





Abstract
Nucleotide variation in the Acph-1 gene region was analyzed in a natural population of Drosophila subobscura from Bizerte (Tunisia). The lines studied differed in their gene arrangement for segment I of the O chromosome: 21 lines were O3+4+8, 21 were O3+4+23, and 3 were O3+4. According to chromosomal phylogenies, O3+4 is a central arrangement from which O3+4+8 and O3+4+23 originated. Strong genetic differentiation at Acph-1 was detected among the different arrangements, which is reflected in strong linkage disequilibrium between the variants at informative polymorphic sites and the type of arrangement. Estimates of silent nucleotide diversity are slightly lower within O3+4+23 (πsilent = 0.0166) than within O3+4+8 (πsilent = 0.0228) or O3+4 (πsilent = 0.0234). In contrast, nonsynonymous nucleotide diversity estimates (around 0.1%) are similar in the three arrangements. Most nonsynonymous rare variants are singletons, which results in highly significant Tajima's neutrality tests within the young O3+4+8 and O3+4+23 arrangements. This test is not significant for nonsynonymous mutations within a large Spanish O3+4 sample. In addition, a significant and marginally significant excess of nonsynonymous polymorphism was detected by the McDonald and Kreitman test within O3+4+23 and O3+4+8, respectively. This excess results in a rather high neutrality index (NI = 5.25) when both arrangements are jointly analyzed, in contrast to its value within the old O3+4 arrangement (NI = 1.74). The pattern of variation at Acph-1 within the young arrangements is unusual for nuclear genes and has the same characteristics previously detected in most genes of the mitochondrial genome. Assuming that most nonsynonymous mutations at Acph-1 are under weak negative selection, a smaller effective size of the young arrangements relative to O3+4 might explain the observed results. The relatively low frequency of O3+4+8 and O3+4+23 in the distribution area of D. subobscura, the more recent origin of these arrangements relative to O3+4 and the suppression of recombination in heterokaryotypes might contribute to the relatively small effective size of the young arrangements. Therefore, present results indicate that the differences in effective size and recombination caused by chromosomal arrangements are modulating nonsynonymous variation at Acph-1.
Introduction
Deviations from the neutral model of molecular evolution (Kimura 1983) caused by a significant excess of nonsynonymous polymorphism have been extensively reported in the mitochondrial genome (mtDNA). Several organisms such as Drosophila, mice, marsh rats, and human present this excess of amino acid variation in mtDNA genes (Ballard and Kreitman 1994; Nachman, Boyer, and Aquadro 1994; Rand, Dorfsman, and Kann 1994; Nachman et al. 1996; Rand and Kann 1996; Hasegawa, Cao, and Yang 1998; Kennedy and Nachman 1998; Wise, Sraml, and Easteal 1998). Different selective factors have been proposed to explain this excess of nonsynonymous polymorphism. First, balancing selection maintaining protein variation or, alternatively, positive selection fluctuating over time or space might contribute to an excess of amino acid variation (Gillespie 1991). Second, mild negative selection acting on replacement mutations can cause these mutations to be maintained in the population at low frequency, but rarely to become fixed (Ohta 1992). Third, a relaxation of selection acting in the past might maintain previously nontolerated nonsynonymous polymorphism (Takahata 1993).
In contrast to mtDNA genes, only a few nuclear Drosophila genes exhibit a significant excess of nonsynonymous polymorphism. In Drosophila melanogaster, this excess has been detected at Gld (Hamblin and Aquadro 1997), Pgi (McDonald and Kreitman unpublished, cited in Moriyama and Powell 1996), Pgm (Verrelli and Eanes 2000), and bcd (Baines et al. 2002). Indeed, an excess of nonsynonymous fixed differences is a much more common result in this species (Moriyama and Powell 1996; Fay, Wyckoff, and Wu 2002). The paucity of nuclear genes showing an excess of nonsynonymous polymorphism led Weinreich and Rand (2000) to suggest that the lack of recombination in the mtDNA and the distribution of the selective effects of newly arisen mutations might explain the commonly detected excess of nonsynonymous polymorphism in mitochondrial genes.
Although most studies of nucleotide variation in Drosophila nuclear genes correspond to species of the melanogaster group, some surveys have been carried out in species of the obscura group. Only the analysis of nucleotide variation in the Acph-1 gene of D. subobscura has revealed an excess of nonsynonymous polymorphism (Navarro-Sabaté, Aguadé, and Segarra 1999a). In this case, however, the region under study maps at section 91C, very close to one of the breakpoints of the O3 inversion. As a consequence, variation in the Acph-1 gene was analyzed separately for lines differing by their chromosomal arrangement for segment I (sections 91–99) of the O chromosome: Ost and O3+4. The excess of nonsynonymous polymorphism was significant in the Ost arrangement, but not in O3+4, which suggested an effect of chromosomal inversion polymorphism on amino acid variation, at least at Acph-1. In addition, assuming that most nonsynonymous mutations at Acph-1 are under weak negative selection, the contrasting results between O3+4 and Ost are consistent with a much smaller effective size of Ost relative to O3+4 (Navarro-Sabaté, Aguadé, and Segarra 1999a).
Chromosomal phylogeny in D. subobscura (Krimbas 1992) indicates that O3+4 is a central arrangement from which several arrangements, such as O3+4+8 and O3+4+23, originated, giving rise to the O[3+4] inversion complex. Therefore, it can be inferred unambiguously that these two arrangements are younger than O3+4. The presence of such a complex system of gene arrangements in segment I of the O chromosome of D. subobscura offers the opportunity to further analyze the putative effect of chromosomal inversions on nonsynonymous variation. Indeed, differences in the effective size of these arrangements are expected as a result of differences in their frequency, and even in their age. The effective size of young arrangements might be strongly affected by the extreme bottleneck produced in their origin, which should cause a reduction in the long-term effective size of the arrangement.
Here, we have studied nucleotide polymorphism at Acph-1 in the young O3+4+8 and O3+4+23 arrangements. The quite low average frequency of these two arrangements in the distribution area of D. subobscura and their derived character suggest that their effective sizes should be lower than the effective size of the old O3+4 arrangement. These differences in the effective size and the well-established role of inversions as suppressors of recombination might affect nonsynonymous variation if nonsynonymous mutations are under weak selection, as shown in the case of Ost. The results obtained support this prediction. A significant and marginally significant excess of nonsynonymous polymorphism is detected by the McDonald and Kreitman test (McDonald and Kreitman 1991) in the young arrangements O3+4+23 and O3+4+8, respectively. In addition, highly significant Tajima's D statistics (Tajima 1989) for nonsynonymous mutations were detected within both gene arrangements. Therefore, variation at Acph-1 within the young arrangements O3+4+23 and O3+4+8 presents the same pattern previously detected for most mitochondrial genes of D. melanogaster. According to these results, it can be inferred that differences in effective size and recombination caused by chromosomal arrangements are modulating nonsynonymous variation in a nuclear gene.
Materials and Methods
Fly Samples
Forty-five isochromosomal lines of D. subobscura were established from flies collected at Bizerte (Tunisia) in 1996. Most of these lines had been previously analyzed for variation at the rp49 gene region (Rozas et al. 1999). Lines differed in their arrangement for segment I of the O chromosome: 21 lines were O3+4+8, 21 were O3+4+23, and 3 were O3+4. All O3+4+23 lines presented an additional inversion (inversion 6) in segment II; therefore, their complete gene arrangement for the O chromosome was O3+4+23+6.
