





Abstract
Preclinical studies show seliciclib (R-roscovitine) suppresses neoplastic corticotroph proliferation and pituitary adrenocorticotrophic hormone (ACTH) production.
To evaluate seliciclib as an effective pituitary-targeting treatment for patients with Cushing disease (CD).
Two prospective, open-label, phase 2 trials, conducted at a tertiary referral pituitary center, included adult patients with de novo, persistent, or recurrent CD who received oral seliciclib 400 mg twice daily for 4 consecutive days each week for 4 weeks. The primary endpoint in the proof-of-concept single-center study was normalization of 24-hour urinary free cortisol (UFC; ≤ 50 µg/24 hours) at study end; in the pilot multicenter study, primary endpoint was UFC normalization or ≥ 50% reduction in UFC from baseline to study end.
Sixteen patients were consented and 9 were treated. Mean UFC decreased by 42%, from 226.4 ± 140.3 µg/24 hours at baseline to 131.3 ± 114.3 µg/24 hours by study end. Longitudinal model showed significant UFC reductions from baseline to each treatment week. Three patients achieved ≥ 50% UFC reduction (range, 55%-75%), and 2 patients exhibited 48% reduction; none achieved UFC normalization. Plasma ACTH decreased by 19% (P = 0.01) in patients who achieved ≥ 48% UFC reduction. Three patients developed grade ≤ 2 elevated liver enzymes, anemia, and/or elevated creatinine, which resolved with dose interruption/reduction. Two patients developed grade 4 liver-related serious adverse events that resolved within 4 weeks of seliciclib discontinuation.
Seliciclib may directly target pituitary corticotrophs in CD and reverse hypercortisolism. Potential liver toxicity of seliciclib resolves with treatment withdrawal. The lowest effective dose requires further determination.
Cushing disease (CD) results from hypercortisolism due to excess adrenocorticotrophic hormone (ACTH) production by a pituitary corticotroph cell adenoma. The disease is rare, with a global prevalence of 2.2 cases per 100 000 persons (1). Despite advances in neurosurgery and radiotherapy techniques, as well as approval of new medications, overall morbidity and mortality remains unacceptably high (2, 3). Therapeutic goals for CD are to normalize cortisol levels while preserving pituitary function, reverse clinical features of hypercortisolism, and control disease (2, 3). As long-term failure rates of transsphenoidal adenoma resection can be as high as 55% to 70% within 10 years, repeat surgery and/or radiation are often required; in these patients, CD is incurable (4, 5).
Multimodal care for persistent or recurrent CD, including pituitary-directed radiation, medical treatment, and surgical adrenalectomy, is usually not sufficient to control pituitary disease, and may be associated with serious sequelae (2–5). For example, adrenal steroidogenesis inhibitors, including the recently approved levoketoconazole and osilodrostat, decrease glucocorticoid synthesis and/or adrenal androgen production and secretion and are effective at controlling cortisol excess (6–8). However, the risk for adrenal insufficiency and adverse sequelae from mineralocorticoid-induced hypokalemia can limit their use (2).
The somatostatin receptor ligand pasireotide, currently the only pituitary-targeted medical therapy approved for CD, lowers corticotroph ACTH production with subsequent reduction of adrenal cortisol production, but is associated with hyperglycemia and/or diabetes development in more than 50% of patients (9–11). There is a clear unmet need for safe and effective pituitary-targeting medical therapies that normalize ACTH and cortisol levels.
Multifactorial pathways including dysregulated signaling, tumor suppressor genes, and cell cycle regulators are all implicated in the pathogenesis of pituitary ACTH-secreting tumors (12–14). We showed that E2F1 induces pituitary tumor transforming gene (PTTG) expression in human pituitary tumors (15), and that PTTG overexpression in corticotroph tumor cells is associated with lineage specific cyclin E upregulation and G1/S disruption (16). Corticotroph lineage specific amplification of cyclin E/E2F1 signals contribute to uncontrolled POMC transcription and autonomous ACTH production (17). Our preclinical screening in zebrafish and subsequent validation in mouse models as well as in human cell cultures showed that seliciclib (R-roscovitine), a small-molecule, selective cyclin-dependent kinase inhibitor, inhibits cell proliferation, POMC transcription, and ACTH production in neoplastic corticotrophs. Thus, seliciclib may directly target pituitary ACTH overproduction and reverse hypercortisolism in patients with CD (16, 17).
Here, we report on 9 patients treated in 2 prospective, open-label phase 2 clinical trials evaluating the safety and efficacy of oral seliciclib in patients with de novo, persistent, or recurrent CD.
Methods
We conducted a proof-of-concept single-center, prospective, open-label, phase 2 clinical trial at the Cedars-Sinai Pituitary Center funded by a National Institutes of Health grant to determine whether seliciclib could safely normalize urinary free cortisol (UFC) levels by reducing pituitary corticotroph tumor ACTH production in patients with de novo, persistent, or recurrent CD (ClinicalTrials.gov NCT02160730). We planned to enroll 16 patients, and per our statistical design described below, we reasoned that seliciclib would be considered promising in the treatment of ACTH-producing pituitary tumors if at least 3 of the 16 responded.
After the first 4 patients completed treatment and interim analysis showed promising safety and efficacy, we received funding from the Food and Drug Administration (FDA) Office of Orphan Products Development to launch a pilot multicenter phase 2 clinical trial of seliciclib using the same inclusion and exclusion criteria as for the proof-of-concept study (ClinicalTrials.gov NCT03774446). Given the rarity of CD, to maximize the recruitment pool, we closed the proof-of-concept single-center study before launching the multicenter pilot study. This second study was closed after 5 patients were treated, as described below.
