





Abstract
Autosomal dominant osteopetrosis (ADO) is a rare genetic disorder resulting from impaired osteoclastic bone resorption. Clinical manifestations frequently include fractures, osteonecrosis (particularly of the jaw or maxilla), osteomyelitis, blindness, and/or bone marrow failure. ADO usually results from heterozygous missense variants in the Chloride Channel 7 gene (CLCN7) that cause disease by a dominant negative mechanism. Variants in the T-cell immune regulator 1 gene (TCIRG1) are commonly identified in autosomal recessive osteopetrosis but have only been reported in 1 patient with ADO.
Here, we report 3 family members with a single heterozygous missense variant (p.Gly579Arg) in TCIRG1 who have a phenotype consistent with ADO. Three of 5 protein prediction programs suggest this variant likely inhibits the function of TCIRG1.
This is the first description of adult presentation of ADO caused by a TCIRG1 variant. Similar to families with ADO from CLCN7 mutations, this variant in TCIRG1 results in marked phenotype variability, with 2 subjects having severe disease and the third having very mild disease. This family report implicates TCIRG1 missense mutations as a cause of ADO and demonstrates that the marked phenotypic variability in ADO may extend to disease caused by TCIRG1 missense mutations.
Autosomal dominant osteopetrosis (ADO), also known as Albers-Schönberg disease, is a rare genetic disorder (incidence of approximately 1:20 000). The underlying pathophysiology includes impaired osteoclastic bone resorption, which contributes to the formation of dense but brittle bones. Clinical manifestations frequently include fractures, osteonecrosis (particularly of the jaw or maxilla), osteomyelitis, blindness, and/or bone marrow failure (1, 2). ADO usually results from heterozygous missense variants in the Chloride Channel 7 gene (CLCN7), which cause disease by a dominant negative mechanism (2, 3).
Because of the incomplete penetrance and a mild phenotype in some individuals with ADO, it is sometimes referred to as the “benign” form of the disease (4) to distinguish it from severe “malignant” autosomal recessive osteopetrosis. However, ADO is not benign because most patients suffer complications from their disease (1, 5-7). For example, our previous cross-sectional study in 94 patients with CLCN7 mutations demonstrated high rates of hip and femur fractures, occurring in 16% of children and 49% of adults with ADO, along with other complications such as vision loss and bone marrow suppression. However, ADO demonstrates widely varying severity and, although some have severe complications, up to one third of patients with such mutations appear to be unaffected carriers (1, 2).
Osteoclasts are multinucleated giant cells responsible for bone resorption. Resorption involves forming an invaginated plasma membrane structure (ie, the “ruffled border”) through which bone mineral matrix is digested and dissolved with acidification and enzyme activity (8, 9). To achieve acidification, protons are actively transported across the membrane of the ruffled border through action of the osteoclast-specific vacuolar-type H+−ATPase (V-ATPase) “proton pump” (10). An essential subunit of V-ATPase, the a3 subunit, is encoded by the T-cell immune regulator 1 gene (TCIRG1). CLCN7 encodes the chloride voltage-gated channel 7, which regulates the flow of chloride ions, providing the chloride conductance necessary for efficient proton pumping by V-ATPase (11). Loss of function variants in TCIRG1 and CLCN7 account for a majority of autosomal recessive osteopetrosis (ARO), with TCIRG1 variants alone responsible for more than 50% of cases (12-14).
Missense variants in CLCN7 frequently cause ADO; however, we found only a single case report of a missense variant in TCIRG1 causing ADO (15). This case presented at 3 years of age and was reported to have spontaneous fractures of the radius and femur, along with right eye blindness from optic atrophy. Genetic analysis of TCIRG1 demonstrated heterozygous adenine substitution for guanine at position c.725 in exon 8, resulting in a His242Arg substitution in the protein (15). Because only this 1 patient with TCIRG1 ADO has been reported, it is unknown whether TCIRG1 missense variants can also give rise to the marked variation in phenotypic severity seen in patients with CLCN7 variants.
Here, we report in-depth studies of 3 family members having a TCIRG1 missense variant resulting in an ADO phenotype. Two of the affected individuals were severely affected, whereas the third was only mildly affected, demonstrating marked differences in phenotypic severity in ADO caused by this TCIRG1 variant.
Methods
We studied 3 subjects with ADO: II-1, II-2, and III-1. Subjects were enrolled in an Osteopetrosis Natural History Study designed to assess the clinical, biochemical, and radiographic manifestations of the disease. In brief, we obtained detailed medical history, physical examination, serum and urine biochemistry, skeletal radiograph surveys, dual-energy X-ray absorptiometry (DXA), quantitative computed tomography (QCT) of the spine, high-resolution peripheral QCT (HRpQCT) of the tibia, and evaluation of physical function. Genetic testing was performed commercially (Invitae Corporation, San Francisco, CA, or Blueprint Genetics, Marlborough, MA).
