





Abstract
Vault RNAs (vtRNAs) are small, about 100 nt long, polymerase III transcripts contained in the vault particles of eukaryotic cells. Presumably due to their enigmatic function, they have received little attention compared with most other noncoding RNA (ncRNA) families. Their poor sequence conservation makes homology search a complex and tedious task even within vertebrates. Here we report on a systematic and comprehensive analysis of this rapidly evolving class of ncRNAs in deuterostomes, providing a comprehensive collection of computationally predicted vtRNA genes. We find that all previously described vtRNAs are located at a conserved genomic locus linked to the protocadherin gene cluster, an association that is conserved throughout gnathostomes. Lineage-specific expansions to small vtRNA gene clusters are frequently observed in this region. A second vtRNA locus is syntenically conserved across eutherian mammals. The vtRNAs at the two eutherian loci exhibit substantial differences in their promoter structures, explaining their differential expression patterns in several human cancer cell lines. In teleosts, expression of several paralogous vtRNA genes, most but not all located at the syntenically conserved protocadherin locus, was verified by reverse transcriptase–polymerase chain reaction.
Introduction
Vaults are large ribonucleoprotein particles in the cytoplasm of many eukaryotic cells. They have been found as highly conserved ribonucleoproteins in several eukaryotes including many deuterostomes and the slime mold (Dictyostelium discoideum), whereas prominent model organisms such as the fruit fly (Drosophila), the roundworm (Caenorhabditis), rockcress (Arabidopsis), and baker's yeast (Saccharomyces cerevisiae) seem to lack vaults (Rome et al. 1991; Vasu and Rome 1995) . Vault particles have received considerable attention because of their large size (about three times that of a ribosome with a weight of about 13 MDa [Dickenson et al. 2007]). They have a characteristic hollow barrel-like shape with an unusual symmetry (Stewart et al. 2005; Anderson et al. 2007; Kato et al. 2008; Tanaka et al. 2009) and a relatively simple molecular composition (Kedersha et al. 1990; Gottesman et al. 2002; van Zon et al. 2003; Steiner et al. 2006; Berger et al. 2009). Major vault protein (MVP, also known as for “Lung Resistance-related Protein”), the major constituent of vault particles, appears to be sufficient to form the ultrastructure of the vault particle, which is dynamic enough to allow the incorporation of the two other known protein components (TEP1 and VPARP [Smith 2001]) after formation of the particle (Poderycki et al. 2006).
Despite numerous reports on vaults’ expression and composition, the function of these complexes is still poorly understood. Due to their subcellular localization in the cytoplasm as well as their association with the nuclear membrane and nuclear pore complex (Abbondanza et al. 1998), a role in intracellular—in particular nucleocytoplasmic—transport processes has been suggested by van Zon et al. (2006). It is interesting to note in this context that MVP is related to a bacterial toxic anion resistance protein family (Suprenant et al. 2007). Because MVP is frequently found to be overexpressed in a variety of drug-resistant cancer cells, it was speculated that vaults may be involved in drug sequestration (Izquierdo et al. 1996; Scheffer et al. 2000). In fact, there is evidence that vault particles contribute to extrusion of anthracyclines from their target site, the nucleus (Kitazono et al. 1999; Kitazono et al. 2001). However, other reports using knockout or small interfering RNA–mediated downregulation of MVP failed to confirm such a role (Mossink et al. 2002; van Zon et al. 2003; Huffman and Corey 2005).
About 5% of the mass of vault particles consists of vault RNAs (vtRNAs). These RNAs are short polymerase III transcripts with a length varying between about 80 and 150 nt. Mammalian vtRNA genes share characteristic upstream elements that are reminiscent of small nuclear RNA (snRNA) promoters (Kickhoefer et al. 2003). So far, their function within the vault complex remains elusive, although they have been shown to bind certain chemotherapeutic drugs (Gopinath et al. 2005; Mashima et al. 2008). A recent study, furthermore, reported that vtRNAs—together with a handful of other transcripts—are dramatically overexpressed following an Epstein–Barr virus (EBV) infection in human B cells (Mrázek et al. 2007).
To date, few examples of vtRNAs have been studied experimentally. They exhibit little sequence conservation beyond their box A and box B internal polymerase III promoter elements. In the human genome, three expressed vtRNAs, hvg1–hvg3, located in a cluster on Chr.5 (van Zon et al. 2001), have been known for a long time. A fourth putative human vtRNA was recently reported (Mrázek et al. 2007; Nandy et al. 2009). In contrast, mouse and rat have only a single vtRNA gene (Kickhoefer et al. 1993; Kickhoefer et al. 2003; Vilalta et al. 1994). In other classes, only two vtRNAs from the bullfrog (Rana catesbeiana) have been sequenced (Kickhoefer et al. 1993). In addition, the existence of vtRNAs in the sea urchin (Strongylocentrotus purpuratus) was shown (Stewart et al. 2005). Its sequence, however, has not been determined. Recently, vtRNAs were used to benchmark the performance of the pattern-based homology search tool fragrep2 (Mosig, Chen, and Stadler 2007). This computational study reported a single vtRNA candidate in the chicken genome and a few candidate sequences in teleost fishes.
Here we report on a comprehensive investigation of vtRNAs in Deuterostomia, describing evolutionary patterns of this ncRNA family in terms of genomic location, patterns of sequence conservation, and structural constraints.
Homology Search Strategy
The identification of vtRNA genes across currently available sequences of deuterostomic genomes is a nontrival computational problem. Because of their high sequence variability, vtRNAs have been annotated only in some mammals and in african clawed frogs (Xenopus tropicalis) by means of ENSEMBL's noncoding RNA (ncRNA) annotation pipeline. Some additional mammalian vtRNA sequences can be retrieved by BlastN using the known human vtRNAs as queries. However, some of the Eutherian homologs are already close to BlastN's detection limit even with ENSEMBL's relaxed parameter settings for “distant homologies.” For example, the horse sequence ranks fourth among all hits with an E values of only 0.022. Blast-based searches beyond mammals have not been successful at all.
The poor sequence conservation and the substantial length variation make it impossible in practice to use a single simple sequence-based search method. Our strategy, which we explain in detail in this section, is therefore to first retrieve candidate sequences with as much sensitivity as possible. These candidates are then “filtered” using multiple lines of evidence to retain only those sequences that are virtually certain vtRNA homologs. The new homologs then provide an additional information to guide the search for further candidates. As part of our study, we queried the genomes included in ENSEMBL and PRE.ENSEMBL (version 50, summer 2008) releases, the genomes of basal deuterostomes (lancelet [Branchiostoma floridae], sea urchin [S. purpuratus], and sea squirts [Ciona savignyi]) as provided through the University of California Santa Cruz (UCSC) genome browser, as well as shotgun traces of the ongoing genome projects for acorn worms (Saccoglossus) and several Mammalia. In addition, the National Center for Biotechnology Information (NCBI) Web interface was employed to query the various sections of the GenBank and the Trace Archive.
The first step is an iterated BlastN search (NCBI BlastN, version 2.2.17) with E ≤ 10 − 3 for known mammalian and teleost vtRNAs, namely Homo sapiens, Mus musculus, Danio rerio, Tetraodon nigroviridis, and X. tropicalis. Candidate hits were retrieved with 100 nt flanking sequence to ensure that the entire vtRNA is covered. A multiple alignment of query sequences and BlastN hits was then computed to check whether the hits show homology over the full length of the known vtRNAs (see detailed methodology below). Incomplete sequences and obvious retropseudogenes (recognizable by long poly-A stretches at their 3′ end) were rejected. In some cases, we found a few nearly identical sequences in the same genome. Of these, we retained only a single representative for the purpose of homology search because extra copies do not provide additional information and would bias profile alignments. The “good” candidates were added to both the query set and the multiple sequence alignment, and the Blast search was repeated with the additional sequences (see below). After several iterations, no new mammalian sequences were found.
The same procedure was employed to retrieve an extended set of teleost vtRNA sequences starting from the candidate sequences reported by Mosig, Chen, and Stadler (2007). In this case, already the second iteration did not result in additional BlastN hits.