In Situ Hybridization
Polytene chromosomes slides were obtained after dissection of the salivary glands from third-instar larvae heterozygous for different arrangements of segment I: O3+4/Ost, O3+4/O3+4+23, O3+4/O3+4+8, and O3+4+23/O3+4+8. The DNA of the recombinant phage λDsubAcph-1.1 (Navarro-Sabaté, Aguadé, and Segarra 1999b) was labeled with biotin-16-dUTP and used as probe. Prehybridization, hybridization, and detection conditions were as described in Segarra and Aguadé (1992).
DNA Sequencing
Genomic DNA of the isochromosomal lines was purified according to Ashburner (1989). The complete Acph-1 gene region, approximately 2.2 kb long, was amplified by polymerase chain reaction (PCR). Amplification primers and PCR conditions were as described in Navarro-Sabaté, Aguadé, and Segarra (1999a). The PCR products were purified with QIAquick columns (Qiagen) and used as templates for sequencing with primers designed at approximately every 300 nucleotides. Both DNA strands were completely sequenced. The cycle sequencing kit from PerkinElmer was used to perform the sequencing reactions according to the manufacturer's instructions. The reaction products were run on an Abi Prism 377 automated DNA sequencer.
Complete sequences of the 45 Bizerte lines were multiply aligned manually with the SeqApp (version 1. 9) program (Gilbert 1993), and the sequence of line B2ST (Navarro-Sabaté, Aguadé, and Segarra 1999b) was used as the reference sequence. A multiple alignment including the 53 Spanish lines previously analyzed for the Acph-1 gene region (Navarro-Sabaté, Aguadé, and Segarra 1999a; European Molecular Biology Laboratory (EMBL) accession numbers AJ389424 to AJ389476) was also obtained. Alignments were edited with the MacClade program (Maddison and Maddison 1992) prior to analysis.
Data Analysis
Data analysis was performed with the DnaSP, version 3.4, program (Rozas and Rozas 1999). Nucleotide polymorphism was estimated as: S (the number of segregating sites), k (the average number of pairwise nucleotide differences), π (nucleotide diversity; Nei 1987) and 𝛉 (the heterozygosity per site expected in a population at mutation-drift equilibrium given the observed S value; Watterson 1975). Genetic differentiation between arrangements or between populations was estimated by the average number of nucleotide differences per site (dxy; Nei 1987). The
Different neutrality tests (Tajima 1989; McDonald and Kreitman 1991) were performed to determine whether the level and pattern of nucleotide variation detected within gene arrangement conformed to neutral model expectations. Statistical significance of the Tajima's D observed values was obtained by coalescent simulation with no recombination (Hudson 1990) after 10,000 replicates. The probability for each test statistic was obtained as the frequency of replicates with a value lower than the observed statistic (one-tailed test).
The gene genealogy of the Acph-1 sequences was reconstructed by the Neighbor-Joining method (Saitou and Nei 1987) implemented in the Mega program, version 2.1 (Kumar et al. 2001). Genetic distances between sequences were estimated according to the Kimura two-parameter model (Kimura 1980) to correct for multiple hits. Sites with polymorphic indels were excluded from the analysis (complete deletion option). Bootstrap percentages for the different nodes were obtained after 500 replicates.
The sequences reported here have been deposited in the EMBL database library under accession numbers AJ563964 to AJ564008.
Results
In Situ Hybridization
The location of the Acph-1 gene in the inversion loop of the heterokaryotypes for the arrangements studied is shown in figure 1. In most nuclei of O3+4/Ost heterokaryotypes, the hybridization signal appears in the asynaptic region of one of the inversion loops; this is in agreement with the location of Acph-1 inside and very close to one of the breakpoints of inversion 3. In O3+4/O3+4+23 and O3+4+23/O3+4+8 heterokaryotypes, the hybridization signal is detected inside the inversion loop and close to one of its ends, but in most nuclei the signal appears in the synaptic region of the loop. Finally, Acph-1 maps in a rather central position of the inversion loop present in O3+4/O3+4+8 heterokaryotypes.
Nucleotide Variation
The multiple alignment of the 45 sequences from the Bizerte sample had 2,169 sites. Nine polymorphic indels with one, two, or seven nucleotides were detected in noncoding regions. These polymorphic indels presented two or three variants segregating in the data set. All polymorphisms with three variants affected the length of runs of a single nucleotide. In addition, line TB21 presented in the third exon of the coding region a 16-nt long deletion that causes a frameshift mutation. After excluding all sites with alignment gaps, a total of 184 nucleotide polymorphisms were detected among the 2,135 sites of the data set (fig. 2). Three different nucleotides were segregating in six sites; thus the minimum number of mutations was 190.
Estimates of nucleotide polymorphism within each chromosomal arrangement are presented in table 1. These estimates were obtained independently for noncoding sites (flanking regions plus introns), silent sites (noncoding sites plus synonymous sites in the coding region), and synonymous and nonsynonymous sites of the coding region. Line TB21 with a frameshift mutation in the coding region was excluded from this analysis, because, for this line, the classification of sites in these different categories is ambiguous. For the O3+4+8 lines from Bizerte, estimated nucleotide diversity was similar (both for all sites and for silent sites) to that present within the O3+4 arrangement either from Bizerte or from Spain (π = 0.0123 and πsilent = 0.0219; Navarro-Sabaté, Aguadé, and Segarra 1999a). In contrast, variation within O3+4+23 is slightly lower when all sites, silent sites, or only synonymous sites in the coding region are considered. In contrast, nucleotide diversity at nonsynonymous sites is similar in the three gene arrangements.
Linkage disequilibrium analysis between pairs of informative sites within each arrangement was also performed. In O3+4+8, 162 of 1,431 comparisons (11%) were significant (P < 0.05) by the χ2 test, but only 2% of the comparisons were significant when the Bonferroni procedure (Weir 1996) to correct for multiple tests was applied. In O3+4+23 the extent of linkage disequilibrium was similar: the percentages of significant comparisons were 12% (99 of 820 comparisons), and only 1% with the Bonferroni correction. The lack of strong linkage disequilibrium is consistent with the rather high recombination detected within gene arrangements. Estimates of the recombination parameter (Hudson 1987) were C = 0.092 within O3+4+8 and C = 0.029 within O3+4+23. In contrast, the minimum number of recombination events (Hudson and Kaplan 1985) detected in the data set was 19 within O3+4+8 and 14 within O3+4+23.