DXA (Norland Elite; Norland at Swissray, Fort Atkinson, WI) was performed to assess whole-body areal bone mineral density (aBMD) and body composition, and hip and spine aBMD. HR-pQCT (XtremeCT II; Scanco Medical, Brüttisellen, Switzerland) was performed as have previously described (16). Tibial bone length was measured using anthropometric calipers. After performance of a scout view, a reference line was placed at the center of the tibia joint surface. Scan stacks (168 tomographic slices with 61-µm isotropic voxel size) were centered 7.3% (distal tibia) and 30% (tibial diaphysis) of tibia length proximal to the reference line. Total volumetric BMD values were expressed as Z scores relative to reference data obtained using the same HR-pQCT scanner (16).
Volumetric BMD (vBMD) of L1-3 was measured by QCT as previously reported (5). In brief, vBMD was measured using a helical CT scanner (IU: Brilliance CT 64 Channel and iCT 128; Philips Medical Systems, Best, Netherlands; Harbor-UCLA: Revolution CT, GE Healthcare, Waukesha, Wisconsin, US) and QCT calibration phantom (Image Analysis QCT-Bone Mineral Phantom; Image Analysis Inc, Columbia, KY) with the subject supine on the calibration phantom. CT imaging was performed with continuous axial slices from L1 through L3 based on scout images. CT imaging parameters used included 120 kV, 300 mAs, 2-mm slice thickness, and no dose modulation. Image analysis was performed manually with the center axial slice of each vertebral body (L1, L2, and L3) selected and manual region of interest drawn within the vertebral body.
Physical function testing of dominant hand grip strength (Jamar Plus+ digital hand dynamometer; Sammons Preston, Bolingbrook, IL) and the number of chair stands completed in 30 seconds were performed as we have previously described (17). Outcomes were converted to age- and sex-matched Z scores relative to our published reference data (17). Time to walk 4 m from a stationary start at normal speed (usual gait speed) was measured with a stopwatch and converted to speed (m/s). A speed below 1.0 m/s is associated with declines in health and life expectancy (18). Age- and sex-matched Z scores were generated from published reference data (19). Distance walked in 6 minutes was assessed on a 20-m-long looped course. Self-reported physical function was assessed using the physical function domain of the NIH Patient Reported Outcomes Measurement Information System (PROMIS-PF), performed via computerized adaptive testing. PROMIS-PF scores are standardized and expressed as T scores with a population mean of 50 and SD of 10 (20).
All procedures were performed in accordance with the Declaration of Helsinki and with prior approval of the Indiana University institutional review board. Written informed consent was obtained from all participants.
Case Reports
The family tree of our subjects is illustrated in Fig. 1. Both parents of subjects II-1 and II-2 are deceased and thus unavailable for genotyping and testing. The mother died of breast cancer at age 73 years and father from acute coronary syndrome at age 69 years. Neither were known to have or exhibit signs of osteopetrosis.
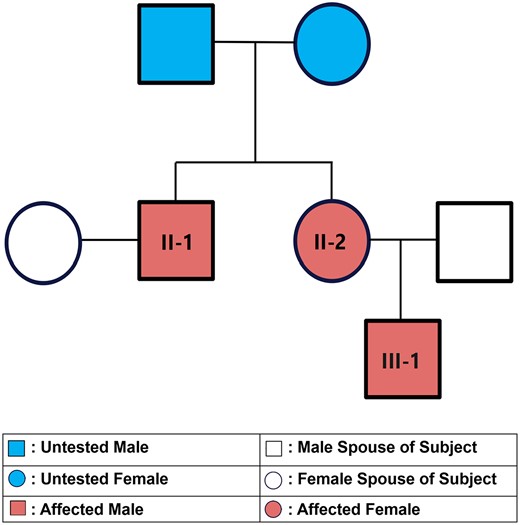
Abbreviated pedigree.