This initial step was then complemented by GotohScan (Hertel et al. 2009) for all noneutherian genomes. This tool implements a semiglobal dynamic programming algorithm with an affine gap cost model (Gotoh 1982) and is not only much more sensitive than BlastN but also computationally much more costly. Using mammalian queries, most teleost vtRNA sequences can be detected as top hits with E values < 1 and vice versa.
The purely sequence-based search was then complemented by two descriptor-based approaches. First, we used fragrep2 to search for combinations of conserved sequence motifs. In order to derive search patterns for fragrep2, we constructed separate alignments for subgroups of our candidates: two eutherian subgroups corresponding to two distinct genomic locations, an alignment of noneutherian tetrapods and an alignment of teleost sequences.
Multiple sequence alignments were constructed manually using the emacs editor in ralee mode (Griffiths-Jones 2005) to integrate separate predictions obtain from both pure sequence alignment methods, RNA secondary structure prediction and combined sequence structure predictions. The reason for the tedious manual approach is that none of the available software tools performs particularly well on all parts of the vtRNAs: ClustalW (Thompson et al. 1994) computes global sequence alignments and works well for sequences of similar length but produced poor results in the presence of large insertions. Dialign2 (Morgenstern 1999; Morgenstern et al. 2006), on the other hand, is a block-based approach without gap penalties that is good at recognizing conserved blocks but performs poorly on regions dominated by many substitutions. The conservation of RNA secondary structure features provides an additional line of evidence. The initial structural annotation was obtained by RNAalifold (Hofacker et al. 2002) for subgroups of closely related sequences for which unambiguous alignments could be obtained. As an alternative, we computed combined sequence structure alignments by means of locarna (version 1.3.3; Will et al. 2007) based on initial base pairing probability matrices produced by RNAfold -p, the Vienna RNA Package (Hofacker et al. 1994) implementation of McCaskill (1990) algorithm. Although locarna typically gives good predictions in highly structured regions, it performs rather poorly in variable regions.
In the second step, we used two descriptor-based methods to search in those genomes where the simpler sequence-based methods did not detect plausible candidates. The fragrep2 approach (Mosig, Guofeng, et al. 2007) looks for the co-occurrence of short approximate sequence patterns within certain distance constraints. Here, the position-specific weight matrices encoding each individual short pattern can be matched with a limited number of insertions and deletions. Matches were filtered as described below and—if accepted—incorporated in the alignments and used to derive modified search patterns. Again, we iterated the search until no further candidates were found. The final search patterns are provided in the Supplementary Material online.
In the final step, we combined three alignments of tetrapod sequences (for the two eutherian loci and for the non-eutherian tetrapods) to a single alignment and manually constructed descriptors for rnabob (ftp://selab.janelia.org/pub/software/rnabob/), a fast implementation of RNAmot (Gautheret et al. 1990) which searches for combined RNA secondary structure patterns. Again, candidates were manually inserted into the multiple alignment and used to refine the query patterns for both fragrep2 and rnabob (for descriptor files, see Supplementary Material online). Both fragrep2 and rnabob return a list of hits that satisfy the search criteria. The fragrep2 algorithm furthermore computes a P value. In the case of rnabob, the number of mismatches can be used to rank the hits.
In each iteration, the candidate sequences were evaluated and filtered. We only accept sequences that satisfy the following criteria:
Sequence conservation. The conserved sequence motifs in both the 5′ and 3′ regions are present.
Terminator. The sequence terminates with a typical polymerase III-terminator motif (consisting mainly of a run of T's; Guffanti et al. 2004).
Secondary structure. The 5′ and 3′ ends of the sequence together can form the conserved stem-loop structure described in detail in the Results.
Reciprocity. Used as a query, the candidate retrieves already known vtRNAs as top candidates from related genomes.
Consistency. Searches for multiple distant query sequences find the same candidate among the top-ranking hits. In the same vain, BlastN finds orthologous sequences whenever closely related species (such as the two Cionas) are available.
Furthermore, we interpret additional lines of information as corroboration of homology:
Synteny. Conserved genomic location relative to flanking protein-coding genes whose homology can be established more easily.
Phylogenetic plausibility. In simple ClustalW-derived distance trees, the candidate falls at or close to the expected phylogenetic position.
Clustering. If paralogs of a candidate can be detected by Blast in the same genome, most of these map to a single genomic location.
Promoters. Polymerase III-promoter elements can be identified upstream of the candidate sequence.
The entire iterative procedure is summarized in figure 1.

Overview of the iterative search strategy. Starting from separate seed sets for eutheria (Hs: Homo sapiens, Mm: Mus musculus, Rn: Rattus norvegicus), teleosts (Dr: Danio rerio, Tn: Tetraodon nigroviridis), and a frog (Xt: Xenopus tropicalis; as representative of other gnathostomes), BlastN with E < 10−3is used until no further candidates are found (dashed arrow). Multiple sequence alignments then serve for the construction of search patterns. Genomic DNA is searched using fragrep and rnabob with these patterns. Additionally, GotohScan is employed to search for more distant GotohScan homologs. Initial candidate sequences are then filtered based on sequence and structure motifs, consistency and additional criteria detailed in the text. The “filtered”candidates are added to the candidate sets. The entire procedure is integrated until no further filtered candidates are found.
Finally, we checked the candidates against the most restrictive group-specific fragrep2 pattern provided in the Supplementary Material online. Each of these patterns can be found in all group members but no other sequences from the same genomes. The fragrep2 program computes P values for its hits. Table 1 shows that each group-specific pattern is highly significant. In addition, each of these patterns is sufficient to find some of the known as well as top-ranking candidate sequences from the other groups.
Validation of fragrep2 Patterns
| Group | Best P Value | Worst P Value | ||
| Basal | 2.6 × 10−6 | Branchiostoma | 1.1 × 10−5 | Petromyzon |
| deuterostomes | ||||
| Gnathostomes | 6.2 × 10−9 | Platypus | 8.5 × 10−9 | Xenopus |
| Teleosts | 2.3 × 10−7 | Zebrafish | 4.1 × 10−7 | Mchenga |
| Eutheria | 2.1 × 10−5 | Echinops | 4.0 × 10−5 | Pteropus |
| VTRNA2 | ||||
| Eutheria | 3.0 × 10−7 | Rat | 2.9 × 10−6 | Chimp |
| VRNRA1 | ||||
| Group | Best P Value | Worst P Value | ||
| Basal | 2.6 × 10−6 | Branchiostoma | 1.1 × 10−5 | Petromyzon |
| deuterostomes | ||||
| Gnathostomes | 6.2 × 10−9 | Platypus | 8.5 × 10−9 | Xenopus |
| Teleosts | 2.3 × 10−7 | Zebrafish | 4.1 × 10−7 | Mchenga |
| Eutheria | 2.1 × 10−5 | Echinops | 4.0 × 10−5 | Pteropus |
| VTRNA2 | ||||
| Eutheria | 3.0 × 10−7 | Rat | 2.9 × 10−6 | Chimp |
| VRNRA1 | ||||
Note.—For each of the five group-specific patterns in the Supplementary Material online, the best and worst P value for fragrep2 is listed. No other hits in the same genomes were found.
Validation of fragrep2 Patterns
| Group | Best P Value | Worst P Value | ||
| Basal | 2.6 × 10−6 | Branchiostoma | 1.1 × 10−5 | Petromyzon |
| deuterostomes | ||||
| Gnathostomes | 6.2 × 10−9 | Platypus | 8.5 × 10−9 | Xenopus |
| Teleosts | 2.3 × 10−7 | Zebrafish | 4.1 × 10−7 | Mchenga |
| Eutheria | 2.1 × 10−5 | Echinops | 4.0 × 10−5 | Pteropus |
| VTRNA2 | ||||
| Eutheria | 3.0 × 10−7 | Rat | 2.9 × 10−6 | Chimp |
| VRNRA1 | ||||
| Group | Best P Value | Worst P Value | ||
| Basal | 2.6 × 10−6 | Branchiostoma | 1.1 × 10−5 | Petromyzon |
| deuterostomes | ||||
| Gnathostomes | 6.2 × 10−9 | Platypus | 8.5 × 10−9 | Xenopus |
| Teleosts | 2.3 × 10−7 | Zebrafish | 4.1 × 10−7 | Mchenga |
| Eutheria | 2.1 × 10−5 | Echinops | 4.0 × 10−5 | Pteropus |
| VTRNA2 | ||||
| Eutheria | 3.0 × 10−7 | Rat | 2.9 × 10−6 | Chimp |
| VRNRA1 | ||||
Note.—For each of the five group-specific patterns in the Supplementary Material online, the best and worst P value for fragrep2 is listed. No other hits in the same genomes were found.