Written informed consent was obtained from each participant before inclusion in each study. The Cedars-Sinai Institutional Review Board (IRB) approved the proof-of-concept single-center study and served as the single IRB of record for the pilot multicenter study. Both studies were monitored by an independent Data Safety Monitoring Board (DSMB).
Study Design
Based on available data from phase 1 and phase 2 studies of seliciclib in other settings (18–21), we selected a dose schedule of 400 mg twice daily given for 4 consecutive days each week for 4 weeks as used in other trials with this drug (20, 21). As was demonstrated with the 15-day, single-arm, open-label, proof-of-concept phase 2 trial of pasireotide (10), we determined that a short 4-week trial would be sufficient to demonstrate seliciclib safety and efficacy, with the advantage of not compromising plans for standard of care in patients destined to undergo pituitary or adrenal surgery.
For our proof-of-concept study, we planned to treat 16 patients in a single arm. For the pilot multicenter study, we planned a Bayesian adaptive design to evaluate 2 of 3 potential doses/schedules of seliciclib in separate cohorts to determine the lowest effective dose. In the first cohort (Cohort A), we planned to treat 13 patients with the same dosing regimen used in the proof-of-concept study, ie, 400 mg twice daily for 4 consecutive days each week for 4 weeks. Efficacy and safety outcomes of the first cohort would then be used to determine the dose/schedule given in the second cohort. If Cohort A were declared a success, 16 patients would be enrolled in Cohort B and treated with 200 mg 3 times daily for 4 consecutive days each week for 4 weeks to determine whether this lower dose is also as efficacious but with a more tolerable safety profile. Alternatively, if Cohort A did not show efficacy, 13 patients would be enrolled in Cohort C and treated with 800 mg per day given in 3 divided doses as 400/200/200 mg for 4 consecutive days each week for 4 weeks. As seliciclib was shown to have a short half-life in patients with solid tumors (19) and the dose-response relationship (ED 50) in CD patients is unknown, Cohort C would evaluate whether a more frequent schedule is effective and has a tolerable side effect profile.
Study Participants
For both studies, adults at least 18 years old with de novo, persistent, or recurrent CD were eligible for enrollment. CD was defined as confirmed pituitary origin of ACTH production as evidenced by persistent hypercortisolemia established by 2 consecutive 24-hour UFC levels ≥1.5 × upper limit of normal (ULN), and normal or elevated ACTH levels and pituitary macroadenoma (> 1 cm) on MRI or inferior petrosal sinus sampling (IPSS) central-to-peripheral ACTH gradient > 2 at baseline and > 3 after corticotrophin-releasing hormone (CRH) stimulation regardless of pituitary adenoma size. Recurrent or persistent CD was defined as pathologically confirmed resected pituitary ACTH-secreting tumor or IPSS central-to-peripheral ACTH gradient > 2 at baseline and > 3 after CRH stimulation, and 2 consecutive 24-hour UFC levels >1.5 × ULN beyond postsurgical week 6. Patients on medical treatment for CD were required to undergo washout before screening assessments were performed: 2 weeks for ketoconazole, metyrapone, mifepristone, and short-acting pasireotide, and 4 weeks for cabergoline and long-acting pasireotide. Washout was also required for patients taking CYP3A4 strong inducers or inhibitors; duration varied between drugs and was a minimum of 5 to 6 times the drug half-life.
Pregnant women and patients with unstable visual fields for ≥ 6 months or abutment or compression of the optic chiasm on magnetic resonance imaging (MRI) were excluded. Patients with ectopic ACTH-secreting tumors or Cushing syndrome or hypercortisolism due to non-pituitary ACTH secretion, cyclic or pseudo-Cushing, or an inherited syndrome were also excluded. Patients with a history of poorly controlled diabetes mellitus (glycated hemoglobin [HbA1c] > 8%) or clinically significant cardiovascular dysfunction were excluded, as were patients with a history of liver disease including cirrhosis, chronic active hepatitis B and C, or chronic persistent hepatitis, or patients with alanine transaminase (ALT) or aspartate transaminase (AST) > 1.5 × ULN, serum total bilirubin > 1 × ULN, or serum albumin < 0.67 × lower limit of normal at screening.
For the proof-of-concept single-center study, eligible patients were identified and recruited at the Cedars-Sinai Pituitary Center, a tertiary referral center for patients with pituitary disorders. For the pilot multicenter study, eligible patients were identified and recruited at 5 centers in the greater Los Angeles area: Cedars-Sinai Medical Center, Lundquist Institute at Harbor-UCLA Medical Center, Providence Saint John's Health Center, UCLA Medical Center, and University of Southern California. Patients were then screened, enrolled, and treated with seliciclib at Cedars-Sinai.
Study Visits and Assessments
All study visits for both trials were conducted at Cedars-Sinai. After the Screening Visit (Day −60 to −1), eligible patients underwent testing at Baseline (Visit 1, Day 0), and were seen weekly for 4 weeks (Visit 2, Day 1; Visit 3, Day 8; Visit 4, Day 15; Visit 5, Day 22; Visit 6, Day 29), and then finally for a Study Completion Visit. Seliciclib was dispensed at Visits 2 to 5 along with a dosing diary to ensure compliance with the oral medication. Blood draws for safety laboratory tests, complete blood count with differential, complete metabolic panel, and coagulation panel (prothrombin and INR) were obtained at Visit 1 and Visits 3 to 5. Hormone profile was performed at Visit 1 and Visit 6, and an additional thyroid panel was performed at Visits 3 to 5. Fasting lipid profile and HbA1c was measured at Visits 1 and 6. Plasma ACTH and serum cortisol levels were obtained at each weekly visit every 3 hours between 09:00 hours to 15:00 hours from samples derived by intravenous catheter, or via blood draw every 3 hours if an intravenous catheter could not be placed. The area under the concentration (C)-time (T) (AUC0-6h) of plasma ACTH and serum cortisol were calculated using the formula AUC = (C1 + C2)/2 × (T2-T1). All laboratory blood tests were processed at Cedars-Sinai Medical Center. Pituitary MRI was performed at Screening or within 28 days prior to Screening as well as at Visit 6.