Subject II-1 Case Report
Subject II-1, now a 45-year-old man, had a skeletal survey at age 13 after learning his sister had ADO. The survey demonstrated characteristic ADO findings. He subsequently had 4 metatarsal fractures requiring surgery at age 21 years. These fractures were discovered after he started having pain while mowing his lawn. He then had 2 femur fractures: an atraumatic left femoral shaft fracture at age 31 years (leg pain after tripping, but not falling, when walking through a doorway) and an atraumatic right femoral neck fracture at age 32 years (pain after stepping down from a stool without falling). At age 33 years, he developed osteomyelitis of the jaw requiring prolonged treatment with IV antibiotics. He had nephrolithiasis twice, at ages 34 and 39 years. On the day of the research study evaluation, he exhibited visible right jaw swelling and chronic jaw pain. He had evidence of osteonecrosis of the jaw and 4 lower right teeth were missing. Complete blood count showed moderate anemia with hemoglobin of 12.8 g/dL (Table 1). Total creatine kinase (CK) was 41 U/L with creatine kinase-BB isoenzyme (CK-BB) levels being elevated, comprising 13% of total CK (normal for age, 0%).
Complete blood count results
| Subject II-1 | Subject II-2 | Subject III-1 | Normal range | |
|---|---|---|---|---|
| Red blood cell count (million/mm3) | 4.3 | 2.85a | 4.38 | 3.71-5.17 |
| Hemoglobin (g/dL) | 12.8 | 8.8b | 13.8 | 12.0-15.0 |
| White blood cell count (thousand/mm3) | 6.6 | 3.1a | 3.5a | 3.6-10.6 |
| Subject II-1 | Subject II-2 | Subject III-1 | Normal range | |
|---|---|---|---|---|
| Red blood cell count (million/mm3) | 4.3 | 2.85a | 4.38 | 3.71-5.17 |
| Hemoglobin (g/dL) | 12.8 | 8.8b | 13.8 | 12.0-15.0 |
| White blood cell count (thousand/mm3) | 6.6 | 3.1a | 3.5a | 3.6-10.6 |
aCytopenia demonstrated.
bAnemia demonstrated.
Complete blood count results
| Subject II-1 | Subject II-2 | Subject III-1 | Normal range | |
|---|---|---|---|---|
| Red blood cell count (million/mm3) | 4.3 | 2.85a | 4.38 | 3.71-5.17 |
| Hemoglobin (g/dL) | 12.8 | 8.8b | 13.8 | 12.0-15.0 |
| White blood cell count (thousand/mm3) | 6.6 | 3.1a | 3.5a | 3.6-10.6 |
| Subject II-1 | Subject II-2 | Subject III-1 | Normal range | |
|---|---|---|---|---|
| Red blood cell count (million/mm3) | 4.3 | 2.85a | 4.38 | 3.71-5.17 |
| Hemoglobin (g/dL) | 12.8 | 8.8b | 13.8 | 12.0-15.0 |
| White blood cell count (thousand/mm3) | 6.6 | 3.1a | 3.5a | 3.6-10.6 |
aCytopenia demonstrated.
bAnemia demonstrated.
Skeletal survey illustrated classic ADO features; multilevel vertebral endplate sclerosis and femoral cortical thickening (Fig. 2). DXA scan revealed a lumbar spine aBMD of 3.03 g/cm2 (Z score +9.62). Hip aBMD was not interpretable because of bilateral hip plate and screws (Table 2). vBMD of L1-3 by QCT was 503 mg/cm3 (Z score +12.6). HRpQCT demonstrated a relatively normal structure of the tibial diaphysis and distal tibia (Fig. 3), yet the distal tibia had visibly more trabecular bone and elevated total vBMD (Z score +2.9).
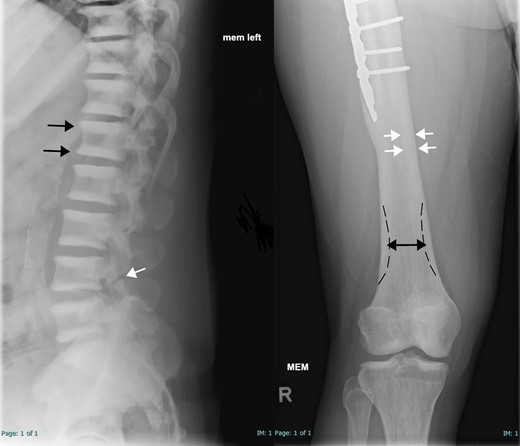
Skeletal survey of subject II-1. Left: Lateral radiograph of the lower thoracic and lumbar spines demonstrated multilevel endplate sclerosis in the vertebral bodies (parallel bands of increased density; black arrows) without appreciable central sclerotic focus. This is commonly described as the “rugger jersey spine.” A chronic L4 pars interarticularis defect is also noted (white arrow). Right: Anteroposterior radiograph of the distal right femur and knee demonstrates femoral cortical thickening (white arrows) and a borderline Erlenmeyer flask shape (club-shaped flaring of long bone metaphysis; double-headed black arrow with dashed black line indicating expected shape). No appreciable bone-in-bone appearance is present. Partial visualization of fixation hardware is visible (#).
