used in this Study
| Multigene Family a | Function a | miRNA | Number of Target Genes | Number of Nontarget Genes |
| SBP | Transcription factor | miR156 | 11 | 5 |
| MYB | Transcription factor | miR159 | 7 | 114 |
| miR828 | 2 | — | ||
| miR858 | 8 | — | ||
| ARF | Transcription factor | miR160 | 3 | 22 |
| miR167 | 2 | — | ||
| NAC | Transcription factor | miR164 | 7 | 72 |
| HAP2 | Transcription factor | miR169 | 7 | 3 |
| SCL | Transcription factor | miR171 | 3 | 19 |
| AP2 | Transcription factor | miR172 | 6 | 12 |
| TCP | Transcription factor | miR319 | 5 | 5 |
| TIR1/F-box | Hormone signaling | miR393 | 4 | 6 |
| GRF | Transcription factor | miR396 | 7 | 2 |
| Laccase | Metabolism | miR397 | 3 | 34 |
| miR408 | 1 | — | ||
| miR857 | 1 | — | ||
| Total | 77 | 294 |
| Multigene Family a | Function a | miRNA | Number of Target Genes | Number of Nontarget Genes |
| SBP | Transcription factor | miR156 | 11 | 5 |
| MYB | Transcription factor | miR159 | 7 | 114 |
| miR828 | 2 | — | ||
| miR858 | 8 | — | ||
| ARF | Transcription factor | miR160 | 3 | 22 |
| miR167 | 2 | — | ||
| NAC | Transcription factor | miR164 | 7 | 72 |
| HAP2 | Transcription factor | miR169 | 7 | 3 |
| SCL | Transcription factor | miR171 | 3 | 19 |
| AP2 | Transcription factor | miR172 | 6 | 12 |
| TCP | Transcription factor | miR319 | 5 | 5 |
| TIR1/F-box | Hormone signaling | miR393 | 4 | 6 |
| GRF | Transcription factor | miR396 | 7 | 2 |
| Laccase | Metabolism | miR397 | 3 | 34 |
| miR408 | 1 | — | ||
| miR857 | 1 | — | ||
| Total | 77 | 294 |
According to Jones-Rhoades et al. (2006), Gustafson et al. (2005), Rajagopalan et al. (2006), and Fahlgren et al. (2007).
The target versus nontarget comparison was performed for polymorphism within A. thaliana using the whole-genome polymorphism data (Clark et al. 2007). As shown in figure 2A, we found that the level of polymorphism (π; Tajima 1983) in nontarget genes is almost identical to that of the genomic average. In contrast, polymorphism levels are significantly reduced in target genes compared with nontarget genes (P < 0.05 for nonsynonymous and P < 10−4 for synonymous; permutation test). Because we compared genes in the same gene families that are presumed to share similar functions, the observed difference is unlikely to be explained by functional differences between target and nontarget genes. A more likely explanation is that selection on the expression is different between target and nontarget genes.
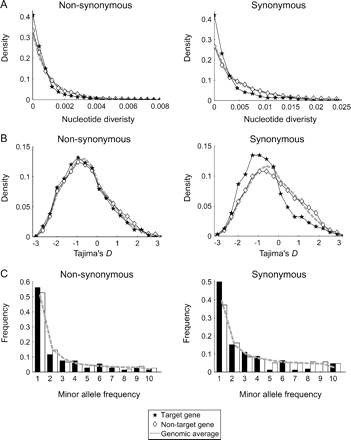
The pattern of polymorphisms in the target and nontarget genes of miRNAs in Arabidopsis thaliana. (A) Density distribution of nucleotide diversity π. Filled stars and open diamonds are for target and nontarget genes, respectively. The genomic average is shown by a dashed line. (B) Density distribution of Tajima's D statistic. (C) Frequency distribution of minor allele frequencies. Filled and open bars are for target and nontarget genes, respectively.
If selective constraint is stronger in target genes due to expression, then it is expected that we would observe footprint of selection especially at synonymous sites. Target and nontarget genes do vary at synonymous sites. The average π for target genes at synonymous sites is roughly half of that for nontarget genes, whereas only about 25% reduction is observed at nonsynonymous sites. Furthermore, we found that Tajima's (1989),D statistic is highly skewed toward left only at synonymous sites (P < 0.005; fig. 2B) because of an excess of synonymous singletons (fig. 2C). This skew should be due to strong negative selection at synonymous sites. It has been known that an excess of minor alleles is a genome-wide phenomenon in A. thaliana, which is due to demographic history of this species (Innan and Stephan 2000, Nordborg et al. 2005, Schmid et al. 2005), but demography alone does not explain the stronger skew at synonymous sites because demography equally affects synonymous and nonsynonymous sites and cannot increase minor alleles only at synonymous sites.