At each weekly visit, patients were given empty containers for 24-hour UFC collections on 3 consecutive days in the week following the study visit, as well as collection kits for twice-weekly late-night salivary cortisol testing, with instructions to bring all collections to Cedars-Sinai or a local Quest Diagnostic location. All 24-hour UFC and salivary cortisol tests were processed by Quest Diagnostics.
Clinical assessments for adverse events (AEs) were assessed weekly at Visits 2 to 6 and again at the Study Completion Visit. Electrocardiogram was performed at Visit 1 and at Visits 3 to 6. AEs were graded using National Cancer Institute Common Toxicity Criteria for Adverse Events (NCI-CTC) version 4.0. For any AE grade ≥ 3, study drug was held, and participants were referred for follow-up and treatment as appropriate until recovery. Serious AEs, whether or not related to the study drug, were reported within 7 days to the FDA, the Cedars-Sinai Office of Research Compliance, the independent DSMB, and Cyclacel Inc.
Endpoints
For the proof-of-concept single-center study, the primary endpoint was normalized mean 24-hour UFC at study end, defined as ≤ 50 µg/24 hours. Given that random intra-subject UFC variability in CD may be up to 50% (22), mean 24-hour UFC reduction by ≥ 50% from baseline to study end was set as a secondary endpoint. For the pilot multicenter study, after consultation with FDA Office of Orphan Products Development, the protocol was amended so that the 2 UFC measures served as co-primary endpoints. Thus, efficacy was declared if patients achieved either normalized 24-hour UFC at study end, defined as ≤ 50 µg/24 hours, or 24-hour UFC reduced by ≥ 50% from baseline to study end.
For both studies, secondary endpoints included percentage change in mean plasma ACTH, 24-hour UFC, serum cortisol, late-night salivary cortisol levels, mean HbA1c, fasting blood glucose, blood electrolytes, and tumor size between baseline and within 5 days of study end; change in clinical signs and symptoms of hypercortisolism, measured as percentage change in mean weight, body mass index, and blood pressure between baseline and study completion visit; change in hematologic and blood chemistry safety endpoints, measured as percentage change in total sodium, potassium, blood urea nitrogen, serum creatinine, estimated glomerular filtration rate, total protein, liver enzymes (ALT, AST, total bilirubin, albumin, alkaline phosphatase), total cholesterol, low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL), triglycerides, and coagulation between baseline and within 5 days of study end; and number of patients with a change in electrocardiogram results between baseline and within 5 days of study end.
Statistics
Because each study was closed after only a small number of patients was treated, it was not possible to perform statistical analysis as originally planned. However, since inclusion and exclusion criteria, endpoints, and dosing regimens were the same for both protocols, we considered the trials exchangeable and combined results for assessment.
Subjects requiring dose reduction for safety were considered for analysis on an intention-to-treat basis. Statistical analyses were conducted on subjects who received at least one study dose and with at least one 24-hour UFC assessment.
Numerical variables were summarized using mean and SD or median and range and/or percentage change from baseline. The primary outcome UFC was analyzed across time using a longitudinal model (5 time points) with Toeplitz covariance structure. Least squares mean differences from baseline across the treatment weeks were reported. Secondary endpoints were analyzed by Student paired t test. A two-sided 0.05 significance level was used throughout. SAS version 9.4 (SAS Institute, Cary, North Carolina) was used for statistical calculations.
Results
Four patients were treated in the proof-of-concept single-center study and 5 patients were treated in the pilot multicenter study before each trial was closed (Table 1).
Patient characteristics
| Single-center study | Multicenter study | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| Age, years | 43 | 66 | 47 | 30 | 58 | 47 | 19 | 42 | 49 |
| Sex | Female | Female | Female | Female | Female | Female | Female | Female | Female |
| BMI, kg/m2 | 44.1 | 27.5 | 22.5 | 53.8 | 41.6 | 27.2 | 36.0 | 37.0 | 34.0 |
| Duration of disease, years | 9 | 0.25 | 6 | 5 | 10 | 4 | 0.5 | 5 | 0.9 |
| Tumor diameter at diagnosis, mm | 4 | 4 | >10 | 25 | NA | NA | NV | NV | >15 |
| Baseline tumor diameter, mm | NV | 4 | 2.2 | NV | 7.3 | 10.8 | 2.1 | NV | NV |
| Baseline mean UFC, µg/24 hoursa | 143 | 111 | 368 | 311 | 106.9 | 147.7 | 494.4 | 98.3 | 258.5 |
| Hormone deficiencies | |||||||||
| Hypopituitarism | No | No | No | Yes | No | No | No | No | No |
| Hypothyroidism | Yes | Yes | No | Yes | No | No | No | No | Yes |
| Hypogonadism | No | Yes | Yes | Yes | Yes | No | Yes | No | PM |
| Diabetes insipidus | No | No | No | No | No | No | No | Yes | No |
| Surgical history | |||||||||
| Before seliciclib, n | 2 | 0 | 1 | 2 | 2 | 1 | 0 | 2 | 1 |