Methods
Analysis of Upstream Regions
The promoter structure of the vtRNA genes and candidates was investigated using the meme suite (Bailey et al. 2006). The meme program (Bailey and Elkan 1994) implements an expectation–maximization algorithm for discovering significantly overrepresented approximate sequence patterns in a set of nonaligned input sequences. The mast program is then used to detect occurrences on the meme-derived patterns in novel sequences.
For our analysis, we used 500 nt of 5′ flanking sequence, the vtRNA gene itself, and 50 nt of 3′ flanking sequence. This sequence interval is chosen to cover the known promoter elements (distal sequence element [DSE] and proximal sequence element [PSE]) upstream and the terminator region downstream of the VTRNA gene. Patterns were learned from DNA surrounding the experimentally known vtRNAs and their syntenically conserved homologs in Mammalia (set A). Independently, a meme alignment was also performed at the syntenically conserved locus in teleosts (set B) and on all vtRNA candidates from amphioxus, tunicates, lamprey (Petromyzon marinus), shark (Callorhinchus milii), Latimeria menadoensis, frog (X. tropicalis), lizard (Anolis carolinensis), and chicken (Gallus gallus; set C). For eutheria, several motif lengths and different models were explored with largely consistent results with respect to the similarities between candidates from the syntenically conserved vtRNA loci. Defaults were used for all other parameters. Full input sets and associated parameters are documented in the Supplementary Material online.
In order to compare the meme-motifs with known features of polymerase III promoters, we extracted the corresponding sequence motifs from the literature on vtRNAs and other polymerase III transcripts (Geiduschek and Tocchini-Valentini 1988; Kickhoefer et al. 1993; Kickhoefer et al. 2003; Vilalta et al. 1994; van Zon et al. 2001; Englert et al. 2004).
Motifs identified by meme then served as an input for mast searches (Bailey and Gribskov 1998) against vt RNA homologs with unknown genomic location, in particular shotgun traces and contigs of low-coverage genomes.
Human vtRNA Expression
Cell Culture
MCF-7, HEK-293, PC3, Du-145, and HeLa cells (ATCC) were grown in Dulbecco's modified Eagle's medium/high glucose with 10% FCS (Biochrom), 100 units/ml of penicillin, and 100 μg/ml of streptomycin (PAA). LNCaP cells (ATCC) were grown in RPMI1640 supplemented with 10% FCS (Biochrom), 100 units/ml of penicillin and 100 μg/ml of streptomycin (PAA), and 10 mM N-2-hydroxyethylpiperazine-N¢-2-ethanesulfonic acid (Biochrom). RWPE-1 cells (ATTC) were grown in keratinocyte serum-free medium (Gibco-BRL) supplemented with 5 ng/ml human recombinant EGF (Gibco-BRL) and 0.05 mg/ml bovine pituitary extract (Gibco-BRL). All cells were cultured at 37 °C in a humidified atmosphere of 5% CO2 in air.
Real-Time Quantitative Polymerase Chain Reaction
Total RNA was extracted from the different fractions by using TRIzol reagent according to the manufacturer's instructions (Invitrogen, Carlsbad, CA). Sequences of primers that were used to carry out the quantitative real-time polymerase chain reaction (qRT-PCR) are listed in the Supplementary Material online. In all, 5 μl total RNA of each fraction was reverse transcribed using random hexamer primers and the High Capacity Reverse Transcription kit (Applied Biosystems). The cDNA was diluted 1:12.5 and served as the template for qRT-PCR analysis using the TaqMan 9700 System (Applied Biosystems) with FAST SYBR green mastermix (Applied Biosystems). Sequences of primers are listed in the Supplementary Material online. All amplicons were confirmed by sequencing. For each vtRNA assay, a standard curve was calculated to check the polymerase chain reaction (PCR) efficiency. Expression of the vtRNA in the different cell lines was normalized to genomic DNA.
vtRNAs in Short Read Sequencing Libraries
The data sets analyzed here for vtRNA fragments were produced in the context of other projects and have been (HeLa data [Friedländer et al. 2008]) or will be published in that context. In brief, total RNA was isolated from the frozen prefrontal cortex tissue using the TRIzol (Invitrogen) protocol with no modifications. Low molecular weight RNA was isolated, ligated to the adapters, amplified, and sequenced following the Small RNA Preparation Protocol (Illumina) with no modifications.
BGI cortex, rep1, and rep2 libraries represent three technical replicates of the same three pooled samples from prefrontal cortex of humans, chimpanzees, and rhesus macaques. In each case, prior to low molecular weight RNA isolation, total RNA from 20 male human individuals aged between 14 and 58 years, 5 chimpanzees aged between 7 and 44 years, and 5 rhesus macaques aged between 4 and 10 years was combined in equal amounts. Replication was carried out by independent processing of the mixed sample of 20 individuals starting from the low molecular weight RNA isolation step. Cerebellum library: total RNA from five male human individuals aged between 20 and 56 years, five chimpanzees aged between 7 and 44 years, and five rhesus macaques aged between 4 and 10 years was combined in equal amounts. Aging library: 14 sequencing lanes of sample containing RNA from the prefrontal cortex of 12 humans aged from 0 to 98 years were analyzed.
Short Read Mapping
The mapping of large libraries containing hundreds of thousands of short, inaccurate sequences to large mammalian genomes cannot be performed reliably and efficiently by commonly used heuristics such as Blat (Kent 2002) or Blast (Altschul et al. 1997). This is due to limitations in both computational resources and accuracy. We therefore used segemehl, a new mapping tool based on enhanced suffix arrays (Abouelhoda et al. 2004), which was developed by Hoffmann et al. (Forthcoming). It uses an alternative heuristics based on the matching statistics (Chang and Lawler 1990) to incorporate not only mismatches but also insertions and deletions.
We also mapped all deep sequencing libraries directly against our four candidate sequences using the Soap program (Li et al. 2008) allowing up to one mismatch position and a seed size of 8.
Expression of Teleost vtRNAs
Genomic DNA and total RNA were isolated from fish liver tissue using DNAzol and TRIzol reagents (Invitrogen), respectively, following the manufacturer's protocols. Concentrations of DNA and RNA samples were determined by A260 measurement using the Nanodrop ND-1000 Nanodrop (Nanodrop Technologies). Putative teleost fish vtRNAs were PCR amplified from genomic DNA (0.5 μg/50 μl reaction) with Taq DNA polymerase (New England Biolabs) and gene-specific primers at 1 μM final concentration. Each primer (listed in the Appendix) was designed to anneal specifically to the respective teleost vtRNA genes.
The PCR was carried out with 1 cycle at 95 °C for 2 min, followed by 35 cycles of 94 °C for 20 s, 58 °C for 20 s, and 72 °C for 15 s, and finished with a final elongation at 72 °C for 2 min. The PCR products were gel purified and cloned into pZero vector (Invitrogen) for sequencing confirmation of the specific vtRNA genes amplified.
Reverse Transcriptase–Polymerase Chain Reaction
The expression of individual vtRNAs was verified by reverse transcriptase–polymerase chain reaction (RT-PCR). From 2 μg of total RNA, medaka (Oryzias latipes) or zebrafish (D. rerio), cDNA libraries were prepared using Thermoscript reverse transcriptase (Invitrogen) and a random hexamer primer following the manufacturer's instruction. Gene-specific primers were used to PCR amplify the putative vtRNA sequences from the cDNA libraries under conditions similar to the PCR condition for genomic DNA samples, except 3–5 additional cycles. The RT-PCR products were cloned into pZero vector and sequenced. The Mock RT reactions with reverse transcriptase enzyme omitted served as the negative control.