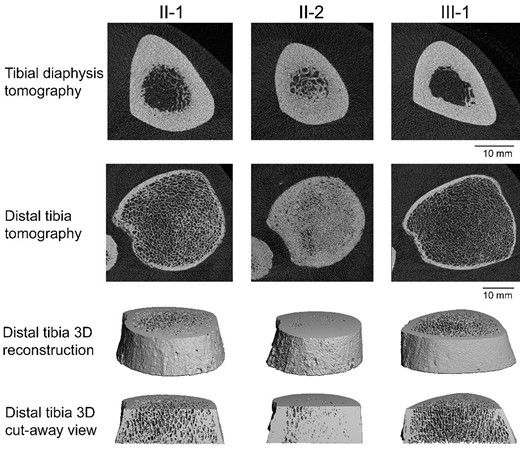
High-resolution peripheral quantitative computed tomography images of the tibia in each family member. Two-dimensional (top 2 rows) and 3-dimensional (bottom 2 rows) views demonstrate the phenotypic diversity in microarchitecture. Subject II-2 (middle column) had significant residual trabecular bone in the medullary cavity at the diaphysis and near solid bone at the distal tibia. Subject II-1 (left column) had some residual trabecular bone at the tibial diaphysis and visibly thicker trabeculae and cortex at the distal tibia when compared to the completely normal looking images of subject III-1 (right column).
DXA, spine QCT, and physical function data
| Subject II-1 | Subject II-2 | Subject III-1 | ||||
|---|---|---|---|---|---|---|
| DXA | Z score | Z score | Z score | |||
| Lumbar spine aBMD (g/cm2) | 3.0 | +9.6 | 4.1 | +17.3 | 1.7 | +2.7 |
| Total hip aBMD (g/cm2) | N/Aa | 3.6 | +22.1 | 1.3 | +1.9 | |
| Spine QCT | ||||||
| vBMD L1-L3 (mg/cm3) | 503.2 | +12.6 | 1057.3 | +31.1 | 227.8 | +1.8 |
| Physical function | ||||||
| Usual gait speed (m/s) | .86 | −1.5 | .65 | −2.3 | 1.21 | +.2 |
| 6-minute walk test (m) | 426 | −1.7 | 310 | −3.4 | 498 | −1.2 |
| Hand grip strength (kg) | 40.8 | −.5 | 20.9 | −.9 | 48.0 | +.1 |
| Repeat chair stands in 30 seconds (n) | 9 | −2.5 | 3b | −6.1b | 12 | −1.6 |
| PROMIS-PF T-scorec | 39.5 | 30.9 | 46.3 | |||
| Subject II-1 | Subject II-2 | Subject III-1 | ||||
|---|---|---|---|---|---|---|
| DXA | Z score | Z score | Z score | |||
| Lumbar spine aBMD (g/cm2) | 3.0 | +9.6 | 4.1 | +17.3 | 1.7 | +2.7 |
| Total hip aBMD (g/cm2) | N/Aa | 3.6 | +22.1 | 1.3 | +1.9 | |
| Spine QCT | ||||||
| vBMD L1-L3 (mg/cm3) | 503.2 | +12.6 | 1057.3 | +31.1 | 227.8 | +1.8 |
| Physical function | ||||||
| Usual gait speed (m/s) | .86 | −1.5 | .65 | −2.3 | 1.21 | +.2 |
| 6-minute walk test (m) | 426 | −1.7 | 310 | −3.4 | 498 | −1.2 |
| Hand grip strength (kg) | 40.8 | −.5 | 20.9 | −.9 | 48.0 | +.1 |
| Repeat chair stands in 30 seconds (n) | 9 | −2.5 | 3b | −6.1b | 12 | −1.6 |
| PROMIS-PF T-scorec | 39.5 | 30.9 | 46.3 | |||
Abbreviations: aBMD, areal bone mineral density; DXA, dual-energy X-ray absorptiometry; N/A, not available; PROMIS-PF, Patient-Reported Outcomes Measurement Information System—Physical Function; QCT, quantitative computed tomography; vBMD, volumetric bone mineral density.
aNot applicable because of the presence of surgical hardware bilaterally.
bLimited by pain 7 of 10.
c50 = normal T score.