Genetic Differentiation Between Arrangements
The dxy and
Genetic exchange between arrangements can explain the relatively high number of shared polymorphisms. Despite the lack of fixed differences and the presence of shared polymorphisms, genetic differentiation between arrangements prevails because of the relatively high number of almost fixed differences between them. For instance, in 17 of the 49 polymorphisms shared between O3+4+8 and O3+4+23, the most frequent variant differs between the two arrangements. Genetic differentiation between arrangements may also be detected by the analysis of linkage disequilibrium between nucleotide polymorphic sites and the type of chromosomal arrangement. Indeed, the presence of fixed or almost fixed variants between arrangements at a particular site should cause a strong association between the variants at this site and the type of chromosomal arrangement. This analysis was performed for all informative polymorphic sites and the chromosomal arrangement, in a sample including the O3+4+23 and O3+4+8 lines studied. The extent of linkage disequilibrium was estimated by the percentage of comparisons that were significant after applying the χ2 test and by the average r2 value. Despite the lack of fixed differences and the presence of shared polymorphisms between O3+4+23 and O3+4+8 (likely due to gene conversion), 28 of 69 comparisons (about 41%) were significant when all 21 lines of each arrangement O3+4+23 and O3+4+8 were considered (table 3). This rather high percentage contrasts with that around 10% detected within arrangement, which reflects that recombination is restricted in heterokaryotypes. Therefore, the maintenance of both chromosomal arrangements in the population contributes to an overall reduction of recombination in the Acph-1 gene region, as shown by the high percentage of significant associations between the alternative variants of the informative sites and the chromosomal arrangement.
The analysis of linkage disequilibrium between nucleotide polymorphic sites and gene arrangements was extended to the Ost and O3+4 arrangements from the Spanish population (Navarro-Sabaté, Aguadé, and Segarra 1999a). As stated above, the rationale for this analysis is that high levels of linkage disequilibrium in joint samples of two arrangements (relative to linkage disequilibrium within each arrangement) indicate a strong genetic differentiation between them and thus a strong reduction of recombination in heterokaryotypes. For the results to be comparable, the analysis was conducted with samples where the two arrangements considered were equifrequent. The highest genetic differentiation was found in the samples including the Ost lines and the lowest differentiation in the sample with the O3+4 and O3+4+8 lines (table 3).
The extent of genetic differentiation between arrangements is also reflected in the gene genealogy reconstructed from variation at Acph-1. The D. subobscura lines group in two clear clusters (fig. 3). One cluster includes all Ost lines, and the other groups all lines of the O[3+4] inversion complex. Bootstrap percentages supporting the Ost and O[3+4] clusters are 51 and 70, respectively. Within the O[3+4] cluster, some clustering of the lines according to their gene arrangement is observed, although the bootstrap support is in all cases very low. Thus, 19 of the 21 O3+4+23 lines and 13 of the 21 O3+4+8 lines group in unique clusters. In contrast, O3+4 lines group mainly in two clusters: one that includes only O3+4 lines (11 lines), and another that includes 17 O3+4, 4 O3+4+8 and 1 O3+4+23 lines.
Replacement Polymorphism
The ACPH-1 preprotein has 447 residues in all lines except TB21. The frameshift mutation present in this line results in a truncated preprotein with 130 residues; thus this line can be considered a null allele for Acph-1. When this line is aligned in frame with the other lines, a single amino acid replacement (site 1537 in fig. 1) is detected, and no additional changes that disrupt the coding region (as nonsense mutations) are present. After excluding this null allele from the analysis, a total of 22 polymorphic residues were detected in the Bizerte sample: 2 within O3+4, 11 within O3+4+23, and 10 within O3+4+8 (fig. 4). These amino acid polymorphisms result in three different protein haplotypes within O3+4, 10 within O3+4+23, and 10 within O3+4+8. The G/V (glycine/valine) polymorphism at residue 387 is shared by the O3+4+8 and O3+4+23 arrangements. However, the presence of a V in TB167 (O3+4+23) can be explained by a gene conversion tract detected in this line. In contrast, three different variants were detected in this residue within O3+4+23. The rarest variant in all the polymorphic amino acid residues, except in residue 387, was present only once in the sample. According to the sequence of D. guanche (Navarro-Sabaté, Aguadé, and Segarra 1999b), it can be inferred that all nonsynonymous singleton variants are due to derived mutations.
Only three of the amino acid polymorphisms detected in Bizerte (at residues 205, 241, and 387) were previously detected in the Spanish sample (Navarro-Sabaté, Aguadé, and Segarra 1999a). Among them, the R/S (arginine/serine) polymorphism at residue 205 and the N/K (asparagine/lysine) polymorphism at residue 241 were identified as those responsible for the change in mobility between the Acph-1100 and Acph-1>100, and between the Acph-1100 and Acph-1054 electromorphs, respectively, within O3+4.
Neutrality Tests
The Tajima test (Tajima 1989) was used to contrast whether the pattern of variation within each gene arrangement is consistent with predictions of the neutral model. This test was performed independently for silent and for nonsynonymous mutations (table 4). The low number of O3+4 lines from Bizerte prevented performance of this analysis within this chromosomal arrangement in this population. Thus, neutrality tests for this gene arrangement were performed with the Spanish random sample (Navarro-Sabaté, Aguadé, and Segarra 1999a). Even under the conservative assumption of no recombination, the test for nonsynonymous mutations is highly significant within the young arrangements: O3+4+23 and O3+4+8. In contrast, the test is not significant for nonsynonymous mutations within O3+4, the oldest gene arrangement. None of the tests performed in the different arrangements was significant for silent mutations. The negative value of the significant Tajima's D statistic indicated a significant excess of low frequency (mostly singletons) nonsynonymous mutations in the data set. Although Tajima's D was also negative for silent sites within O3+4+23 and O3+4+8, the relative excess of low frequency silent mutations was not significant in these arrangements. In addition, the number of singleton and non-singleton polymorphisms differs significantly for nonsynonymous and synonymous mutations both within O3+4+23 (χ2 = 11.27, 1 df, P = 0.0008) and O3+4+8 (χ2 = 5.37, 1 df, P = 0.0205). The same test is not significant within O3+4 (χ2 = 0.13, 1 df, P = 0.7156). These results clearly indicate a different pattern of variation for silent and nonsynonymous mutations in the young arrangements (O3+4+23 and O3+4+8).
The McDonald and Kreitman (1991) test of neutrality (MK test) was also performed within each gene arrangement using the sequence of D. guanche (Navarro-Sabaté, Aguadé, and Segarra 1999b) in the interspecific comparison (table 5). This test is based on the comparison of the ratio of nonsynonymous to synonymous polymorphisms (Pnonsyn/Psyn) and the ratio of nonsynonymous to synonymous fixed differences between species (Fnonsyn/Fsyn). Both ratios are expected to be equal under strict neutrality and putative deviations from this expectation can be determined by a G-test of independence. The test was significant (P = 0.0374) within O3+4+23 and marginally significant (P = 0.0647) within O3+4+8. In fact, the Pnonsyn/Psyn ratio within gene arrangements (0.28 and 0.24 within O3+4+8 and O3+4+23, respectively) was much higher than the Fnonsyn/Fsyn ratio (0.06 in both comparisons). This deviation is also reflected in the neutrality index (NI; Rand and Kann 1996) estimated within O3+4+8 and O3+4+23 (table 5). This index is expected to be 1 under strict neutrality, and values higher than 1 indicate that the Pnonsyn/Fnonsyn ratio is higher than the Psyn/Fsyn ratio.