| After seliciclib, n | No | Yes | No | No | No | No | Yes | No | No |
| Radiotherapy before seliciclibb | No | No | No | Yes | No | No | No | No | No |
| Medical therapy history | |||||||||
| Ketoconazole | Yesc | No | No | Yesc | Yesc | No | No | Yesc | No |
| Pasireotide LAR | Yesc | No | Yesd | No | No | No | No | No | No |
| Cabergoline | Yesc | No | No | No | Yesc | No | No | No | No |
| Seliciclib dose regimen | 400 mg BID × 4 days/week × 2 weeks, break for 2 weeks, then 200 mg TID × 4 days/week × 2 weeks | 400 mg BID × 4 days/week × 1 week, break for 1 week, then 200 mg BID × 4 days/week × 1 week, then 400 mg BID x 4 days/week × 2 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 2 weeks then withdrawn | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 1 week then withdrawn |
| Single-center study | Multicenter study | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| Age, years | 43 | 66 | 47 | 30 | 58 | 47 | 19 | 42 | 49 |
| Sex | Female | Female | Female | Female | Female | Female | Female | Female | Female |
| BMI, kg/m2 | 44.1 | 27.5 | 22.5 | 53.8 | 41.6 | 27.2 | 36.0 | 37.0 | 34.0 |
| Duration of disease, years | 9 | 0.25 | 6 | 5 | 10 | 4 | 0.5 | 5 | 0.9 |
| Tumor diameter at diagnosis, mm | 4 | 4 | >10 | 25 | NA | NA | NV | NV | >15 |
| Baseline tumor diameter, mm | NV | 4 | 2.2 | NV | 7.3 | 10.8 | 2.1 | NV | NV |
| Baseline mean UFC, µg/24 hoursa | 143 | 111 | 368 | 311 | 106.9 | 147.7 | 494.4 | 98.3 | 258.5 |
| Hormone deficiencies | |||||||||
| Hypopituitarism | No | No | No | Yes | No | No | No | No | No |
| Hypothyroidism | Yes | Yes | No | Yes | No | No | No | No | Yes |
| Hypogonadism | No | Yes | Yes | Yes | Yes | No | Yes | No | PM |
| Diabetes insipidus | No | No | No | No | No | No | No | Yes | No |
| Surgical history | |||||||||
| Before seliciclib, n | 2 | 0 | 1 | 2 | 2 | 1 | 0 | 2 | 1 |
| After seliciclib, n | No | Yes | No | No | No | No | Yes | No | No |
| Radiotherapy before seliciclibb | No | No | No | Yes | No | No | No | No | No |
| Medical therapy history | |||||||||
| Ketoconazole | Yesc | No | No | Yesc | Yesc | No | No | Yesc | No |
| Pasireotide LAR | Yesc | No | Yesd | No | No | No | No | No | No |
| Cabergoline | Yesc | No | No | No | Yesc | No | No | No | No |
| Seliciclib dose regimen | 400 mg BID × 4 days/week × 2 weeks, break for 2 weeks, then 200 mg TID × 4 days/week × 2 weeks | 400 mg BID × 4 days/week × 1 week, break for 1 week, then 200 mg BID × 4 days/week × 1 week, then 400 mg BID x 4 days/week × 2 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 2 weeks then withdrawn | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 1 week then withdrawn |
Abbreviations: BID, twice daily; BMI, body mass index; NA, not available; NV, not visible; PM, postmenopausal; TID, 3 times daily.
Upper limit of normal = 50 µg/24 hours.
Completed 3 years before trial enrollment.
Administered as standard of care and enrolled after washout.
Administered as part of a clinical trial > 2 years before enrollment.
Patient characteristics
| Single-center study | Multicenter study | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| Age, years | 43 | 66 | 47 | 30 | 58 | 47 | 19 | 42 | 49 |
| Sex | Female | Female | Female | Female | Female | Female | Female | Female | Female |
| BMI, kg/m2 | 44.1 | 27.5 | 22.5 | 53.8 | 41.6 | 27.2 | 36.0 | 37.0 | 34.0 |
| Duration of disease, years | 9 | 0.25 | 6 | 5 | 10 | 4 | 0.5 | 5 | 0.9 |
| Tumor diameter at diagnosis, mm | 4 | 4 | >10 | 25 | NA | NA | NV | NV | >15 |
| Baseline tumor diameter, mm | NV | 4 | 2.2 | NV | 7.3 | 10.8 | 2.1 | NV | NV |
| Baseline mean UFC, µg/24 hoursa | 143 | 111 | 368 | 311 | 106.9 | 147.7 | 494.4 | 98.3 | 258.5 |
| Hormone deficiencies | |||||||||
| Hypopituitarism | No | No | No | Yes | No | No | No | No | No |
| Hypothyroidism | Yes | Yes | No | Yes | No | No | No | No | Yes |
| Hypogonadism | No | Yes | Yes | Yes | Yes | No | Yes | No | PM |
| Diabetes insipidus | No | No | No | No | No | No | No | Yes | No |
| Surgical history | |||||||||
| Before seliciclib, n | 2 | 0 | 1 | 2 | 2 | 1 | 0 | 2 | 1 |
| After seliciclib, n | No | Yes | No | No | No | No | Yes | No | No |
| Radiotherapy before seliciclibb | No | No | No | Yes | No | No | No | No | No |
| Medical therapy history | |||||||||
| Ketoconazole | Yesc | No | No | Yesc | Yesc | No | No | Yesc | No |
| Pasireotide LAR | Yesc | No | Yesd | No | No | No | No | No | No |
| Cabergoline | Yesc | No | No | No | Yesc | No | No | No | No |
| Seliciclib dose regimen | 400 mg BID × 4 days/week × 2 weeks, break for 2 weeks, then 200 mg TID × 4 days/week × 2 weeks | 400 mg BID × 4 days/week × 1 week, break for 1 week, then 200 mg BID × 4 days/week × 1 week, then 400 mg BID x 4 days/week × 2 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 2 weeks then withdrawn | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 1 week then withdrawn |
| Single-center study | Multicenter study | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| Age, years | 43 | 66 | 47 | 30 | 58 | 47 | 19 | 42 | 49 |