Northern Blot Analysis
The northern blotting was carried out as previously described (Xie et al. 2008) with minor modifications. Briefly, 20 μg of total RNA and in vitro transcribed vtRNA (0.1 and 1 ng) were resolved on a 6% polyacrylamide/8 M urea denaturing gel and electrotransferred to a Hybond-XL membrane (Amersham Biosciences) at 0.5 A for 2 h. The riboprobes for northern blotting analysis and the size markers, medaka (MEDAKA1_s3838_742) and zebrafish (ZFISH7_14_454804) vtRNAs, were prepared by T7 in vitro transcription using PCR DNA as template (Xie et al. 2008). PCR primer sequences are listed in the Supplementary Material online. The riboprobes were labeled internally with [α-32P] UTP in a T7 transcription reaction using a Maxiscript kit (Ambion). After transfer, the membrane was hybridized with riboprobes (1 × 106 cpm/ml) at 65 °C overnight in Ultrahyb buffer (Ambion) and washed twice at 65 °C in 1x standard saline citrate (SSC)/0.2% sodium dodecyl sulfate (SDS) for 10 min and twice in 0.2x SSC/0.1% SDS for 20 min. The blot was analyzed and quantitated using a phosphorimager, Bio-Rad FX Pro.
Results
Homology Search
Mammalian vtRNAs
To date, three expressed human vtRNA genes (hvg1–hvg3) forming a small cluster on Chr.5 have been described (van Zon et al. 2001). Orthologs of these three human vtRNA clusters can easily be found in the chimp (Pan troglodytes) genome. In contrast, both orangutan (Pongo pygmaeus) and macaque (Macaca mulatta) have two copies instead of three. A sequence alignment clearly shows that human hvg2 and hvg3 are very recent duplicates. This vtRNA cluster is located between the ZMAT2 and PCDHA (protocadherin-α) genes. Because the protocadherin-α cluster, which encodes a family of synaptic adhesion molecules, has received quite a bit of attention in recent years (Noonan et al. 2004; Sugino et al. 2004; Tada et al. 2004; Yu et al. 2007; Yu et al. 2008), we will refer to these genomic regions as the pcdh locus. In addition, a single closely related pseudogene has been found on human Chr.X (van Zon et al. 2001).
Most mammals have a single vtRNA copy at the pcdh locus, including in particular the experimentally determined mouse and rat vtRNAs (fig. 2). In some cases, it has expanded locally into a multicopy cluster, notably in primates (as mentioned above), pika (Ochotona princeps), guinea pig (Cavia porcellus), and shrew (Sorex araneus). The opossum (Monodelphis domestica) also exhibits a (recent) tandem duplication of the vtRNA at the pcdh locus.
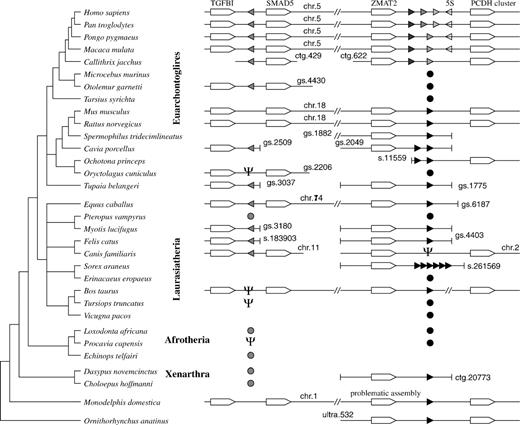
Overview of the putative functional vtRNA genes in Mammalia. The two syntenically conserved mammalian vtRNA loci are usually located on the same chromosome. Most investigated genomes are not assembled to chromosomes; in these cases, “Contig”(ctg.), “Scaffold”(s.), “GeneScaffold”(gs.), or “UltraContig”(ultra.) numbers are provided. Filled triangles indicate vtRNAs and their reading direction relative to the adjacent genes (the reading direction of the database entries is not indicated). Pseudogenes are marked by psi symbol. In primates, a 5S rRNA is located adjacent to the vtRNAs (open triangle). For Afrotheria and Xenarthra, the presence of recognizable homologs is indicated. The current assemblies do not allow to determine whether they are located at the syntenic positions. Circles indicate vtRNA homologs with promoter element characteristic for one of the two loci (for details, see fig. 3). The phylogeny of the eutherian superfamilies follows (Kriegs et al. 2006). The figure represents the genomic data from ENSEMBL 50 and the NCBI Trace Archive in fall 2008.
In the dog (Canis familiaris) genome, we found only a degraded pseudogene of the vtRNA at the pcdh locus. Instead, we detected a single alternative sequence by BlastN, which is located (in antisense direction) between TGFB1 and SMAD5. Using this sequence as query, we recovered homologs at the syntenic position throughout all eutheria (the lack of Blast hits in the earliest branching eutherian, the armadillo [Dasypus novemcinctus] may be due to the low coverage of the genome). It is interesting to note that—with the exception of some laurasiatheria—both the TGFB1–SMAD5 and the ZMAT2–PCHDA locus are found on the same chromosome, barely 0.5 Mb separated from each other.
To our surprise, the corresponding Blast hits are annotated as mir-886 in human (Landgraf et al. 2007) and macaque (Yue et al. 2008). Sequence alignments, however, quite clearly identify this sequence as a vtRNA homolog. This was already recognized by Mrázek et al. (2007): A transcript called CBL-3 matching our prediction was identified as a putative fourth vtRNA in EBV-infected cells. In an independent study, Nandy et al. (2009) investigated this molecule in detail and demonstrated that it indeed associates with the vault complex.
In response to the work of Nandy et al. (2009) and the present study, the HUGO Gene Nomenclature Committee (HGNC; http://www.genenames.org/) will, in its next release, revise the nomenclature of vtRNA genes to reflect their well-conserved genomic locations. The new HGNC gene symbols are VTRNA1-1, VTRNA1-2, and VTRNA1-3 for the hvg-1 through hvg-3 at the canonical locus. The newly identified vtRNA gene linked to SMAD5 is then called VTRNA2-1, whereas the pseudogene on the X chromosome will be denoted VTRNA3-1P. The first number identifies the genomic locus, whereas the number after the dash identifies the individual members of vtRNA clusters. From here on, we will therefore refer to the pcdh and SMAD5-linked loci as VTRNA1 and VTRNA2 locus or cluster, respectively.
The VTRNA2-1 gene is typically somewhat better conserved than its paralogs at the VTRNA1 locus. So far, no duplicates have been found at the VTRNA2 locus in any of the investigated mammalian genomes. The locus harbors a highly derived, probably pseudogenized sequence in the rabbit (Oryctolagus cuniculus) and in the cow (Bos taurus) genomes. The VTRNA2-1 has been lost completely in both mouse and rat.
Our data show that the origin of the VTRNA2 locus predates the divergence of Afrotheria, Laurasiatheria, and Euarchontoglires. A search in the NCBI Trace Archive returned two putative vtRNAs from the sloth Choloepus hoffmanni, whereas only a single vtRNA sequence is found in the survey genome of D. novemcinctus. A ClustalW alignment and Neighbor-Joining analysis tentatively places one of the two sloth sequences together with the human VTRNA2-1, suggesting that Xenarthra also possess both vtRNAs in both loci. This result is supported independently by the observation that one of the sloth vtRNAs has promoter elements characteristic for the VTRNA1 locus, whereas the promoter of the other copy matches the VTRNA2 promoters of other eutheria, see below and figure 3. In contrast, no VTRNA2 gene can be identified in both the opossum (M. domestica) and the platypus (Ornithorhynchus anatinus) genome, placing the Mammalia-specific vtRNA duplication at the root of the Eutheria.
Several nonexpressed mouse, rat, and human vtRNA pseudogenes are explicitly discussed in the literature (van Zon et al. 2001; Kickhoefer et al. 2003). In most species, a BlastN (E < 10 − 3) search returns no or very few hits that can be expanded to full-length vtRNA candidates outside of the canonical loci. Most of these hits are easily identified as pseudogenes because they match only parts of human vtRNA queries and/or only imperfectly to the conserved stem-loop structure. In the guinea pig, however, VTRNA1 spawned a larger family of pseudogenes with about 100 members, including many highly degraded sequences. The VTRNA2 locus, on the other hand, is the origin of several dozens of likely pseudogenes in primates. Many of these are listed as vtRNAs by the ENSEMBL genome annotations because this annotation pipeline at present cannot distinguish reliably between functional loci and pseudo-genes.