The MK tests indicated a stronger deviation from predictions of the neutral model within the young arrangements (O3+4+8 and O3+4+23) than within O3+4. In addition, neutrality indexes much higher than 1 were found within O3+4+8 and O3+4+23, which indicates that the deviation from neutrality is in the same direction in both young arrangements—i.e., an excess of nonsynonymous polymorphism (or an excess of synonymous fixed differences). As a consequence, the MK test is highly significant when all the lines with the young arrangements (O3+4+8 and O3+4+23) are jointly analyzed (table 5). In addition, the neutrality index equal to 5.25 for the young arrangements is clearly higher than that detected in the oldest arrangement O3+4 (NI = 1.74). A G test of independence revealed a marginally significant difference between the numbers of synonymous and nonsynonymous polymorphisms in the young arrangements (O3+4+8 and O3+4+23) and in O3+4 (G = 2.74, 1 df, P = 0.098). The same test revealed a significant difference between Ost (NI = 9.01) and O3+4 (Navarro-Sabaté, Aguadé, and Segarra 1999a).
Discussion
Nucleotide Variation Within and Between Arrangements
Nucleotide variation at Acph-1 has been analyzed in four chromosomal arrangements segregating in natural populations of D. subobscura: O3+4, Ost, O3+4+8, and O3+4+23. Because of the mutational process and the suppression of recombination in inversion heterokaryotypes, some genetic differentiation between arrangements is expected. However, differentiation may be counterbalanced by genetic exchange between arrangements. It has been proposed that the level of genetic exchange should be higher for markers located in central positions of the inversion loop than for markers located near the breakpoints (Rozas and Aguadé 1994; Navarro et al. 1997; Navarro, Barbadilla, and Ruiz 2000; Andolfatto, Depaulis, and Navarro 2001). Indeed, near breakpoints, genetic exchange should be possible only by gene conversion, whereas in the central region of the inversion loop not only gene conversion but also double crossover can contribute to the genetic exchange. The genetic exchange is considered to be eventually null at the inversion breakpoints themselves where chromosomes cannot establish synapsis properly (Wesley and Eanes 1994; Andolfatto, Wall, and Kreitman 1999; Cáceres et al. 1999; Cáceres, Puig, and Ruiz 2001).
Empirical data obtained for markers located at different positions relative to the breakpoints of, for instance, the In(3L)Payne inversion of D. melanogaster support the proposed differential genetic exchange along the inversion (Hasson and Eanes 1996). Acph-1 is located inside and rather close to one of the ends of the inversion loop formed in O3+4+23/O3+4 and O3+4+23/O3+4+8 heterokaryotypes. This is not the case for the O3+4/O3+4+8 heterokaryotypes, in which Acph-1 maps in a central position of the inversion loop (fig. 1). Consequently, genetic differentiation at Acph-1 is expected to be higher between O3+4+23 relative to both O3+4 and O3+4+8 than between the two latter arrangements. Although the dxy estimates are similar in the three comparisons between these arrangements, the data on linkage disequilibrium between nucleotide polymorphic sites and the type of arrangement (table 3) are consistent with this expectation. Linkage disequilibrium in the O3+4 and O3+4+8 joint sample (and thus genetic differentiation between O3+4 and O3+4+8) is lower than that detected in the O3+4 and O3+4+23 or in the O3+4+23 and O3+4+8 joint samples. In addition, the highest level of linkage disequilibrium was found in the samples including the Ost lines. This result was expected because Acph-1 maps very close to one of the breakpoints of inversion 3 and would thus be asynaptic in heterokaryotypes between Ost and any of the O[3+4] arrangements (fig. 1 and results not shown). Therefore, the level of genetic exchange between Ost and the other chromosomal arrangements is expected to be considerably reduced at Acph-1.
Prevalence of genetic differentiation despite the presence of genetic exchange between arrangements is also reflected in the gene genealogy inferred from nucleotide variation at Acph-1. The clustering of lines into two main groups (Ost and O[3+4]) is consistent with restricted genetic exchange between these chromosomal classes. In contrast, O[3+4] lines do not completely cluster according to their gene arrangement, which is consistent with a higher level of genetic exchange within O[3+4] than between Ost and O[3+4]. However, genetic exchange within O[3+4] has not been high enough to erase completely the original chromosomal relationships among lines, as reflected in the partial clustering of lines by gene arrangement. A similar result was reported earlier, according to variation at the rp49 gene region (Rozas et al. 1999) that is tightly linked to Acph-1.
Excess of Nonsynonymous Polymorphism
Under predictions of the neutral model, the ratio between the number of polymorphic variants within species and the number of fixed differences between species is expected to be similar for synonymous and nonsynonymous mutations. This prediction results in a neutrality index equal to 1. Neutrality indexes considerably higher than 1 have been detected in O3+4+23 and O3+4+8. Indeed, this index is 5.25 when both young arrangements are jointly analyzed. This deviation from strict neutrality is statistically significant, as revealed by the MK test (table 5). Also, a significant MK test associated with a neutrality index higher than 1 (NI = 9.01) was previously detected in the Ost arrangement (Navarro-Sabaté, Aguadé, and Segarra 1999a). In contrast, the MK test is not significant for the old O3+4 arrangement. Neutrality indexes higher than 1 have been interpreted mainly as resulting from an excess of nonsynonymous polymorphism. Alternatively, they could be due to an excess of fixed synonymous differences, but this latter explanation should hold for all chromosomal arrangements and thus should not cause differences between arrangements. The contrasting results obtained by the MK test in O3+4 versus O3+4+23, O3+4+8, and Ost indicate that different constraints act on nonsynonymous mutations in the two sets of arrangements.
The different constraints might be a priori attributable to differences in the strength of positive selection favoring or maintaining amino acid variation in the studied arrangements. In fact, an excess of replacement polymorphism is expected from positive selection fluctuating over time or space (Gillespie 1991). The excess of nonsynonymous polymorphism detected at Pgm in D. melanogaster (Verrelli and Eanes 2000) has been attributed to this kind of selection. Also, positive selection was proposed to explain the different ratio of replacement to silent polymorphism detected at fused-1 in standard X chromosomes and in chromosomes carrying a centric fusion between the X chromosome and the fourth chromosome of D. americana (Vieira, McAllister, and Charlesworth 2001). It does not seem likely, however, that positive selection can explain the results observed at Acph-1. All amino acid polymorphisms within O3+4+23 and all but one within O3+4+8 are singletons, which results in highly significant Tajima's D statistics for nonsynonymous variation (table 4). In addition, a significant negative Tajima's D was detected within Ost (Navarro-Sabaté, Aguadé, and Segarra 1999a and present data). If positive selection were maintaining nonsynonymous variation at Acph-1, then a higher proportion of nonsynonymous polymorphisms segregating at intermediate frequencies would be expected.
The excess of nonsynonymous polymorphism detected at Acph-1 can be more likely explained according to the nearly neutral model of molecular evolution (Ohta 1992), and assuming that most replacement mutations at Acph-1 are slightly deleterious. Apart from the variants responsible for the polymorphism at residues 205 and 241 of the ACPH-1 protein, all singleton nonsynonymous variants detected within O3+4+8 and O3+4+23 are derived and absent from the other arrangements. Therefore, it can be inferred that these variants appeared by mutation and were kept at low frequency within each arrangement by weak negative selection; i.e. nonsynonymous mutations are slightly deleterious. The highly significant negative Tajima's D statistic values for nonsynonymous mutations also support this argument. Tajima's D statistic is, however, highly sensitive to demographic factors such as population expansions and population subdivision. The presence of the different chromosomal arrangements clearly causes genetic substructuring at Acph-1, which in turn may affect Tajima's D statistic. Nevertheless, demographic factors are expected to affect different regions of the genome or different kinds of sites to a similar extent. The different pattern of variation detected for nonsynonymous and silent mutations is not consistent with demographic explanations.