| Sex | Female | Female | Female | Female | Female | Female | Female | Female | Female |
| BMI, kg/m2 | 44.1 | 27.5 | 22.5 | 53.8 | 41.6 | 27.2 | 36.0 | 37.0 | 34.0 |
| Duration of disease, years | 9 | 0.25 | 6 | 5 | 10 | 4 | 0.5 | 5 | 0.9 |
| Tumor diameter at diagnosis, mm | 4 | 4 | >10 | 25 | NA | NA | NV | NV | >15 |
| Baseline tumor diameter, mm | NV | 4 | 2.2 | NV | 7.3 | 10.8 | 2.1 | NV | NV |
| Baseline mean UFC, µg/24 hoursa | 143 | 111 | 368 | 311 | 106.9 | 147.7 | 494.4 | 98.3 | 258.5 |
| Hormone deficiencies | |||||||||
| Hypopituitarism | No | No | No | Yes | No | No | No | No | No |
| Hypothyroidism | Yes | Yes | No | Yes | No | No | No | No | Yes |
| Hypogonadism | No | Yes | Yes | Yes | Yes | No | Yes | No | PM |
| Diabetes insipidus | No | No | No | No | No | No | No | Yes | No |
| Surgical history | |||||||||
| Before seliciclib, n | 2 | 0 | 1 | 2 | 2 | 1 | 0 | 2 | 1 |
| After seliciclib, n | No | Yes | No | No | No | No | Yes | No | No |
| Radiotherapy before seliciclibb | No | No | No | Yes | No | No | No | No | No |
| Medical therapy history | |||||||||
| Ketoconazole | Yesc | No | No | Yesc | Yesc | No | No | Yesc | No |
| Pasireotide LAR | Yesc | No | Yesd | No | No | No | No | No | No |
| Cabergoline | Yesc | No | No | No | Yesc | No | No | No | No |
| Seliciclib dose regimen | 400 mg BID × 4 days/week × 2 weeks, break for 2 weeks, then 200 mg TID × 4 days/week × 2 weeks | 400 mg BID × 4 days/week × 1 week, break for 1 week, then 200 mg BID × 4 days/week × 1 week, then 400 mg BID x 4 days/week × 2 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 2 weeks then withdrawn | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 4 weeks | 400 mg BID × 4 days/week × 1 week then withdrawn |
Abbreviations: BID, twice daily; BMI, body mass index; NA, not available; NV, not visible; PM, postmenopausal; TID, 3 times daily.
Upper limit of normal = 50 µg/24 hours.
Completed 3 years before trial enrollment.
Administered as standard of care and enrolled after washout.
Administered as part of a clinical trial > 2 years before enrollment.
In the proof-of-concept multicenter study, 7 patients were consented and 3 were excluded before the first dose was dispensed: 2 withdrew for logistical reasons unrelated to the study and 1 showed disease progression on screening MRI and decided not to continue with the study. Of the 4 patients who completed 4 weeks of treatment, 2 required dose adjustment. One showed grade ≤ 2 elevated liver enzymes after 2 weeks; treatment was withheld for 2 weeks, and then resumed at 75% dose (200 mg 3 times daily) for 2 weeks. The other patient showed an elevated creatinine level at the beginning of Week 2; treatment was withheld for 1 week and resumed at 50% dose (200 mg twice daily) for 1 week then at full dose (400 mg twice daily) for 2 weeks.
In the pilot multicenter study, 9 patients were consented, and 4 were excluded before starting treatment with seliciclib: 1 had macrocytic anemia and another had elevated ALT/AST that met exclusion criteria between Screening and Baseline visits, both of unknown causes. The third patient was determined at screening to have urinary incontinence that precluded weekly urine collections, and the fourth was determined to have cyclic Cushing syndrome after multiple discordant 24-hour UFC assessments. Of the 5 patients treated with seliciclib in the multicenter study, 3 completed 4 weeks of therapy. Two were withdrawn after 1 to 2 weeks because of grade 4 liver toxicity, as described below.
After carefully reviewing all accumulated safety data, FDA regulatory authorities recommended a lower dose for future enrollment to minimize liver toxicity. The 400 mg twice daily dose was discontinued, the pilot multicenter study was closed, and a new phase 2 clinical trial investigating a lower dose will be open for recruitment.
Patient Characteristics
Table 1 shows baseline characteristics of the 9 patients treated with seliciclib on the 2 studies. Median age was 47 years (range, 19-66). All were female. Median disease duration was 5 years (range, 4 months to 10 years), including 2 patients diagnosed within 6 months of starting trial enrollment. Maximal tumor diameter on pituitary MRI at initial diagnosis was > 10 mm in 3 patients, 4 mm in 2 patients, and non-visible in 2 patients; tumor size at diagnosis was not available in the remaining 2 patients.
Two patients were treated with seliciclib prior to undergoing pituitary surgery, while 7 patients were treated for recurrent or persistent disease. Of these, 3 patients were treated after 1 pituitary surgery and 4 patients after 2 surgeries. One patient had pituitary irradiation for residual disease 3 years before trial enrollment. Five patients were treated with at least 1 medical therapy for CD. Of these, 4 were treated with ketoconazole, 1 with cabergoline, 1 with pasireotide LAR, and 1 with pasireotide LAR followed by cabergoline. At baseline, all but 1 had at least 1 pituitary hormone deficiency. Five patients had central hypogonadism, 4 had central hypothyroidism, 1 had panhypopituitarism, and 1 had diabetes insipidus.