![Conserved elements associated with mammalian vtRNAs. The diagram combines a subset of the patterns discovered by MEME with three different parameter settings. Previously described polymerase III upstream elements (DSE, cyclic AMP response element [a part of the larger PSE], and the TATA box) are indicated. The vtRNA itself is delimited by the box A containing motif 2 (blue) on its 5′side and by the elements 1 and 3 (cyan and red), which contain the box B and the polymerase III terminator signal, on its 3′side. Primate vtRNA-1 genes have a second copy of the box B and terminator downstream of the experimentally determined end of the transcript. The extended structure of the mouse and rat vtRNAs (Kickhoefer et al. 2003) is also clearly visible. Almost all vtRNA genes at the VTRNA1 locus exhibit elements 6 (green), 8 (gray), and the DSE containing motif 11 (violet), whereas the distal region of the novel locus is characterized by three unrelated motifs 5 (yellow), 9 (dark green), and 12 (olive).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/mbe/26/9/10.1093_molbev_msp112/2/m_molbiolevolmsp112f03_4c.jpeg?Expires=1750247912&Signature=WLTapGe0ROXydVL2bxp4vJUx3v3RTdfLvTblBa2BSKHG595QGj0FdEwlKzaoqDZe8a0V88TaGNfcFEaK-GMwtGRdKraXQvHe8XM8xv-nFKktvNzLECqhgrsqqXT9Jh5~1nZrLf9pScdhEJGBu6COzbQbraRx0vV7zIKGWKzd1~O7hnIBsWTtf6yHYtkhbij9Jbud6lA51kMbPTgN4Z0i9BeOX5GtcuP110UVHbFCbfrWcA4ntjzrBqDU6KQFp39tAldkvtpBd5qxEDwlQ33ZZPCzcE9xqJfRUNeqO4JAhXy0-bj-NmDEWMYAe--EbMHEjhWfN13bcTg7hfUuq6MrSg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Conserved elements associated with mammalian vtRNAs. The diagram combines a subset of the patterns discovered by MEME with three different parameter settings. Previously described polymerase III upstream elements (DSE, cyclic AMP response element [a part of the larger PSE], and the TATA box) are indicated. The vtRNA itself is delimited by the box A containing motif 2 (blue) on its 5′side and by the elements 1 and 3 (cyan and red), which contain the box B and the polymerase III terminator signal, on its 3′side. Primate vtRNA-1 genes have a second copy of the box B and terminator downstream of the experimentally determined end of the transcript. The extended structure of the mouse and rat vtRNAs (Kickhoefer et al. 2003) is also clearly visible. Almost all vtRNA genes at the VTRNA1 locus exhibit elements 6 (green), 8 (gray), and the DSE containing motif 11 (violet), whereas the distal region of the novel locus is characterized by three unrelated motifs 5 (yellow), 9 (dark green), and 12 (olive).
At present, several mammalian genomes are sequenced to 2x coverage only, entailing significant problems for their assembly. In particular, it is not possible to determine the exact number of vtRNAs in these genomes. There is no evidence that vtRNAs outside the VTRNA1 and VTRNA2 loci are syntenically conserved, indicating that there is no selection pressure to maintain these copies. We therefore conjecture that most or all of them are nonexpressed pseudogenes.
Figure 3 summarizes conserved sequence elements associated with vtRNAs at the two syntenically conserved eutherian loci. Consensus sequences and sequence logos of the conserved elements are also listed in the Supplementary Material online. Although the tandem copies at the VTRNA1 locus have highly similar upstream regions in primates, there are substantial differences in the sequence motifs compared with the VTRNA2 locus. Although meme uncovers different sequence elements depending on parameter settings, the distinction between VTRNA1 and VTRNA2 is clearly visible for almost all parameter choices. The sequence motifs in figure 3 include most of the well-known polymerase III associated elements that have been described for both vtRNAs and snRNAs (see e.g., Vilalta et al. 1994; van Zon et al. 2001; Domitrovich and Kunkel 2003; Kickhoefer et al. 2003; Englert et al. 2004; Wise et al. 2007). Interestingly, the VTRNA1 promoters have a TATA box, which is derived in rodents (Vilalta et al. 1994; Kickhoefer et al. 2003). In contrast, no TATA box was found for the VTRNA2 genes.
The rich promoter structure and the clear distinction between two eutherian loci allow us to use these sequence motifs to identify functional vtRNA genes also in cases where the genomic location could not be determined based on flanking genes. This includes low-coverage genomes such as those of the bushbaby (Otolemur garnettii) or the elephant (Loxodonta africana) and shotgun traces of genome projects in progress. Positive predictions are indicated by colored circles in figure 2; the corresponding sequences have been compiled in the Supplementary Material online.
Other Tetrapod vtRNAs
Beyond mammals, genomes are available for only four additional tetrapods: a frog (X. tropicalis), two birds (chicken [G. gallus] and zebrafinch [Taeniopygia guttata]), and a lizard (A. carolinensis). The frog vtRNAs form a cluster downstream of the ZMAT2 homolog. Whereas only a single candidate is found in the chicken genome (Mosig, Chen, and Stadler 2007), a cluster consisting of six potentially expressed vtRNAs is found in the lizard genome (fig. 4). No convincing candidate was found in the genome of the zebrafinch T. guttata. This may be due to the preliminary status of the genome assembly.

vtRNA locus in the lizard and frog genomes. Pictures taken from the UCSC genome browser.
Although somewhat rearranged in chicken and lizard, the genomic locations of the tetrapod vtRNA candidates are clearly syntenic to the mammalian VTRNA1 locus. The identity of the Xenopus sequence, furthermore, is unambiguous because it is almost identical to the two experimentally known bullfrog vtRNAs (Kickhoefer et al. 1993). The chicken candidate, also identified in Mosig, Guofeng, et al. (2007), matches the unannotated 96 nt ncRNA GGN147 (GenBank accession number EU240351 from a direct submission by Zhang Y, Wang J, Huang SJ, Zhu XP, Deng W, Yang N, Chen Y, Wu RM, Chen RS, Zhu DH: “Systematic identification of Gallus gallus small non-coding RNAs.”)
Latimeria and Shark
In addition to genome projects, we also profit from the interest in the protocadherin gene cluster. This locus was sequenced in the coelacanth L. menadoensis (Noonan et al. 2004) and more recently also in the elephant shark (C. milii; Yu et al. 2008). Searching the region upstream of the pcdh genes with Gotohscan resulted in a single shark vtRNA and a pair of hits in the coelacanth, one of which is probably the functional vtRNA, whereas the other appears to be a truncated pseudogene. Because the entire region surrounding the protocadherin cluster is syntenically conserved between shark and human (Yu et al. 2008), we can conclude that the VTRNA1 locus was present already in the ancestral jawed vertebrate.
Teleost vtRNAs
Multiple vtRNA candidates were found for all five available teleost genomes. By their genomic location, they can be grouped into two classes. The first group, which most likely comprises functional vtRNA genes, is located at the pcdh2 locus, one of the two paralogous copies of the ancestral pcdh locus in teleosts (Tada et al. 2004; Yu et al. 2007) that arose through the fish-specific genome duplication (FSGD; Taylor et al. 2003; Vandepoele et al. 2004). In contrast, the other paralog of the protocadherin locus does not contain a recognizable vtRNA in any of the investigated teleost genomes.
Reconstruction of the ancestral karyotypes based on comparisons of teleost fish and tetrapod genomes has suggested that several major chromosomal rearrangements occurred in the fish lineage within a short period after the FSGD (Kasahara et al. 2007). In particular, several local changes have modified the surrounding of the two paralogous pcdh loci, see also figure. 5. The analysis of the teleost loci is complicated by the fact that the fugu genome shows a substantial amount of misassembly within the protocadherin loci, presumably in part due to the similarity between many protocadherin genes (Yu et al. 2007). The aberrant fugu candidate vtRNA at this position, which lacks almost the entire variable loop region, however, was confirmed by independent sequencing of the pcdh2 locus by Yu et al. (2007). It remains surprising that the fugu and tetraodon vtRNA candidates are much more different than one would expect for these closely related species. This may be due to rapid evolution of the protocadherin region, as observed also within primates, or due to persisting inaccuracies in the current genome assemblies of one or both species.