The pattern of variation in the young O3+4+23 and O3+4+8 arrangements, and even in Ost, has the same signature as previously reported for most genes of the mitochondrial genome (mtDNA) in D. melanogaster and other organisms. This signature has been mainly explained according to the nearly neutral model and assuming that nonsynonymous mutations are weakly selected. Although theoretical models based on weak selection have recently been developed (McVean and Charlesworth 2000; Comeron and Kreitman 2002), the study of the joint effects of selection, drift, and recombination on weakly selected mutations requires further attention. However, as discussed by Weinreich and Rand (2000), two characteristics of the mitochondrial genome that may contribute to the detected pattern of variation are the lack of recombination and its lower effective size relative to the nuclear genome. Therefore, it is tempting to argue that these characteristics may also contribute to the pattern of variation detected at Acph-1 in the young O3+4+23 and O3+4+8 arrangements, and even in Ost. The lack of recombination in mtDNA might be related to the reduction in recombination at Acph-1 in heterokaryotypes. In addition, a reduction in recombination can be viewed as a reduction in effective size as a consequence of the Hill-Robertson effect (Hill and Robertson 1966). In contrast, the effective size of these arrangements might be low given their frequency and their young character. In any case, a small effective size results in a longer persistence of slightly deleterious mutations in the population (i.e., negative selection against slightly deleterious mutations is relaxed).
The frequencies of O3+4+8 and O3+4+23 in Bizerte are 0.58 and 0.31, respectively (Rozas et al. 1999). These frequencies are in the upper limit of the frequencies of these arrangements in the Palearctic region. O3+4+23 has a very localized distribution and it is present at appreciable frequencies only around Tunisia and in England. O3+4+8 has a wider distribution, but its frequency rarely is larger than 0.1 except in the north of Africa, where it reaches the highest frequency in Tunisia. Therefore, it can be inferred that the average frequencies of these arrangements in the whole distribution area of D. subobscura are lower than the frequencies in Bizerte, with the lowest average frequency for O3+4+23. The frequency of O3+4 is 0.79 in the Spanish sample, but only 0.11 in Bizerte (Rozas et al. 1999). The lack of genetic differentiation between both populations indicates that migration is high enough to homogenize the gene content within arrangement (see also Rozas et al. 1995). O3+4 is an arrangement that reaches its highest frequencies (up to 0.9) in Iran and the Canary Islands (Krimbas 1992). Only in Tunisia and the north of Europe is its frequency lower than 0.1. In northwest Africa and the Mediterranean, frequencies from 0.2 to 0.7 are common. Therefore, the average frequency of O3+4 in the distribution area of D. subobscura is expected to be slightly but not much lower than its frequency in Spain. The different average frequencies of these arrangements in the distribution area of D. subobscura allows us to predict that the effective size of O3+4 is considerable higher than effective sizes of O3+4+8 and O3+4+23. In addition, these are the youngest arrangements, which may be why their long-term effective size was smaller than that of O3+4. It has to be also considered that recombination at Acph-1 is only likely in homokaryotypes, which are expected to be rare for low-frequency arrangements. In this sense, the threefold difference in the recombination parameter between O3+4+8 and O3+4+23 (see Results) would be consistent with the expected frequencies of the O3+4+8 and O3+4+23 homokaryotypes in Bizerte (0.34 and 0.1, respectively).
All arrangements except O3+4 exhibit an excess of nonsynonymous polymorphism at Acph-1. This excess should be also expected in other genes associated with the different arrangements. Previous results at rp49 do not support this expectation, because in this gene no nonsynonymous polymorphism was detected in any of the studied gene arrangements (Rozas et al. 1999). However, it has to be considered that the distribution of the selective coefficients acting against nonsynonymous mutations may differ among genes depending on the functional constraints of the encoded proteins. Constraints on the ACPH-1 protein would likely be relatively low, as indicated by the rather high nonsynonymous divergence at Acph-1 (Navarro-Sabaté, Aguadé, and Segarra 1999b) and the detection of one line (TB21) with a null allele. The low functional constraints on the ACPH-1 protein could have greatly contributed to the detected results. The effect of chromosomal polymorphism on nonsynonymous variation would thus vary with the level of constraints of the encoded protein, and such an effect might be negligible in genes subject to strong purifying selection, like rp49.
Present address: Servei de Radioisòtops, Campus Bellvitge, Universitat de Barcelona, Spain.
David Rand, Associate Editor
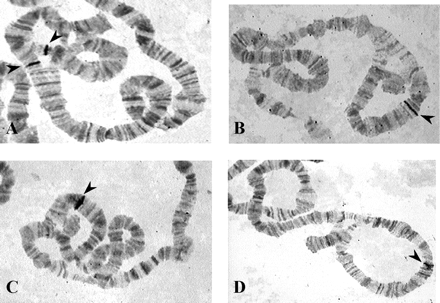
Location of the Acph-1 gene revealed by in situ hybridization in the different heterokaryotypes for the arrangements studied of D. subobscura: A, O3+4/Ost. B, O3+4/O3+4+23. C, O3+4+23/O3+4+8. D, O3+4/O3+4+8. Hybridization signals are indicated with arrowheads
Nucleotide polymorphic sites in the 45 lines of D. subobscura from Bizerte (Tunisia) after excluding all sites with alignment gaps. The numbering of sites is given as in Navarro-Sabaté, Aguadé, and Segarra (1999a) using as reference the B2ST sequence (EMBL accession number Y18839). Dots indicate nucleotides identical to the reference sequence. Sites located in the exons of the coding region are in bold. Asterisks indicate replacement polymorphisms in the sample from Bizerte. The identification of each line is given as in Rozas et al. (1999). Lines are grouped according to their gene arrangement, which is shown in parentheses after the identification names. The last row of the alignment shows the information for the polymorphic sites in the D. guanche sequence (EMBL accession number Y18841). Gaps in this sequence are indicated by dashes
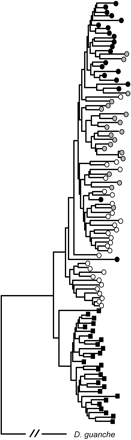
Gene genealogy reconstructed by the Neighbor-Joining method (Saitou and Nei 1987) from variation at the Acph-1 gene region in 99 D. subobscura lines using D. guanche as the outgroup. Genetic distances were estimated by the 2-parameter method (Kimura 1980) with the complete deletion option. Black squares indicate Ost lines. Open, gray and black circles correspond to O3+4, O3+4+8, and O3+4+23 lines, respectively

Replacement polymorphism at the ACPH-1 preprotein in the 45 lines of D. subobscura from Bizerte (Tunisia). Fixed differences relative to the D. guanche sequence (EMBL accession number Y18841) are also shown in the last row of the alignment. The numbering at the top is given as in figure 2, using as reference the B2ST sequence (EMBL accession number Y18839). Numbers in boldface correspond to amino acid residues in the ACPH-1 preprotein. Dots indicate amino acids identical to the reference sequence. The amino acid sequence of line TB21 is altered from residue 119 (data not shown) as the result of a frameshift mutation that results in a premature stop codon (black square). Replacement polymorphisms shared with the Spanish lines are boxed
Estimates of Polymorphism Within Gene Arrangement.