DXA, spine QCT, and physical function data
| Subject II-1 | Subject II-2 | Subject III-1 | ||||
|---|---|---|---|---|---|---|
| DXA | Z score | Z score | Z score | |||
| Lumbar spine aBMD (g/cm2) | 3.0 | +9.6 | 4.1 | +17.3 | 1.7 | +2.7 |
| Total hip aBMD (g/cm2) | N/Aa | 3.6 | +22.1 | 1.3 | +1.9 | |
| Spine QCT | ||||||
| vBMD L1-L3 (mg/cm3) | 503.2 | +12.6 | 1057.3 | +31.1 | 227.8 | +1.8 |
| Physical function | ||||||
| Usual gait speed (m/s) | .86 | −1.5 | .65 | −2.3 | 1.21 | +.2 |
| 6-minute walk test (m) | 426 | −1.7 | 310 | −3.4 | 498 | −1.2 |
| Hand grip strength (kg) | 40.8 | −.5 | 20.9 | −.9 | 48.0 | +.1 |
| Repeat chair stands in 30 seconds (n) | 9 | −2.5 | 3b | −6.1b | 12 | −1.6 |
| PROMIS-PF T-scorec | 39.5 | 30.9 | 46.3 | |||
| Subject II-1 | Subject II-2 | Subject III-1 | ||||
|---|---|---|---|---|---|---|
| DXA | Z score | Z score | Z score | |||
| Lumbar spine aBMD (g/cm2) | 3.0 | +9.6 | 4.1 | +17.3 | 1.7 | +2.7 |
| Total hip aBMD (g/cm2) | N/Aa | 3.6 | +22.1 | 1.3 | +1.9 | |
| Spine QCT | ||||||
| vBMD L1-L3 (mg/cm3) | 503.2 | +12.6 | 1057.3 | +31.1 | 227.8 | +1.8 |
| Physical function | ||||||
| Usual gait speed (m/s) | .86 | −1.5 | .65 | −2.3 | 1.21 | +.2 |
| 6-minute walk test (m) | 426 | −1.7 | 310 | −3.4 | 498 | −1.2 |
| Hand grip strength (kg) | 40.8 | −.5 | 20.9 | −.9 | 48.0 | +.1 |
| Repeat chair stands in 30 seconds (n) | 9 | −2.5 | 3b | −6.1b | 12 | −1.6 |
| PROMIS-PF T-scorec | 39.5 | 30.9 | 46.3 | |||
Abbreviations: aBMD, areal bone mineral density; DXA, dual-energy X-ray absorptiometry; N/A, not available; PROMIS-PF, Patient-Reported Outcomes Measurement Information System—Physical Function; QCT, quantitative computed tomography; vBMD, volumetric bone mineral density.
aNot applicable because of the presence of surgical hardware bilaterally.
bLimited by pain 7 of 10.
c50 = normal T score.
Regarding physical function, his average gait speed was slow at .86 m/s (Z score −1.5) and he was able to walk 426 m in 6 minutes. His hand grip strength reached 40.8 kg (Z score −.5) and he was able to complete 9 repeat chair stands in 30 seconds (Z score −2.5). His self-reported physical function (PROMIS-PF T-score) was just over 1 SD below normal at 39.5.
Subject II-2 Case Report
Subject II-2 is a 51-year-old woman who was diagnosed with ADO at age 17 years and has had >20 fractures throughout her life. She had 3 atraumatic metatarsal fractures by age 14 years and a right subtrochanteric proximal femur fracture requiring open reduction and internal fixation after slipping on ice at age 35 years. She later suffered a left foot stress fracture at age 39 years after climbing stairs and an atraumatic right 5th metatarsal fracture at age 42 years. At age 43 years, she had 2 left rib fractures followed by a left metatarsal fracture at age 45 years. At age 46 years, she had a traumatic right acromion and right intercondylar fracture, and at age 47 years, had 2 right-sided rib fractures. The rib fractures were discovered in the setting of chest pain experienced while straining when constipated. At age 48 years, she was sitting at a table when she started having new left leg pain. An atraumatic left femur fracture was revealed requiring open reduction and internal fixation. At age 49 years, she had a repeat right acromion fracture, with no trauma attributed, and 3 new right metatarsal fractures. Before her 50th birthday, she suffered a left tibial fracture while undergoing left knee replacement. She started using a walker at age 50 years. She developed osteomyelitis of the jaw at age 51 years and has osteonecrosis of the jaw at 2 sites. Her complete blood count showed moderate normocytic anemia with hemoglobin of 8.8 g/dL (Table 1). Total CK was 76 U/L, whereas CK-BB levels were elevated, composing 33% of total CK.
Her skeletal survey showed near-complete vertebral endplate sclerosis with an anvil shape within the vertebral bodies and extensive femoral cortical thickening with intramedullary space involvement (Fig. 4). On DXA scan, her lumbar spine aBMD was 4.13 g/cm2 (Z score +17.3) with a total hip aBMD of 3.63 g/cm2 (Z score +22.1). vBMD of L1-3 by QCT was 1057.3 mg/cm3 (Z score +31.1). HRpQCT revealed relatively solid bones with abnormal microarchitecture at the distal tibia and remnant trabeculae within the marrow space at the tibial diaphysis (Fig. 3). The distal tibia had elevated total vBMD (Z score +10.0).