Seliciclib Effect on 24-hour UFC
All 9 subjects treated with seliciclib demonstrated reduced mean 24-hour UFC levels from baseline to study end (range, 11% to 75%; Fig. 1). Five patients had near or > 50% UFC reduction (range, 48%-75%). None achieved normalization of 24-hour UFC. Overall, mean ± SD UFC levels decreased from 226.4 ± 140.3 µg/24 hours (4.5 ± 2.8 × ULN) at baseline to 131.3 ± 114.3 µg/24 hour (2.6 ± 2.3 × ULN) by study end, corresponding to a 42% UFC reduction with seliciclib treatment. In the 7 patients who completed 4 weeks of treatment, overall mean UFC was reduced by 36%, from 238.8 ± 153.4 µg/24 hours (4.8 ± 3.1 × ULN) at baseline to 151.7 ± 123.4 µg/24 hours (3.0 ± 2.5 × ULN) at study end.
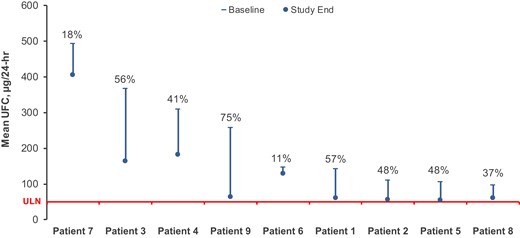
Change in mean UFC from baseline to study end. Percentage decrease in UFC from baseline to study end for each individual patient is depicted. Red line indicates upper limit of Normal (ULN) = 50 µg/24 hours.
In the longitudinal model, each treatment week showed significant decrease of mean UFC from baseline. The largest decrease was at week 1 (141.0 µg/24 hours [95% CI, 70.9-211.0]; P = 0.0003), followed by a small mean rebound during the subsequent 3 weeks that is mainly due to the prominent rebound of one subject (#7) (Fig. 2 and Table 2), although each follow-up week had significantly lower mean UFC compared to baseline (Table 2).
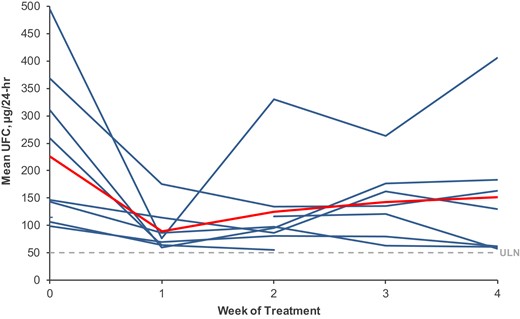
Mean weekly UFC. Blue lines depict weekly mean UFC of individual patients (mUFC), n = 9. Red line depicts mean of weekly mUFC. Dashed line indicates UFC upper limit of Normal = 50 µg/24 hours.
Weekly UFC levels
| Week | N | Mean mUFC ± SD, µg/24 hours | Median mUFC, µg/24 hours | Reduction in mean mUFC from baseline, % |
|---|---|---|---|---|
| 0 | 9 | 226.4 ± 140.3 (4.5 ± 2.8 × ULN) | 146.4 (2.9 × ULN) | — |
| 1 | 8 | 88.9 ± 39.3 (1.8 ± 0.8 × ULN) | 73.2 (1.5 × ULN) | 63 |
| 2 | 8 | 124.4 ± 86.2 (2.5 ± 1.7 × ULN) | 96.6 (1.9 × ULN) | 44 |
| 3 | 7 | 142.8 ± 67.0 (2.9 ± 1.3 × ULN) | 135.0 (2.7 × ULN) | 40 |
| 4 | 7 | 151.7 ± 123.4 (3.0 ± 2.5 × ULN) | 130.2 (2.6 × ULN) | 36 |
| Longitudinal model estimates | ||||
| Week | N | Least squares mean difference (reduction) from baseline | 95% CI | P value |
| 0 | 9 | — | — | — |
| 1 | 8 | 141.0 | 70.9-211.0 | 0.0003 |
| 2 | 8 | 100.3 | 42.1-158.5 | 0.002 |
| 3 | 7 | 88.7 | 13.2-164.2 | 0.023 |
| 4 | 7 | 84.7 | 40.3-129.2 | 0.0006 |
| Week | N | Mean mUFC ± SD, µg/24 hours | Median mUFC, µg/24 hours | Reduction in mean mUFC from baseline, % |
|---|---|---|---|---|
| 0 | 9 | 226.4 ± 140.3 (4.5 ± 2.8 × ULN) | 146.4 (2.9 × ULN) | — |
| 1 | 8 | 88.9 ± 39.3 (1.8 ± 0.8 × ULN) | 73.2 (1.5 × ULN) | 63 |
| 2 | 8 | 124.4 ± 86.2 (2.5 ± 1.7 × ULN) | 96.6 (1.9 × ULN) | 44 |
| 3 | 7 | 142.8 ± 67.0 (2.9 ± 1.3 × ULN) | 135.0 (2.7 × ULN) | 40 |
| 4 | 7 | 151.7 ± 123.4 (3.0 ± 2.5 × ULN) | 130.2 (2.6 × ULN) | 36 |
| Longitudinal model estimates | ||||
| Week | N | Least squares mean difference (reduction) from baseline | 95% CI | P value |
| 0 | 9 | — | — | — |
| 1 | 8 | 141.0 | 70.9-211.0 | 0.0003 |
| 2 | 8 | 100.3 | 42.1-158.5 | 0.002 |
| 3 | 7 | 88.7 | 13.2-164.2 | 0.023 |
| 4 | 7 | 84.7 | 40.3-129.2 | 0.0006 |
The mUFC ULN = 50 µg/24 hours.
Abbreviations: mUFC, mean weekly urinary free cortisol in each individual patient; ULN, upper limit of normal.