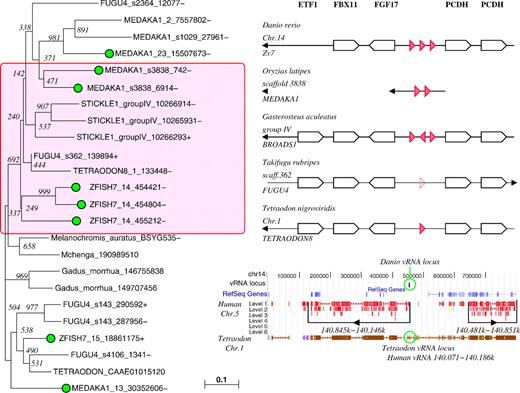
Evolutions and locations of vtRNAs in teleosts. Left panel: Neighbor-Joining tree of the teleost vtRNA candidates, highlighting the sequences located at the pcdh2 locus and their most likely paralogs in the medaka (Oryzias latipes) genome. Additionally, putative vtRNA sequences from two cichlids (Melanochromis auratusand Mchenga conophoros, NCBI Trace Archive) and from the herring (Gadus morrhua, NCBI EST DB) are included. Sequences with experimental evidence for expression are marked with a green bullet. Right panel (top): genomic organization of the pcdh2 locus in the five sequenced teleosts. Right panel (bottom): UCSC genome browser map of zebrafish (Danio rerio) pcdh2 locus showing synteny with the human pcdh region on Chr.5 and with the pcdh2 locus in the pufferfish Tetraodon nigroviridis. Although largely conserved among teleosts, the pcdh2 has been broken up and rearranged locally in teleosts relative to the ancestral state still present in the human genome.
The second group of candidates is more heterogeneous and does not show recognizable syntenic conservation. Some of these sequences are almost certainly pseudogenes, judging from mutations that interrupt the closing stems that otherwise are nearly perfectly conserved. In both medaka (O. latipes) and zebrafish (D. rerio), we verified the expression of the vtRNA genes at the syntenically conserved pcdh2 locus as well as expression of some additional homologs, see Results for Teleost vtRNAs. The analysis of the upstream regions with meme did not identify significant sequence motifs, suggesting that the promoter elements have evolved fairly rapidly in teleosts.
Interestingly, one of the zebrafish vtRNA candidates located in the pcdh2 locus was annotated as a microRNA, mir-733, based on the expression of short RNA resembling a mature microRNA (Kloosterman et al. 2006). In complete analogy to the human mir-866, this is probably a mis-annotation, see also Teleost vtRNAs in the Results.
Basal Deuterostomes
A collection of related hits was detected in the genome of the lamprey (P. marinus). Using these as queries for a Blast search against the same genome reveals hits to seven contigs (number of hits given in parenthesis): Contig 9902 (1) and two groups of contigs, each covering a genomic region according to sequence alignments, 36465 (3), 33930 (3), 30520 (2) and 11460 (1), 21649 (1), 5840 (1). Consequently, there are only five distinct vtRNA candidates, of which at least three form a rather tightly linked cluster. Due to the fragmented nature of the current lamprey genome assembly, we cannot determine whether some or all the vtRNA candidates are located in the vicinity of the lamprey protocadherin.
Three candidate sequences were found in sea squirt genomes. In the Ciona intestinalis genome, one on chromosome 3p, the other one on 9q. Only the first has a (syntenically conserved) counterpart in C. savignyi. Mutations in the putative terminal stem of the copy on chr.3p in C. intestinalis make it likely that this extra sequence is a pseudogene.
Six candidates were identified in the genome of the lancelet (B. floridae). The sequences are located on scaffold 41 (assembly 2.0) in two tight clusters containing three vtRNAs each. Phylogenetic analysis of the six sequences suggests that the two subclusters arose by independent expansions following an ancestral duplication of the vtRNA. In the lancelet, all homologs of protocadherin detectable by TBlastN are located on different scaffolds than the vtRNA candidates.
The existence of a sea urchin (S. purpuratus) vtRNA was reported previously (Stewart et al. 2005). Our search strategy resulted in two series of tightly clustered hits for the sea urchin and more than a dozen shotgun traces for the acorn worm. In both cases, we then used BlastN to retrieve all putative homologs from the respective genomes. Our candidates are located on scaffolds StPu13288 and StPu11479 of the S. purpuratus genome (Sea Urchin Genome Sequencing Consortium 2006), assembly 2.1. Nearly identical sequences were also found in the 454 data of the sea urchins Strongylocentrotus franciscanus and Allocentrotus fragilis (available at http://www.hgsc.bcm.tmc.edu/projects/seaurchin/). The two S. purpuratus scaffolds share large amounts of almost identical sequence and exhibit large gaps; it is not unlikely, hence, that they represent the same genomic locus. If this assumption is correct, the sea urchin vtRNA genes form a single cluster with a length of ∼20 kb containing 7–10 vtRNA genes and pseudogenes. The acorn worm read M228602676 contains two vtRNA genes or pseudogenes, suggesting that this hemichordate may have a similar vtRNA cluster.
The motifs identified by meme within the upstream regions of the vtRNAs of chordates (except teleosts and mammals) are more similar among the clustered copies than between species. This is consistent with the observation that many polymerase III-transcripts are subject to concerted and/or birth-death-process evolution (Nei and Rooney 2005).
vtRNA Secondary Structures
We constructed separate alignments for the two eutherian loci, teleosts, other gnathostomes, and basal deuterostomes. Figure shows an alignment of the sequence logos derived from these sequence alignments. The internal polymerase III promoter elements, box A and box B (Kickhoefer et al. 1993; Vilalta et al. 1994; Kickhoefer et al. 2003), as well as the terminator signal are clearly visible. Interestingly, we observed the insertion of an `A’ to box A of some of the eutherian vtRNAs. Furthermore, vertebrates have lost a junk of variable sequence immediately downstream of the box B that is present in the more basal lineages.
As reported previously (Mosig, Chen, and Stadler 2007), vtRNAs form a conserved panhandle-like secondary structure with a well-conserved extended stem-loop structure connecting 5′ end and 3′ end of the molecule. This structure also involves the box A sequence. The manually generated alignments and the locarna alignments agree almost perfectly in this region. This highly conserved part of the vtRNA structure was used to derive the secondary structure constraints of the rnabob descriptors.
The box B, on the other hand, does not take part in conserved structural features, albeit in vertebrates, the stem-loop structure overlaps the last 1 or two nt of the box B. In the basal lineages, box B and the 3′ side of the stem-loop structure are separated by at least 10 nt of intervening sequence (fig. 6). The base pairing of box A likely contributes to the sequence conservation in the 3′ region of the vtRNAs. The vtRNAs of mouse and rat are peculiar in that it has a duplicated box B (Kickhoefer et al. 2003). A similar extension is observed in the tupaia.