Skeletal survey of subject II-2. Left: Lateral radiograph of the lower thoracic and lumbar spine demonstrates multilevel anvil-shape appearance of the vertebral bodies (dotted black outline) with near-complete vertebral body sclerosis at multiple levels. Chronic L2, L3, and L4 pars interarticularis defects are noted (white arrows). Right: Anteroposterior radiograph of the left distal femur and knee demonstrates extensive femoral cortical thickening with sclerosis in the mid-distal diaphysis intramedullary region suggestive of intramedullary involvement (white arrows). Erlenmeyer flask shape (double-headed black arrow with dashed black line indicating expected shape) and subtle bone-in-bone appearance at the proximal tibia (black arrows). Femoral fixation hardware is present (#).
Her average gait speed was very slow at .65 m/s (Z score = −2.3) and she was only able to walk 310 m in 6 minutes, requiring a walker. Hand grip strength reached 20.9 kg (Z score −.9) and she could only complete 3 repeat chair stands in 30 seconds (Z score −6.1), with the activity limited by pain. Her self-reported physical function (PROMIS-PF T-score) was 2 SDs below normal at 30.9.
Subject III-1 Case Report
Subject III-1, a 28-year-old man, is the son of subject II-2. He has a considerably milder phenotype compared with his mother and uncle. At age 14 years, after an intense season of baseball, he started having lower back pain while pitching and batting. He was found to have an L5 compression fracture, which resolved with physical therapy. He can currently ambulate and run without limitations or pain. His complete blood count did not show anemia (Table 1). Total CK was 93 U/L, and CK-BB levels were undetectable (undetectable CK-BB levels is normal). Physical examination demonstrated no visible skeletal deformities, and his skeletal survey was essentially normal. His thoracic and lumbar spines show some mild thoracic endplate degenerative changes, but the changes are not considered suggestive of osteopetrosis (Fig. 5). His lumbar spine aBMD was 1.73 g/cm2 (Z score +2.7) with a total hip aBMD of 1.36 g/cm2 (Z score +2.0). vBMD of L1-3 by QCT was 227.86 mg/cm3 (Z score +1.8). HRpQCT revealed completely normal structure of the tibial diaphysis and distal tibia, with a Z score of +.04 for total vBMD at the distal tibia (Fig. 3). His average gait speed was normal at 1.21 m/s (Z score +.2) and he walked 498 m in 6 minutes. He completed 12 chair stands in 30 seconds (Z score −1.8), and his self-reported physical function (PROMIS-PF T-score) was normal at 46.3.

Skeletal survey of subject III-1. Left: Lateral radiograph of the lower thoracic and lumbar spine demonstrates minimal multilevel vertebral body endplate irregularities without sclerosis. Right: Anteroposterior radiograph of the distal femur appears normal without appreciable cortical thickening, bone-in-bone appearance, or Erlenmeyer flask shape.
Genetic Analysis
Genomic DNA was extracted from either peripheral lymphocyte (subject II-1), saliva (subject II-2), or buccal mucosa (subject III-1) and sequenced commercially with Invitae (subjects II-1 and III-1) and Blueprint Genetics (subject II-2). These samples were enriched for targeted regions using a hybridization-based protocol and sequenced using Illumina technology by the Invitae Corporation. Genetic testing of all 3 subjects revealed an allelic variant in exon 15: NM_006019.4(TCIRG1):c.1735G > A (p.Gly579Arg) of the TCIRG1 gene. Thus, the neutral and nonpolar amino acid glycine was replaced by the basic and polar arginine at position 579. This missense variant has a minor allele frequency of <.01 and was classified as a variant of uncertain significance. Variants in CLCN7 were not detected. Both TCIRG1 and CLCN7 genes were covered with targeted regions coverage depths of ≥50× and ≥20× for the Blueprint Osteopetrosis and Dense Bone Dysplasia Panel Plus and the Invitae Skeletal Disorders Panel, respectively. Sequence analysis covered coding exons, 10 to 20 base pairs of adjacent intronic sequence on either side of the coding exons, select noncoding variants with >99% analytical sensitivity and specificity for single nucleotide variants, insertion, and deletions <15 bp in length as well as with 100% sensitivity for copy number variations at a single exon resolution (exon-level deletions and duplications). Additional genes tested by the Blueprint Osteopetrosis and Dense Bone Dysplasia Panel Plus included OSTM1, SNX10, PLEKHM1, TNFRSF11A, TNFSF11, CA2, CTSK, FERMT3, and LEMD3 genes, whereas the Invitae Skeletal Disorders Panel tested 358 genes, including OSTM1, SNX10, TNFRSF11A, TNFSF11, CA2, CTSK, and LEMD3, known osteopetrosis genes.