Weekly UFC levels
| Week | N | Mean mUFC ± SD, µg/24 hours | Median mUFC, µg/24 hours | Reduction in mean mUFC from baseline, % |
|---|---|---|---|---|
| 0 | 9 | 226.4 ± 140.3 (4.5 ± 2.8 × ULN) | 146.4 (2.9 × ULN) | — |
| 1 | 8 | 88.9 ± 39.3 (1.8 ± 0.8 × ULN) | 73.2 (1.5 × ULN) | 63 |
| 2 | 8 | 124.4 ± 86.2 (2.5 ± 1.7 × ULN) | 96.6 (1.9 × ULN) | 44 |
| 3 | 7 | 142.8 ± 67.0 (2.9 ± 1.3 × ULN) | 135.0 (2.7 × ULN) | 40 |
| 4 | 7 | 151.7 ± 123.4 (3.0 ± 2.5 × ULN) | 130.2 (2.6 × ULN) | 36 |
| Longitudinal model estimates | ||||
| Week | N | Least squares mean difference (reduction) from baseline | 95% CI | P value |
| 0 | 9 | — | — | — |
| 1 | 8 | 141.0 | 70.9-211.0 | 0.0003 |
| 2 | 8 | 100.3 | 42.1-158.5 | 0.002 |
| 3 | 7 | 88.7 | 13.2-164.2 | 0.023 |
| 4 | 7 | 84.7 | 40.3-129.2 | 0.0006 |
| Week | N | Mean mUFC ± SD, µg/24 hours | Median mUFC, µg/24 hours | Reduction in mean mUFC from baseline, % |
|---|---|---|---|---|
| 0 | 9 | 226.4 ± 140.3 (4.5 ± 2.8 × ULN) | 146.4 (2.9 × ULN) | — |
| 1 | 8 | 88.9 ± 39.3 (1.8 ± 0.8 × ULN) | 73.2 (1.5 × ULN) | 63 |
| 2 | 8 | 124.4 ± 86.2 (2.5 ± 1.7 × ULN) | 96.6 (1.9 × ULN) | 44 |
| 3 | 7 | 142.8 ± 67.0 (2.9 ± 1.3 × ULN) | 135.0 (2.7 × ULN) | 40 |
| 4 | 7 | 151.7 ± 123.4 (3.0 ± 2.5 × ULN) | 130.2 (2.6 × ULN) | 36 |
| Longitudinal model estimates | ||||
| Week | N | Least squares mean difference (reduction) from baseline | 95% CI | P value |
| 0 | 9 | — | — | — |
| 1 | 8 | 141.0 | 70.9-211.0 | 0.0003 |
| 2 | 8 | 100.3 | 42.1-158.5 | 0.002 |
| 3 | 7 | 88.7 | 13.2-164.2 | 0.023 |
| 4 | 7 | 84.7 | 40.3-129.2 | 0.0006 |
The mUFC ULN = 50 µg/24 hours.
Abbreviations: mUFC, mean weekly urinary free cortisol in each individual patient; ULN, upper limit of normal.
Seliciclib Effect on ACTH and Cortisol
AUC0-6h of plasma ACTH and serum cortisol at baseline and end of study on Day 29 were available for 5 of 7 patients who completed all 4 weeks of treatment. Due to state, county, and institutional COVID-19 restrictions, 2 patients did not undergo serial plasma ACTH and serum cortisol measurements to minimize duration of the weekly clinic visits.
As preclinical data indicate that seliciclib acts directly on ACTH to induce changes in circulating cortisol, we analyzed changes in plasma ACTH and serum cortisol based on 24-hour UFC response. We grouped together 3 patients who achieved near or ≥ 50% reduction in 24-hour UFC (range, 48%-57%), defined as responders, and 2 patients who had UFC reduction of 11% and 41%, defined as non-responders. There were no significant differences in baseline mean ACTH between responders (506.1 ± 240.8 pg/mL/h) and non-responders (389.3 ± 295.9 pg/mL/h). At study end, mean ACTH was reduced to 407.9 ± 185.9 pg/mL/h in responders (P = 0.058) while non-responders had a mean ACTH of 459.3 ± 372.9 pg/mL/h (Table 3). There was no difference in mean AUC0-6h serum cortisol at baseline vs study end in responders (197.8 ± 39.2 vs 185.9 ± 62.6 µg/mL/h) or non-responders (96.5 ± 3.3 vs 103.2 ± 2.6 µg/mL/h). There was no difference in mean late-night salivary cortisol available in 5 patients after 4 weeks of treatment (0.44 ± 0.32 vs 0.38 ± 0.28 µg/dL).
Mean ACTH at baseline and study end
| Mean ACTH ± SD, pg/mL/h | ||||
|---|---|---|---|---|
| Patient # | Baseline | Study End | P Value | |
| Responders | 1, 2, 3 | 506.1 ± 240.8 | 407.9 ± 185.9 | 0.058 |
| Non-responders | 4, 6 | 389.3 ± 295.9 | 459.3 ± 372.9 | 0.21 |
| Mean ACTH ± SD, pg/mL/h | ||||
|---|---|---|---|---|
| Patient # | Baseline | Study End | P Value | |
| Responders | 1, 2, 3 | 506.1 ± 240.8 | 407.9 ± 185.9 | 0.058 |
| Non-responders | 4, 6 | 389.3 ± 295.9 | 459.3 ± 372.9 | 0.21 |
Responders include 3 patients who achieved near or >50% reduction of 24-hour UFC (range, 48%-57%). Non-responders include 2 patients who had UFC reduction of 11% and 41%.