| Coding Region | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Arrangement | Total (2151 sites) | Silent (1,141 sites) | Noncoding (810 sites) | Synonymous (331 sites) | Nonsynonymous (1,010 sites) | ||||
| O3+4 (n = 3) | S | 42 | 40 | 15 | 25 | 2 | |||
| k | 28.0000 | 26.6703 | 10.0000 | 16.6637 | 1.3329 | ||||
| π | 0.0130 | 0.0234 | 0.0123 | 0.0503 | 0.0013 | ||||
| O3+4+23 (n = 20) | S | 114 | 102 | 53 | 49 | 12 | |||
| Singletons (%) | 73 (64%) | 45 (44%) | 33 (62%) | 28 (57%) | 12 (100%) | ||||
| k | 20.1320 | 18.9313 | 9.8630 | 9.0706 | 1.2026 | ||||
| π | 0.0094 | 0.0166 | 0.0122 | 0.0274 | 0.0012 | ||||
| θ | 0.0149 | 0.0252 | 0.0184 | 0.0418 | 0.0033 | ||||
| O3+4+8 (n = 21) | S | 114 | 104 | 59 | 45 | 10 | |||
| Singletons (%) | 57 (50%) | 48 (46%) | 35 (56%) | 13 (29%) | 9 (90%) | ||||
| k | 26.0190 | 24.9080 | 12.1430 | 12.7634 | 1.1110 | ||||
| π | 0.0121 | 0.0218 | 0.0150 | 0.0386 | 0.0011 | ||||
| θ | 0.0147 | 0.0261 | 0.0202 | 0.0386 | 0.0027 | ||||
| Coding Region | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Arrangement | Total (2151 sites) | Silent (1,141 sites) | Noncoding (810 sites) | Synonymous (331 sites) | Nonsynonymous (1,010 sites) | ||||
| O3+4 (n = 3) | S | 42 | 40 | 15 | 25 | 2 | |||
| k | 28.0000 | 26.6703 | 10.0000 | 16.6637 | 1.3329 | ||||
| π | 0.0130 | 0.0234 | 0.0123 | 0.0503 | 0.0013 | ||||
| O3+4+23 (n = 20) | S | 114 | 102 | 53 | 49 | 12 | |||
| Singletons (%) | 73 (64%) | 45 (44%) | 33 (62%) | 28 (57%) | 12 (100%) | ||||
| k | 20.1320 | 18.9313 | 9.8630 | 9.0706 | 1.2026 | ||||
| π | 0.0094 | 0.0166 | 0.0122 | 0.0274 | 0.0012 | ||||
| θ | 0.0149 | 0.0252 | 0.0184 | 0.0418 | 0.0033 | ||||
| O3+4+8 (n = 21) | S | 114 | 104 | 59 | 45 | 10 | |||
| Singletons (%) | 57 (50%) | 48 (46%) | 35 (56%) | 13 (29%) | 9 (90%) | ||||
| k | 26.0190 | 24.9080 | 12.1430 | 12.7634 | 1.1110 | ||||
| π | 0.0121 | 0.0218 | 0.0150 | 0.0386 | 0.0011 | ||||
| θ | 0.0147 | 0.0261 | 0.0202 | 0.0386 | 0.0027 | ||||
Estimates of Polymorphism Within Gene Arrangement.
| Coding Region | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Arrangement | Total (2151 sites) | Silent (1,141 sites) | Noncoding (810 sites) | Synonymous (331 sites) | Nonsynonymous (1,010 sites) | ||||
| O3+4 (n = 3) | S | 42 | 40 | 15 | 25 | 2 | |||
| k | 28.0000 | 26.6703 | 10.0000 | 16.6637 | 1.3329 | ||||
| π | 0.0130 | 0.0234 | 0.0123 | 0.0503 | 0.0013 | ||||
| O3+4+23 (n = 20) | S | 114 | 102 | 53 | 49 | 12 | |||
| Singletons (%) | 73 (64%) | 45 (44%) | 33 (62%) | 28 (57%) | 12 (100%) | ||||
| k | 20.1320 | 18.9313 | 9.8630 | 9.0706 | 1.2026 | ||||
| π | 0.0094 | 0.0166 | 0.0122 | 0.0274 | 0.0012 | ||||
| θ | 0.0149 | 0.0252 | 0.0184 | 0.0418 | 0.0033 | ||||
| O3+4+8 (n = 21) | S | 114 | 104 | 59 | 45 | 10 | |||
| Singletons (%) | 57 (50%) | 48 (46%) | 35 (56%) | 13 (29%) | 9 (90%) | ||||
| k | 26.0190 | 24.9080 | 12.1430 | 12.7634 | 1.1110 | ||||
| π | 0.0121 | 0.0218 | 0.0150 | 0.0386 | 0.0011 | ||||
| θ | 0.0147 | 0.0261 | 0.0202 | 0.0386 | 0.0027 | ||||
| Coding Region | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Arrangement | Total (2151 sites) | Silent (1,141 sites) | Noncoding (810 sites) | Synonymous (331 sites) | Nonsynonymous (1,010 sites) | ||||
| O3+4 (n = 3) | S | 42 | 40 | 15 | 25 | 2 | |||
| k | 28.0000 | 26.6703 | 10.0000 | 16.6637 | 1.3329 | ||||
| π | 0.0130 | 0.0234 | 0.0123 | 0.0503 | 0.0013 | ||||
| O3+4+23 (n = 20) | S | 114 | 102 | 53 | 49 | 12 | |||
| Singletons (%) | 73 (64%) | 45 (44%) | 33 (62%) | 28 (57%) | 12 (100%) | ||||
| k | 20.1320 | 18.9313 | 9.8630 | 9.0706 | 1.2026 | ||||
| π | 0.0094 | 0.0166 | 0.0122 | 0.0274 | 0.0012 | ||||
| θ | 0.0149 | 0.0252 | 0.0184 | 0.0418 | 0.0033 | ||||
| O3+4+8 (n = 21) | S | 114 | 104 | 59 | 45 | 10 | |||
| Singletons (%) | 57 (50%) | 48 (46%) | 35 (56%) | 13 (29%) | 9 (90%) | ||||
| k | 26.0190 | 24.9080 | 12.1430 | 12.7634 | 1.1110 | ||||
| π | 0.0121 | 0.0218 | 0.0150 | 0.0386 | 0.0011 | ||||
| θ | 0.0147 | 0.0261 | 0.0202 | 0.0386 | 0.0027 | ||||
Genetic Differentiation Between Gene Arrangements.
| O3+4/O3+4+8 | O3+4/O3+4+23 | O3+4+8/O3+4+23 | |
|---|---|---|---|
| dxy | 0.0144 | 0.0150 | 0.0136 |
\(\mathit{K}^{{\ast}}_{s}\) | 3.245 | 3.012 | 3.117 |
| P( \(\mathit{K}^{{\ast}}_{s}\) ≥ \(\mathit{K}^{{\ast}}_{s}\) observed) | 0.002 | 0.001 | 0.000 |
| Fixed differences | 0 | 0 | 0 |
| Number of silent shared | 28 | 27 | 49 |
| polymorphisms | |||
| P valuea | 0.000 | 0.000 | 0.000 |
| Expected numberb | 3.576 | 3.751 | 9.565 |
| O3+4/O3+4+8 | O3+4/O3+4+23 | O3+4+8/O3+4+23 | |
|---|---|---|---|
| dxy | 0.0144 | 0.0150 | 0.0136 |
\(\mathit{K}^{{\ast}}_{s}\) | 3.245 | 3.012 | 3.117 |
| P( \(\mathit{K}^{{\ast}}_{s}\) ≥ \(\mathit{K}^{{\ast}}_{s}\) observed) | 0.002 | 0.001 | 0.000 |
| Fixed differences | 0 | 0 | 0 |
| Number of silent shared | 28 | 27 | 49 |
| polymorphisms | |||
| P valuea | 0.000 | 0.000 | 0.000 |
| Expected numberb | 3.576 | 3.751 | 9.565 |
aP value based on the hypergeometric distribution.
bExpected number of silent shared polymorphism based on the hypergeometric distribution.