These sequencing methods also identify variants affecting splicing as they included adjacent intronic sequence on either side of the coding exons. However, no deep intronic sequences were investigated by the methods used in this study; therefore, variants in other part of the introns, if there are any, remains undetermined.
We analyzed the variant using several protein prediction programs to identify the possible impact of this amino acid substitution on the structure and function of the protein. Sorting Intolerant From Tolerant predicts whether an amino acid substitution affects protein function based on sequence homology and the physical properties of amino acids. PolyPhen-2 (Polymorphism Phenotyping v2) predicts the possible impact of an amino acid substitution on the structure and function of a human protein using physical and comparative considerations. Previously, we reported that the Gly579Arg variant is predicted to be deleterious by the Sorting Intolerant From Tolerant and PolyPhen-2 programs, with scores of .02 and .824, respectively (21).
Protein Variation Effect Analyzer (PROVEAN) predicts whether an amino acid substitution has an impact on the biological function of a protein. A PROVEAN score is either equal to or below a predefined threshold (score below −2.5 is considered deleterious, whereas a score above −2.5 is considered neutral). PROVEAN predicted the Gly579Arg variant is neutral with a score of −1.7 (21). Functional Analysis through Hidden Markov Models v2.3 predicts the functional effects of protein missense variants by combining sequence conservation and pathogenicity weights for the overall tolerance of the protein to variants within hidden Markov models. A score less than −1.5 is predicted to be damaging. Functional Analysis through Hidden Markov Models v2.3 predicted the Gly579Arg variant as damaging with a score of −2.09. Screening for Non-Acceptable Polymorphisms (SNAP2) predicts the effect of single amino acid substitutions on protein function based on a machine learning device that distinguishes between neutral and deleterious variants by taking a variety of sequence and variant features into account. A SNAP2 score <40 is neutral and >50 is considered a deleterious effect. SNAP2 predicted the Gly579Arg variant with a score of 58 with an expected accuracy of 75%.
Discussion
TCIRG1 encodes the essential alpha 3 subunit of osteoclast-specific V-ATPase “proton pump,” which is necessary for osteoclast resorption, pit acidification, and osteoclast function. TCIRG1 defects lead to inefficient bone resorption from nonfunctional osteoclasts (22). In a prior report of novel mutations in TCIRG1, most missense mutations identified involved changes to arginine (23). We report 3 individuals with a uniform TCIRG1 missense variant and ADO phenotype, without evidence of a CLCN7 variant. This case series is the first to report a TCIRG1 variant in adolescent/adult presentation of ADO. Variants in the TCIRG1 gene are common in ARO. The specific pathogenic variant, (TCIRG1):c.1735G > A (p.Gly579Arg), seen in our subjects here was previously reported in ARO patients by us (21) and others (23-25).
We previously reported 2 brothers with attenuated ARO who underwent hematopoietic stem cell transplant as young adults, having compound heterozygous variants in TGIRG1: a Gly579Arg (as in our patient discussed previously) and Ala417Thr. One brother was diagnosed at 6 months and the other at age 3 years after a hip fracture. Family history was negative for osteopetrosis. HRpQCT of the distal tibia was performed on 1 of the brothers at age 33 years (several years after transplant) and showed near complete obliteration of the trabecular compartment with extensive cortical porosity (21). This is most comparable to HRpQCT findings of subject II-2 (Fig. 3), who also had impressive obliteration of the trabecular compartment; however, the cortical porosity was not seen. Overall, these data suggest that the substitution of glycine at codon 579 with arginine affects protein structure/function of the a3 subunit of V-ATPase, resulting in the ADO phenotype of our subjects.
A 5-year-old girl with ARO presented with bilateral proximal femoral Salter-Harris type II fractures with associated slipped capital femoral epiphysis, secondary to confirmed autosomal recessive osteopetrosis, also having the p.Gly579Arg variant (maternally inherited) and p.Ala417Thr variant (paternally inherited), which are the same variants reported by Afshariyamchlou et al. This 5-year-old patient had generalized osteosclerotic bones, rugger jersey spine, and optic nerve compression. The authors reported no known family history of osteopetrosis features, although bone density and radiography in the parents was not reported, and we cannot exclude that they had mild disease (26).