Very similar patterns are observed for the nucleotide divergence between A. thaliana and its close relative A. lyrata (Hu et al. 2011). We first confirmed that the A. thaliana target genes' orthologs in A. lyrata should also be target genes of the same miRNAs because we found very few mutations in binding sites (see Methods and Ehrenreich and Purugganan 2008). Then, synonymous and nonsynonymous divergences for the target and nontarget genes were investigated. It was found that the two distributions of nonsynonymous divergences are almost identical to the genome average, whereas synonymous divergence is significantly reduced in the target genes (P < 0.05; fig. 3A). Thus, this analysis of long-term evolution also suggests stronger selective pressure at synonymous sites, probably due to selection.
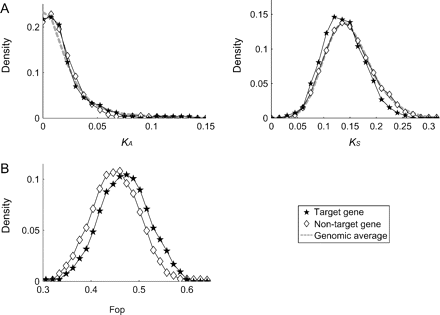
The pattern of divergence and codon usage in the target and nontarget genes of miRNAs in Arabidopsis thaliana. Filled stars and open diamonds are for target and nontarget genes, respectively. The genomic average is shown by a dashed line. (A) Density distributions of nonsynonymous (KA) and synonymous (KS) substitutions between A. thaliana and A. lyrata. (B) Density distribution of Fop.
We obtained a consistent result from the analysis of codon usage bias, which is caused by preferential use of optimal codons to increase the efficiency and accuracy of translation (Ikemura 1985, Akashi 1995, Akashi and Eyre-Walker 1998). Stronger selection on the target genes at the expression level can be supported if they have more codon bias than nontarget genes. To test this, we compared the frequency of optimal codons (Fop) between target and nontarget genes (Ikemura 1985, Wright et al. 2004). As expected, the average Fop value of the target genes was significantly larger than that of the nontarget genes (0.468 vs. 0.452, P < 0.005; fig. 3B).
One might think that our observation of codon usage could be caused by the relaxation of selective constraint in nontarget genes. The evolution of a gene family is a complicated process involving birth and death of copy members (Walsh 2003, Innan201020051986). Pseudogenization of a copy member of a multigene family occurs relatively frequently, and if pseudogenization frequently occurs in nontarget genes, the Fop values will be reduced. However, this explanation does not fully explain our data because the KA/KS ratio in nontarget genes is as low as that in target genes (0.225 vs. 0.203, P > 0.2; permutation test). Thus, our analysis provides no indication that nontarget genes are undergoing pseudogenization. Furthermore, for all investigated genes, we determined whether orthologs are present in the A. lyrata genome; target and nontarget genes are retained between species at roughly the same proportion (85%).
The results of our analyses are consistent in suggesting that there is a stronger footprint of selection at synonymous sites in miRNA-targeted genes (figs. 2 and 3A). The plausible explanation would be that a higher efficiency and accuracy of translation should be favored in target genes, as suggested by the analysis of codon usage bias (fig. 3B). This is a typical pattern of highly expressed genes, and many target genes indeed encode transcription factors (Rhoades et al. 2002) that could be highly expressed. However, high expression of target genes should not simply be the reason why target genes are subject to selection for translation efficiency and accuracy because miRNAs work to reduce expression levels. It is suggested that miRNA should operate to tune the expression pattern of such highly expressed gene on a fine scale. In fact, some miRNAs operate by “tuning” expression and setting a limit on maximum expression levels (Bartel and Chen 2004, Flynt and Lai 2008), and there is empirical evidence that this is true at least for miR164 and miR169 (Cartolano et al. 2007, Sieber et al. 2007). Thus, it is suggested that plants have established a highly sophisticated regulation system to tune expression levels by the joint effect of miRNAs and optimal codons. To further verify our scenario derived from evolutionary analyses, huge amounts of empirical data (especially protein expression data) are needed, which are unfortunately not available at this moment.