Genetic Differentiation Between Gene Arrangements.
| O3+4/O3+4+8 | O3+4/O3+4+23 | O3+4+8/O3+4+23 | |
|---|---|---|---|
| dxy | 0.0144 | 0.0150 | 0.0136 |
\(\mathit{K}^{{\ast}}_{s}\) | 3.245 | 3.012 | 3.117 |
| P( \(\mathit{K}^{{\ast}}_{s}\) ≥ \(\mathit{K}^{{\ast}}_{s}\) observed) | 0.002 | 0.001 | 0.000 |
| Fixed differences | 0 | 0 | 0 |
| Number of silent shared | 28 | 27 | 49 |
| polymorphisms | |||
| P valuea | 0.000 | 0.000 | 0.000 |
| Expected numberb | 3.576 | 3.751 | 9.565 |
| O3+4/O3+4+8 | O3+4/O3+4+23 | O3+4+8/O3+4+23 | |
|---|---|---|---|
| dxy | 0.0144 | 0.0150 | 0.0136 |
\(\mathit{K}^{{\ast}}_{s}\) | 3.245 | 3.012 | 3.117 |
| P( \(\mathit{K}^{{\ast}}_{s}\) ≥ \(\mathit{K}^{{\ast}}_{s}\) observed) | 0.002 | 0.001 | 0.000 |
| Fixed differences | 0 | 0 | 0 |
| Number of silent shared | 28 | 27 | 49 |
| polymorphisms | |||
| P valuea | 0.000 | 0.000 | 0.000 |
| Expected numberb | 3.576 | 3.751 | 9.565 |
aP value based on the hypergeometric distribution.
bExpected number of silent shared polymorphism based on the hypergeometric distribution.
Linkage Disequilibrium Between Informative Polymorphic Sites and Arrangements.
| % of Significant Comparisons (χ2 test) | Average r2 | |
|---|---|---|
| O3+4+23 and O3+4+8 | 40.58 | 0.1135 |
| O3+4+23 and O3+4 | 40.51 | 0.1399 |
| O3+4+8 and O3+4 | 35.80 | 0.1074 |
| O3+4+23 and Ost | 65.59 | 0.3087 |
| O3+4+8 and Ost | 63.33 | 0.2542 |
| O3+4 and Ost | 58.00 | 0.2191 |
| % of Significant Comparisons (χ2 test) | Average r2 | |
|---|---|---|
| O3+4+23 and O3+4+8 | 40.58 | 0.1135 |
| O3+4+23 and O3+4 | 40.51 | 0.1399 |
| O3+4+8 and O3+4 | 35.80 | 0.1074 |
| O3+4+23 and Ost | 65.59 | 0.3087 |
| O3+4+8 and Ost | 63.33 | 0.2542 |
| O3+4 and Ost | 58.00 | 0.2191 |
Linkage Disequilibrium Between Informative Polymorphic Sites and Arrangements.
| % of Significant Comparisons (χ2 test) | Average r2 | |
|---|---|---|
| O3+4+23 and O3+4+8 | 40.58 | 0.1135 |
| O3+4+23 and O3+4 | 40.51 | 0.1399 |
| O3+4+8 and O3+4 | 35.80 | 0.1074 |
| O3+4+23 and Ost | 65.59 | 0.3087 |
| O3+4+8 and Ost | 63.33 | 0.2542 |
| O3+4 and Ost | 58.00 | 0.2191 |
| % of Significant Comparisons (χ2 test) | Average r2 | |
|---|---|---|
| O3+4+23 and O3+4+8 | 40.58 | 0.1135 |
| O3+4+23 and O3+4 | 40.51 | 0.1399 |
| O3+4+8 and O3+4 | 35.80 | 0.1074 |
| O3+4+23 and Ost | 65.59 | 0.3087 |
| O3+4+8 and Ost | 63.33 | 0.2542 |
| O3+4 and Ost | 58.00 | 0.2191 |
Tajima's Test.
| Tajima's D | P(D ≤ Dobs)a | |
|---|---|---|
| Nonsynonymous sites | ||
| Old arrangement | ||
| O3+4b | −0.8410 | 0.2211 |
| Young arrangements | ||
| O3+4+8 | −2.0679 | 0.0052 |
| O3+4+23 | −2.3162 | 0.0000 |
| Silent sites | ||
| Old arrangement | ||
| O3+4b | 0.2050 | 0.6472 |
| Young arrangements | ||
| O3+4+8 | −0.6625 | 0.2652 |
| O3+4+23 | −1.4037 | 0.0580 |
| Tajima's D | P(D ≤ Dobs)a | |
|---|---|---|
| Nonsynonymous sites | ||
| Old arrangement | ||
| O3+4b | −0.8410 | 0.2211 |
| Young arrangements | ||
| O3+4+8 | −2.0679 | 0.0052 |
| O3+4+23 | −2.3162 | 0.0000 |
| Silent sites | ||
| Old arrangement | ||
| O3+4b | 0.2050 | 0.6472 |
| Young arrangements | ||
| O3+4+8 | −0.6625 | 0.2652 |
| O3+4+23 | −1.4037 | 0.0580 |
aP values under no recombination (one-tailed test).
bData from Navarro-Sabaté, Aguadé, and Segarra (1999a).
Tajima's Test.
| Tajima's D | P(D ≤ Dobs)a | |
|---|---|---|
| Nonsynonymous sites | ||
| Old arrangement | ||
| O3+4b | −0.8410 | 0.2211 |
| Young arrangements | ||
| O3+4+8 | −2.0679 | 0.0052 |
| O3+4+23 | −2.3162 | 0.0000 |
| Silent sites | ||
| Old arrangement | ||
| O3+4b | 0.2050 | 0.6472 |
| Young arrangements | ||
| O3+4+8 | −0.6625 | 0.2652 |
| O3+4+23 | −1.4037 | 0.0580 |
| Tajima's D | P(D ≤ Dobs)a | |
|---|---|---|
| Nonsynonymous sites | ||
| Old arrangement | ||
| O3+4b | −0.8410 | 0.2211 |
| Young arrangements | ||
| O3+4+8 | −2.0679 | 0.0052 |
| O3+4+23 | −2.3162 | 0.0000 |
| Silent sites | ||
| Old arrangement | ||
| O3+4b | 0.2050 | 0.6472 |
| Young arrangements | ||
| O3+4+8 | −0.6625 | 0.2652 |
| O3+4+23 | −1.4037 | 0.0580 |
aP values under no recombination (one-tailed test).
bData from Navarro-Sabaté, Aguadé, and Segarra (1999a).