A TCIRG1 variant causing ADO has been reported previously in a 3-year-old boy having a de novo heterozygous p.His24Arg missense variant, which was not detected in his family members (15). He presented with fracture, bony sclerosis, and high bone mass. Of note, this young patient had right optic nerve atrophy, which was not observed in our subjects. In this boy's variant, a basic amino acid (histidine) is replaced by another (arginine). In our variant, basic arginine replaced a neutral nonpolar amino acid glycine.
Our case series, in combination with prior reports of ARO (21, 23-25) because of the p.Gly579Arg variant, indicates that this variant can cause either dominant or recessive osteopetrosis. There is an impressive variability in clinical and radiologic manifestations reported in ADO, even within single kindred (1, 27, 28), so much that the term of “benign osteopetrosis” has become a misnomer (4). We were able to demonstrate this variability specifically in ADO adults with CLCN7 variants in a prior cohort (1). Though nearly all clinically affected adults suffered fractures, the type, number, and severity varied. Nearly one half (49%) of adults suffered a hip or femur fracture, 33% suffered severe fractures (defined as defined as >10 fractures of any type and/or greater than one hip/femur fracture), and nearly 12% had 15 or more fractures (1). The subjects also had a high prevalence of severe visual loss (occurring in 19%) because of optic nerve compression resulting from impaired bone resorption and causing failure of the optic canal to expand during growth. Additionally, 16% of our CLCN7 patients had osteonecrosis or osteomyelitis and 3% developed bone marrow failure (1). Such variability has not been previously demonstrated with a variant in ADO, outside of CLCN7. The 3 members in the current family with a TCIRG1 variant exhibited marked variability in disease severity, with siblings subjects II-1 and II-2 having more severe disease manifestations. Each experienced multiple fractures throughout their lives and subsequent debility. Characteristic radiographic findings were seen in subjects II-1 and II-2, including Erlenmeyer Flask Deformity/club-shaped flaring of long bone metaphyses (Figs. 2 and 4) and rugger jersey spine (normal appearing vertebral midbody sandwiched between dense sclerotic bone) seen in Fig. 2. In contrast, subject III-1 had no fractures attributable to ADO (Fig. 5). This diversity extends to hematology because anemia was present in subjects II-1 and II-2 but not III-1. In terms of bone microarchitecture, subject III-1's HRpQCT findings were unremarkable and markedly different from the extremely abnormal trabecular microarchitecture of subject II-2. Last, subject III-1 performed considerably better on physical function tests, unsurprisingly because he did not have the same skeletal pathologic impediments as his relatives.
It has been demonstrated that patients with ADO tend to have increased CK isoenzyme levels, specifically CK-BB (29, 30). Our subjects with severe disease manifestations, subjects II-1 and II-2, both had elevated CK-BB isoenzyme levels. It is interesting that subject III-1, with mild phenotype (Z score +2.7 in spine), had undetectable CK-BB levels. It would be interesting to assess in a future study the associations between CK-BB serum levels and ADO disease severity.
We have discovered a novel TCIRG1 variant with variable expressivity causing ADO. Our findings demonstrate phenotypic diversity exists in ADO caused by TCIRG1 variants, which was previously only seen in ADO with CLCN7 variants. This phenotypic diversity is overtly shown in anemia, imaging and densitometry assessments, and physical function metrics. This report adds to the growing evidence of various genetic etiologies of ADO and the phenotypic variability seen in this disease, even within families with the same disease-causing variant.
Funding
This study was funded in part by the National Institutes of Health through NIAMS grants R01AR077869 and P30AR072581 (The Indiana Center for Musculoskeletal Health Clinical Research Cores), and by the NCATS award UL1TR002529 (The Indiana Clinical and Translational Sciences Institute) and by philanthropic support from the Ancient Accepted Scottish Rite, Valley of Indianapolis. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or any other organization.
Data Availability
Original data generated and analyzed during this study are included in this published article or in the data repositories listed in References.
References
Abbreviations
- aBMD
areal bone mineral density
- ADO
autosomal dominant osteopetrosis
- ARO
autosomal recessive osteopetrosis
- CK
creatine kinase
- CK-BB
creatine kinase-BB isoenzyme
- DXA
dual-energy X-ray absorptiometry
- HRpQCT
high-resolution peripheral quantitative computed tomography
- PROMIS-PF
Patient-Reported Outcomes Measurement Information System—Physical Function
- PROVEAN
Protein Variation Effect Analyzer
- QCT
quantitative computed tomography
- SNAP2
Screening for Non-Acceptable Polymorphisms
- V-ATPase
vacuolar-type H+−ATPase
- vBMD
volumetric bone mineral density

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}