METHODS
Data
The whole-genome sequence of A. thaliana was obtained from the TAIR database (TAIR8 release; Swarbreck et al. 2008). The genome sequence of A. lyrata, a close relative of A. thaliana, was available from the JGI database (Hu et al. 2011). Some genes have multiple alternative splicing cDNA forms. In our evolutionary analyses, we used randomly chosen forms to represent the gene. We confirmed that the choice of splicing form did not critically affect the results.
The mature sequences of miRNAs were extracted from the miRBase database (Griffiths-Jones et al. 2006). The data of the target genes of the miRNAs were retrieved from the ASRP database (Gustafson et al. 2005) and other studies (Rajagopalan et al. 2006, Fahlgren et al. 2007). Because the two latter studies (Rajagopalan et al. 2006, Fahlgren et al. 2007) were based on an earlier version of the genome sequence (TAIR6 release), we confirmed that miRNA-binding sites were still located on the cDNA sequences of putative target genes in the TAIR8 release. In the ASRP database, miR159 and miR319 are classified as the same miRNA family. However, these two miRNAs could regulate different target gene families (Schwab et al. 2005, Palatnik et al. 2007), and therefore, we treated these as distinct miRNA systems.
Identifying Target and Nontarget Genes
Multigene families regulated by miRNAs were screened by BlastP search (version 2.2.17; Altschul et al. 1997) with cutoff E value ≤ 10−10 following Takuno and Innan (2008). Some members of multigene families are generally conserved only in a portion of amino acid sequence. These sequences often correspond to functional domains, which were confirmed by the PFAM-HMM domain search in the TIGRFAMs database (Haft et al. 2003).
Evolutionary Analyses of miRNA Target Genes
Evolutionary analyses were performed for the gene families listed in table 1. For each gene, polymorphism data for A. thaliana were obtained from the TAIR database (Clark et al. 2007). The data contain the whole-genome DNA sequences of 20 worldwide ecotypes obtained by a tiling DNA microarray method. For each gene, we did not consider sites where the genotype was missing for > 10 or > 15 accessions (the results were essentially the same in both cases). Nucleotide diversity (π) (Tajima 1983) and Tajima's (1989)D statistic at synonymous and nonsynonymous changes were computed for each gene separately when data were available for ≥ 100 synonymous sites.
Interspecific divergence was measured by the rates of synonymous and nonsynonymous substitutions with A. lyrata. The list of 18,330 orthologs between A. thaliana and A. lyrata was kindly provided by J. A. Fawcett. The synonymous and nonsynonymous substitution rates between orthologs were estimated using the Nei and Gojobori (1986) method, with the Jukes and Cantor (1969) correction after an alignment using CLUSTALW (version 1.83; Thompson et al. 1994). It was confirmed that the A. lyrata's orthologs had conserved target sites by reliable alignment around putative binding sites. We found the orthologs of all target genes have nearly identical binding sites (perfect match for 59 genes and one mismatch for 9 genes), which are consistent with Ehrenreich and Purugganan (2008).
Codon usage bias was also computed for all target and nontarget genes by using CodonW (version 1.3; http://www.molbiol.ox.ac.uk/cu/culong.html). The Fop was used as an index (Ikemura 1985). Optimal (or preferred) codons for each amino acid were based on the copy numbers of transfer RNAs in A. thaliana as predicted by Wright et al. (2004).
Statistical Test
We employed a permutation test to examine whether the averages of two categories (i.e., target vs. nontarget genes) are statistically different. Suppose that there are n1 target and n2 nontarget genes. Denote by dobs, the difference of mean between the two categories for a summary statistic. We first pooled all n1 + n2 genes and randomly divided into two groups with sizes n1 and n2. The averages in the two groups were computed, from which the difference (dsim) between the two groups was obtained. This process was repeated 10,000 times, and the null distribution of the difference was obtained. The P value of the observed difference was given by the proportion of random permutations with dsim > dobs.
We are grateful to Brandon Gaut, Naoki Takebayashi, the members of the Innan Lab, and two anonymous reviewers for helpful comments and discussion. We also thank Yves van de Peer and Jeffrey Fawcett for sharing unpublished A. lyrata sequences. S.T. is a JSPS (the Japan Society for the Promotion of Science) Postdoctoral Fellow for Research Abroad. H. I. is also supported by JSPS, Japan Science and Technology Agency, and an internal grant of the Graduate University for Advanced Studies.
References
Author notes
Associate editor: Naoki Takebayashi

{kind=link}
{kind=link}
{kind=link}