McDonald and Kreitman's Test.
| Fixed | Polymorphic | ||
|---|---|---|---|
| O3+4a (old arrangement) | |||
| Synonymous | 35 | 47 | G = 0.611 |
| Nonsynonymous | 3 | 7 | P = 0.434 |
| NI = 1.74 | |||
| O3+4+8 (young arrangement) | |||
| Synonymous | 35 | 46 | G = 3.411 |
| Nonsynonymous | 2 | 10 | P = 0.065 |
| NI = 3.80 | |||
| O3+4+23 (young arrangement) | |||
| Synonymous | 35 | 49 | G = 4.331 |
| Nonsynonymous | 2 | 12 | P = 0.037 |
| NI = 4.29 | |||
| O3+4+8 and O3+4+23 (young arrangements) | |||
| Synonymous | 33 | 66 | G = 6.619 |
| Nonsynonymous | 2 | 21 | P = 0.010 |
| NI = 5.25 |
| Fixed | Polymorphic | ||
|---|---|---|---|
| O3+4a (old arrangement) | |||
| Synonymous | 35 | 47 | G = 0.611 |
| Nonsynonymous | 3 | 7 | P = 0.434 |
| NI = 1.74 | |||
| O3+4+8 (young arrangement) | |||
| Synonymous | 35 | 46 | G = 3.411 |
| Nonsynonymous | 2 | 10 | P = 0.065 |
| NI = 3.80 | |||
| O3+4+23 (young arrangement) | |||
| Synonymous | 35 | 49 | G = 4.331 |
| Nonsynonymous | 2 | 12 | P = 0.037 |
| NI = 4.29 | |||
| O3+4+8 and O3+4+23 (young arrangements) | |||
| Synonymous | 33 | 66 | G = 6.619 |
| Nonsynonymous | 2 | 21 | P = 0.010 |
| NI = 5.25 |
aData from Navarro-Sabaté, Aguadé, and Segarra (1999a).
McDonald and Kreitman's Test.
| Fixed | Polymorphic | ||
|---|---|---|---|
| O3+4a (old arrangement) | |||
| Synonymous | 35 | 47 | G = 0.611 |
| Nonsynonymous | 3 | 7 | P = 0.434 |
| NI = 1.74 | |||
| O3+4+8 (young arrangement) | |||
| Synonymous | 35 | 46 | G = 3.411 |
| Nonsynonymous | 2 | 10 | P = 0.065 |
| NI = 3.80 | |||
| O3+4+23 (young arrangement) | |||
| Synonymous | 35 | 49 | G = 4.331 |
| Nonsynonymous | 2 | 12 | P = 0.037 |
| NI = 4.29 | |||
| O3+4+8 and O3+4+23 (young arrangements) | |||
| Synonymous | 33 | 66 | G = 6.619 |
| Nonsynonymous | 2 | 21 | P = 0.010 |
| NI = 5.25 |
| Fixed | Polymorphic | ||
|---|---|---|---|
| O3+4a (old arrangement) | |||
| Synonymous | 35 | 47 | G = 0.611 |
| Nonsynonymous | 3 | 7 | P = 0.434 |
| NI = 1.74 | |||
| O3+4+8 (young arrangement) | |||
| Synonymous | 35 | 46 | G = 3.411 |
| Nonsynonymous | 2 | 10 | P = 0.065 |
| NI = 3.80 | |||
| O3+4+23 (young arrangement) | |||
| Synonymous | 35 | 49 | G = 4.331 |
| Nonsynonymous | 2 | 12 | P = 0.037 |
| NI = 4.29 | |||
| O3+4+8 and O3+4+23 (young arrangements) | |||
| Synonymous | 33 | 66 | G = 6.619 |
| Nonsynonymous | 2 | 21 | P = 0.010 |
| NI = 5.25 |
aData from Navarro-Sabaté, Aguadé, and Segarra (1999a).
We thank J. Rozas for critical comments on a previous version of the manuscript, as well as D. M. Rand and the anonymous reviewers for their constructive comments. We also thank Serveis Científico-Tècnics, Universitat de Barcelona, for automated sequencing facilities. This work was supported by a predoctoral fellowship from Comissió Interdepartamental de Recerca i Innovació Tecnològica, Catalonia, Spain to À.N.-S. and by grants PB94-923 and PB97-918 from Comisión Interdepartamental de Ciencia y Tecnología, Spain, and 1995SGR-577, 1997SGR-59, and 1999SGR-25 from Comissió Interdepartamental de Recerca i Innovació Tecnològica, Catalonia, Spain, to M.A.
Literature Cited
Andolfatto, P., F. Depaulis, and A. Navarro.
Andolfatto, P., J. D. Wall, and M. Kreitman.
Ashburner, M.
Ballard, J. W. O., and M. Kreitman.
Baines, J. F., Y. Chen, A. Das, and W. Stephan.
Betrán, E., J. Rozas, A. Navarro, and A. Barbadilla.
Cáceres, M., M. Puig, and A. Ruiz.
Cáceres, M., J. M. Ranz, A. Barbadilla, M. Long, and A. Ruiz.
Comeron, J. M., and M. Kreitman.
Fay, J. C., G. J. Wyckoff, and C.-I. Wu.
Gilbert, D.
Hamblin, M. T., and C. F. Aquadro.
Hasegawa, M., Y. Cao, and Z. Yang.
Hasson, E., and W. F. Eanes.
Hill, W. G., and A. Robertson.
Hill, W. G., and A. Robertson.
Hudson, R. R.
Hudson, R. R., D. D. Boos, and N. L. Kaplan.
Hudson, R. R., and N. L. Kaplan.
Kennedy, P., and M. W. Nachman.
Kimura, M.
Kimura, M.
Krimbas, C. B.
Kumar, S., K. Tamura, I. B. Jakobsen, and M. Nei.
Maddison, W. P., and D. R. Maddison.
McDonald, J. H., and M. Kreitman.
McVean, G. A. T., and B. Charlesworth.
Moriyama, E. N., and J. R. Powell.
Nachman, M. W., S. N. Boyer, and C. F. Aquadro.
Nachman, M. W., W. M. Drown, M. Stoneking, and C. F. Aquadro.
Navarro, A., A. Barbadilla, and A. Ruiz.
Navarro, A., E. Betrán, A. Barbadilla, and A. Ruiz.
Navarro-Sabaté, À., M. Aguadé, and C. Segarra.
Navarro-Sabaté, À., M. Aguadé, and C. Segarra.
Rand, D. M., M. Dorfsman, and L. M. Kann.
Rand, D. M., and L. M. Kann.
Rozas, J., and M. Aguadé.
Rozas, J., and R. Rozas.
Rozas, J., C. Segarra, G. Ribó, and M. Aguadé.
Rozas, J., C. Segarra, C. Zapata, G. Álvarez, and M. Aguadé.
Saitou, N., and M. Nei.
Segarra, C., and M. Aguadé.
Tajima, F.
Takahata, N.
Verrelli, B. C., and W. F. Eanes.
Vieira, J., B. F. McAllister, and B. Charlesworth.
Watterson, G. A.
Weinreich, D. M., and D. M. Rand.
Wesley, C. S., and W. F. Eanes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}