![Comparison of consensus secondary structures and sequence logos derived from separate alignments of the deuterostome vtRNAs. (A) Basal deuterostomes (sea urchins [Strongylocentrotus franciscanus, Allocentrotus fragilis, and Strongylocentrotus purpuratus], acorn worm [Saccoglossus kowalevskii), lancelet (Branchiostoma floridae), tunicates, and lamprey (Petromyzon marinus]), (B): teleosts, (C): shark (Callorhinchus milii) and sarcopterygii without eutheria, (D): eutherian vtRNA-4, and (E): eutherian pcdh locus. The upper panel shows the consensus secondary structure computed from locarna-based alignments, the sequence logos were derived from the manually curated sequence-based alignments. For the latter, the base pairs of the consensus structures of the latter are indicated by “<“and “>”pairs. They agree almost exactly with the locarna-based structures. Circles in the secondary structure drawings indicate compensatory substitutions, whereas letters in gray imply that one or two of the aligned sequences cannot form the corresponding base pair. Gaps in the alignment of the logos below are indicated by *, framed blocks are essentially gap less in each of the sequence alignments. For gap-rich regions and the variable loop region of the vtRNA, only the length ranges of intervening sequences are shown. The internal polymerase III promoter elements are indicated (box A: blue and box B: cyan, cp.fig. 3).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/mbe/26/9/10.1093_molbev_msp112/2/m_molbiolevolmsp112f06_4c.jpeg?Expires=1750247912&Signature=kartRq0fnN7JiQPD5lC5V~xZ16yViN67MUCzv-LtmsqE5bL5xEHtcomJhAOcQ2HzN~luj~Y8bk-vKvUAW8J5dDk-cfnu~SDbwFrhcTRLLelwJtmo5WaYrdHjE0Zcghie57buxhAZgB5lcamrEW5pEwJW11vhZpV2zfJdIMJOP67d-kFCsVlQWUTctCcx1yGOSn543JKWKinKuFupk~zmkZjTzSC6XD-8vR1pLoyrHBMPuJb4OVPSz-yavPKXnZqsw6d1RbNJZDmxYF0Epj-bx5vfOGPGkEUQZl6ahWHS5tf4TWoeVwYW7ldCI9~zSHZAph6aQ~6R3fLTzxiGMqYKAg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Comparison of consensus secondary structures and sequence logos derived from separate alignments of the deuterostome vtRNAs. (A) Basal deuterostomes (sea urchins [Strongylocentrotus franciscanus, Allocentrotus fragilis, and Strongylocentrotus purpuratus], acorn worm [Saccoglossus kowalevskii), lancelet (Branchiostoma floridae), tunicates, and lamprey (Petromyzon marinus]), (B): teleosts, (C): shark (Callorhinchus milii) and sarcopterygii without eutheria, (D): eutherian vtRNA-4, and (E): eutherian pcdh locus. The upper panel shows the consensus secondary structure computed from locarna-based alignments, the sequence logos were derived from the manually curated sequence-based alignments. For the latter, the base pairs of the consensus structures of the latter are indicated by “<“and “>”pairs. They agree almost exactly with the locarna-based structures. Circles in the secondary structure drawings indicate compensatory substitutions, whereas letters in gray imply that one or two of the aligned sequences cannot form the corresponding base pair. Gaps in the alignment of the logos below are indicated by *, framed blocks are essentially gap less in each of the sequence alignments. For gap-rich regions and the variable loop region of the vtRNA, only the length ranges of intervening sequences are shown. The internal polymerase III promoter elements are indicated (box A: blue and box B: cyan, cp.fig. 3).
Both the “loop” region and the sequence between box B and the closing stem in the basal lineages are highly variable. Even with the rather dense taxon coverage reported here, at best partial sequence alignments within subgroups can be obtained. The sequence/structure alignment algorithm locarna finds some partially conserved structural features within the loop region of the eutherian vtRNAs, in particular in vtRNA2-1. Interestingly, the regions binding chemotherapeutic drugs are located within this highly variable loop region (Gopinath et al. 2005). The vtRNAs arising from the VTRNA1 locus show massive structural changes in the loop already among primates. Neither sequence nor structure patterns in the loop are sufficiently conserved to be of use for homology search.
VtRNA Expression
Human vtRNAs
The abundance of vault particles differs substantially between different human cell types (van Zon et al. 2003). Here, we have investigated the relative expression of the four vtRNA genes in five different human cancer and two nontumor cell lines in which vault particles are abundant. Expression levels normalized to genomic DNA for each cell line are shown in figure 7. We observe that the four vtRNA genes are expressed in different proportions depending on the cell line.
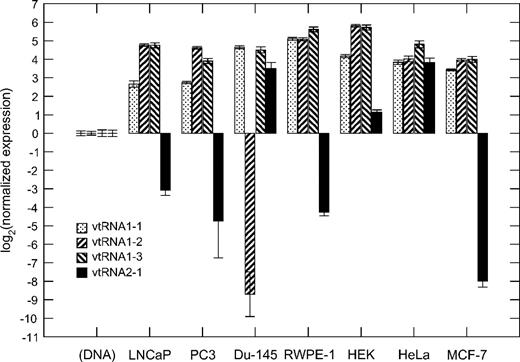
Expression of the four human vtRNA loci in five different cancer and two nontumor cell lines. Expression levels are normalized to a genomic DNA standard curve for each cell line.
Although the expression levels of the three genes at the VTRNA1 locus are similar in most cell lines, the novel VTRNA2 gene shows substantial variations. Interestingly, VTRNA1-2 expression is missing in Du-145 cells, whereas the expression ratio of the most recent duplicates, VTRNA1-2 and VTRNA1-3, is close to 1 in other cell lines.
Evidence from Short RNA Sequencing
The Human microRNA Atlas (Landgraf et al. 2007) contains small RNA sequences that uniquely map to the VTRNA2 locus; hence, this vtRNA has been misannotated as microRNA mir-886. Figure shows deep sequencing of small RNA libraries uncovering multiple uniquely mapped reads at all four vtRNA loci. Although Illumina's Small RNA Protocol is tailored toward detecting small RNAs, the data convincingly demonstrate expression from all four vtRNA loci. In fact, we find a high degree of similarity among the transcription patterns in the human, chimpanzee, and rhesus macaque data sets.
The pattern of short reads that map to VTRNA2-1 is distinctly different from the pattern typically observed for true microRNAs, as shown in the lower panel of figure 8: 1) The starting points of the short reads are more heterogeneous in the vtRNAs. 2) The location of the reads relative to the peaks of the sequence conservation does not match the pattern expected for microRNAs. 3) The distance between the putative mature miR and miR* sequences is very large for a mammalian microRNA.
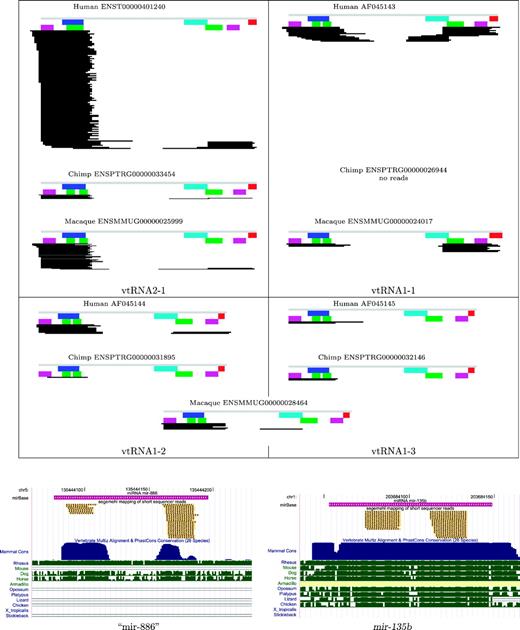
Short read sequencing data. Upper panel: patterns of short reads mapping to the primate vtRNAs. The respective libraries are combined. The gray bar indicates the vtRNA sequence with annotation for box A (blue), box B (cyan), and the terminator signal (red) as in figure 3. The two major conserved stem regions in magenta and green. Expression levels differ significantly between four (three in Macaca) vtRNA genes. The panel was prepared using the custom-made C++ program soap2eps Lower panel: comparison of the vtRNA2-1, which was annotated as mir-886and mir-135b, a bona fide microRNA. The panel was produced with the UCSC genome browser.
The recent study by Nandy et al. (2009), furthermore, shows that vtRNA2-1 cofractionates with the major vault protein MVP in a sucrose gradient, which is a strong indication that vtRNA2-1 indeed is a functional vtRNA.
Expression of Teleost vtRNAs
All predicted genes at the canonical pcdh2 locus are expressed in both zebrafish (D. rerio) and medaka (O. latipes), as verified by RT-PCR. Surprisingly, we also find positive RT-PCR results for fourth zebrafish homolog and for two additional loci in the medaka.
Mapping the four expressed vtRNAs and their homologs to the zebrafish genome reveals that two of these loci are annotated as microRNA (ZFISH7_14_454421- = dre-mir-733) and a predicted putative microRNA (ZFISH7_14_454804- = ENSDARGrespectively. Both loci are expressed. As in the case of the eutherian mir-886, this is most likely a misannotation. In addition to the evidence derived from comparative sequence analysis, we observe that a fragment amplified by PCR with a 3′primer starting well outside the putative mir-733 amplifies a vtRNA-like product.
For both MEDAKA1_s3838_742 and ZFISH7_14_454804-, Northern blots show that the transcripts match the predicted length of 97 and 102 nt, respectively (fig. 9).
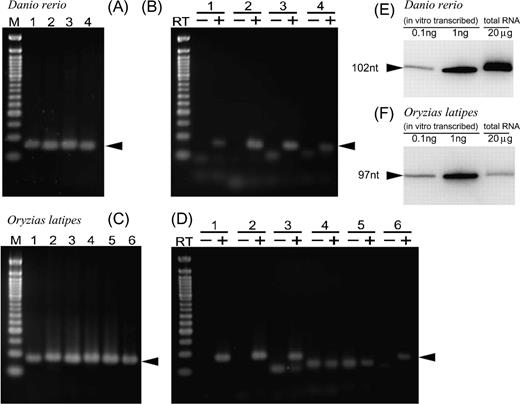
Detection of teleost vtRNA transcripts. Specific primer sets were designed for amplification of each putative teleost vtRNA transcript. (A) PCR amplification of zebrafish (Danio rerio) vtRNA genes from genomic DNA. Lane 1: ZFISH7_14_455212-, lane 2: ZFISH7_14_454804-, lane 3: ZFISH7_14_454421-, and lane 4: ZFISH7_15_18861175+. (B) PCR amplification of zebrafish (D. rerio) vtRNA genes from cDNA library. (C) PCR amplification of medaka (Oryzias latipes) vtRNA genes from genomic DNA. Lane 1: MEDAKA1_s3838_6914-, lane 2: MEDAKA1_s3838_742-, lane 3: MEDAKA1_23_15507673-, lane 4: MEDAKA1_s1029_27961-, lane 5: MEDAKA1_2_7557802-, and lane 6: MEDAKA1_13_30352606. (D) PCR amplification of medaka (O. latipes) vtRNA genes from cDNA library. (RT-): PCR amplification using a cDNA library generated from a mock RT reaction in which the thermoscript reverse transcriptase was omitted. (E and F) Northern blotting analysis of D. rerioZFISH7_14_454804, 102 nt (E), and O. latipesMEDAKA1_s3838_742, 97 nt (F). In each case, 20 μg of total RNAs (right-most lanes) were electrophoresed on a 6% denaturing polyacrylamide gel electrophoresis gel and analyzed by northern blotting. As markers for size estimation and ng, 0.1 and 1 ng (left and middle lanes) of in vitro transcribed medaka vtRNA were included in the analysis.
Discussion
We have performed the first comprehensive computational analysis of the little studied family of vtRNAs. Using an iterative procedure employing Blast, full dynamic programming, and pattern-based search procedures, we have identified more than 100 vtRNA genes in almost all the currently available deuterostome genomes. Expression of the predicted vtRNA genes was verified by PCR in human, zebrafish, and medaka. The identification of vtRNA genes outside of eutherian mammals is complicated by the highly variable loop region in the interior of the gene, which is not at all informative beyond genus level. This leaves only the information within the short 5′ and 3′ terminal regions as basis for search patterns. Current homology software can only be employed to retrieve candidate sequences that the need to be evaluated subsequently with respect to features such as conservation of sequence motifs, structural features, genomic location, and consistency of results among different queries.
In gnathostomes, the functional VTRNA1 genes are usually located upstream of the protocadherin cluster. Within eutheria, however, there is a second locus harboring a functional VTRNA2 gene between the TGFB1 and SMAD5 genes. In several eutherian lineages, only one of the two loci contains a vtRNA gene. This apparent complementation strongly supports the conclusion that the transcript from the VTRNA2 locus is indeed an additional vtRNA. While this contribution was under review, the association of human VTRNA2 with the vault complex has also been demonstrated experimentally (Nandy et al. 2009). Analysis of the external polymerase III-promoter elements revealed that the two eutherian vtRNA types exhibit significantly different upstream elements, potentially explaining why the relative expression of vtRNA paralogs differs substantially among human cell lines. The different promoter structures were then used to identify likely functional vtRNA genes in several species where information on genomic location was unavailable.
The secondary structures of vtRNAs are dominated by the GC-rich terminal stem-loop, which on the 5′ side contain the box A promoter sequence. The stem-loop is subject to a large number of compensatory mutations, which provide further evidence for the inferred consensus structures. In contrast, the intervening loop region is highly variable in length, sequence, and structure. Conserved structural elements are discernible only in the eutheria-specific VTRNA2 family and—to a lesser extent—in the most basal deuterostome lineages.
Both the structural organization and the genomic behavior of vtRNAs is reminiscent of the Y RNAs, another relatively rarely studied class of polymerase III transcripts (Perreault et al. 2007; Mosig, Guofeng, et al. 2007). Both classes of RNAs have a similar size, a similar promoter structure, they appear in a single or a small number of functional copies, and they are typically transcribed from a small gene cluster. Both families are stably associated with their adjacent protein-coding genes for unknown reasons. The only major distinction is that the Y RNA cluster in tetrapods is composed of four or five ancient paralogs dating back to the divergence of Actinopterygii and Sarcopterygii. In contrast, clustered vtRNA paralogs are evolutionarily young and have arisen independently multiple times. Only within primates, there is a clear distinction of VTRNA1-1 versus VTRNA1-2 and VTRNA1-3 sequences, indicating that gene duplication occurred early in the primate lineage. Because the molecular function of vtRNAs remains in the dark, we cannot even speculate what the reasons might be for such lineage-specific evolutionary patterns.
The insights into the peculiarities of the evolutionary history of vtRNAs are an important facet in our understanding of the evolutionarily forces shaping the polymerase III-transcriptome in general. It appears that almost all polymerase III-transcripts are prone to spawning lineage-specific families of repetitive elements. The most famous examples include tRNA-derived B2 elements and 7SL RNA-derived Alus. Similarly, large numbers of repeat-like pseudogenes originating from the different Y RNA paralogs and from the 7SK are found throughout mammals. For vtRNAs, the genome of the guinea pig (C. porcellus) is the only example with a large number of pseudogenes. Most polymerase III-transcripts, and also some of the small pol-II–derived RNAs, including snRNAs (Dávila López et al. 2008; Marz et al. 2008) and snoRNAs (Weber 2006; Shao et al. 2009), behave much like mobile elements. Presumably, this is achieved by frequent gene duplications that often carry the localized promoters with them. In the case of polymerase III-transcripts with mostly internal promoters and snoRNAs, which are processed from introns, this does not pose a restriction. Most other polymerase III-transcripts usually require external promoter elements. Nevertheless, most of them are highly mobile. The U6 snRNA (Marz et al. 2008) and the SmY RNAs (Jones et al. 2009) appear in many species as multicopy genes scattered around in the genome. The genomic anchoring of the vtRNAs and Y RNAs is therefore quite exceptional and calls for an explanation. Due to lack of a functional understanding of both vtRNAs and Y RNAs, however, it is unclear whether synteny is preserved because of functional linkage of the ncRNAs with their neighboring genes.
In this contribution, we have limited our attention to deuterostomes, although vault particles are evolutionarily older. For unknown reasons, they appear to be absent from the two best-studied invertebrate model organisms, Caenorhabditis elegans and Drosophila melanogaster. A cursory TBlastN search indeed revealed no sign of an MVP homolog in any ecdysozoan genome (see also Suprenant et al. 2007). As a consequence, we do not expect to find vtRNAs in these organisms either. Although MVP is present in several other invertebrates (notably in several lophotrochozoans) and in a variety of other eukaryote lineages, this part of the phylogeny appears too distant for direct homology search with currently available methods.
We gratefully acknowledge Liang Zhu (PICB) for help with fragrep2 searches and Manja Marz (Leipzig) for providing U6 snRNA promoter sequences for comparison. This work was supported in part by the Interdisciplinary Center for Clinical Research of the Medical Faculty, University of Leipzig (IZKF, research network funding, ncRNA initiative), a formel.1 grant by the Medical Faculty, University of Leipzig, the Leipzig Interdisciplinary Research Cluster of Genetic Factors, Clinical Phenotypes and Environment funded by the State of Saxony, by a National Science Foundation CAREER Award (MCB0642857) to J.J.-L.C., and by the sixth Framework Programme of the European Union, projects EMBIO (012835) to S.J.P., and SYNLET to J.H. and P.F.S.
References
Author notes
Hervé Philippe, Associate Editor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}