Mean ACTH at baseline and study end
| Mean ACTH ± SD, pg/mL/h | ||||
|---|---|---|---|---|
| Patient # | Baseline | Study End | P Value | |
| Responders | 1, 2, 3 | 506.1 ± 240.8 | 407.9 ± 185.9 | 0.058 |
| Non-responders | 4, 6 | 389.3 ± 295.9 | 459.3 ± 372.9 | 0.21 |
| Mean ACTH ± SD, pg/mL/h | ||||
|---|---|---|---|---|
| Patient # | Baseline | Study End | P Value | |
| Responders | 1, 2, 3 | 506.1 ± 240.8 | 407.9 ± 185.9 | 0.058 |
| Non-responders | 4, 6 | 389.3 ± 295.9 | 459.3 ± 372.9 | 0.21 |
Responders include 3 patients who achieved near or >50% reduction of 24-hour UFC (range, 48%-57%). Non-responders include 2 patients who had UFC reduction of 11% and 41%.
Cardiovascular and Metabolic Secondary Endpoints
Table 4 shows cardiovascular and metabolic secondary endpoints in the 7 patients who completed 4 weeks of treatment. Mean systolic blood pressure decreased from baseline to study end (142.7 ± 18.3 mmHg vs 125.7 ± 8.4 mmHg, P = 0.027); there were no significant changes in mean body weight, diastolic blood pressure, fasting blood glucose, and HbA1c levels from baseline to study end. Mean cholesterol, triglyceride, and LDL levels increased mildly and HDL levels decreased mildly from baseline to study end; these changes were not significant. Three subjects voluntarily discontinued statin treatment while receiving seliciclib.
Cardiovascular and metabolic secondary endpoints
| Baseline | End of study | P Value | |
|---|---|---|---|
| Weight, kg | 91.7 ± 31.1 | 91.9 ± 31.8 | 0.73 |
| sBP, mmHg | 142.7 ± 18.4 | 125.7 ± 8.4 | 0.027 |
| dBP, mmHg | 80.1 ± 11.4 | 76.3 ± 13.4 | 0.20 |
| FBG, mg/dL | 122.2 ± 48.1 | 101.7 ± 24.1 | 0.48 |
| HbA1c, % | 6.6 ± 1.0 | 6.5 ± 1.1 | 0.66 |
| Cholesterol, mg/dL | 227.9 ± 37.9 | 250.0 ± 41.8 | 0.25 |
| Triglycerides, mg/dL | 154.9 ± 83.9 | 170.9 ± 90.2 | 0.51 |
| LDL, mg/dL | 137.6 ± 31.8 | 161.1 ± 31.9 | 0.17 |
| HDL, mg/dL | 59.3 ± 14.8 | 54.6 ± 17.7 | 0.17 |
| VLDL, mg/dL | 31.0 ± 16.8 | 34.3 ± 17.8 | 0.49 |
| Baseline | End of study | P Value | |
|---|---|---|---|
| Weight, kg | 91.7 ± 31.1 | 91.9 ± 31.8 | 0.73 |
| sBP, mmHg | 142.7 ± 18.4 | 125.7 ± 8.4 | 0.027 |
| dBP, mmHg | 80.1 ± 11.4 | 76.3 ± 13.4 | 0.20 |
| FBG, mg/dL | 122.2 ± 48.1 | 101.7 ± 24.1 | 0.48 |
| HbA1c, % | 6.6 ± 1.0 | 6.5 ± 1.1 | 0.66 |
| Cholesterol, mg/dL | 227.9 ± 37.9 | 250.0 ± 41.8 | 0.25 |
| Triglycerides, mg/dL | 154.9 ± 83.9 | 170.9 ± 90.2 | 0.51 |
| LDL, mg/dL | 137.6 ± 31.8 | 161.1 ± 31.9 | 0.17 |
| HDL, mg/dL | 59.3 ± 14.8 | 54.6 ± 17.7 | 0.17 |
| VLDL, mg/dL | 31.0 ± 16.8 | 34.3 ± 17.8 | 0.49 |
Abbreviations: dBP, diastolic blood pressure; FBG, fasting blood glucose; HbA1c, hemoglobin A1c; HDL, high-density lipoprotein; LDL, low-density lipoprotein; sBP, systolic blood pressure; VLDL, very low-density lipoprotein.
Cardiovascular and metabolic secondary endpoints
| Baseline | End of study | P Value | |
|---|---|---|---|
| Weight, kg | 91.7 ± 31.1 | 91.9 ± 31.8 | 0.73 |
| sBP, mmHg | 142.7 ± 18.4 | 125.7 ± 8.4 | 0.027 |
| dBP, mmHg | 80.1 ± 11.4 | 76.3 ± 13.4 | 0.20 |
| FBG, mg/dL | 122.2 ± 48.1 | 101.7 ± 24.1 | 0.48 |
| HbA1c, % | 6.6 ± 1.0 | 6.5 ± 1.1 | 0.66 |
| Cholesterol, mg/dL | 227.9 ± 37.9 | 250.0 ± 41.8 | 0.25 |
| Triglycerides, mg/dL | 154.9 ± 83.9 | 170.9 ± 90.2 | 0.51 |
| LDL, mg/dL | 137.6 ± 31.8 | 161.1 ± 31.9 | 0.17 |
| HDL, mg/dL | 59.3 ± 14.8 | 54.6 ± 17.7 | 0.17 |
| VLDL, mg/dL | 31.0 ± 16.8 | 34.3 ± 17.8 | 0.49 |
| Baseline | End of study | P Value | |
|---|---|---|---|
| Weight, kg | 91.7 ± 31.1 | 91.9 ± 31.8 | 0.73 |
| sBP, mmHg | 142.7 ± 18.4 | 125.7 ± 8.4 | 0.027 |
| dBP, mmHg | 80.1 ± 11.4 | 76.3 ± 13.4 | 0.20 |
| FBG, mg/dL | 122.2 ± 48.1 | 101.7 ± 24.1 | 0.48 |
| HbA1c, % | 6.6 ± 1.0 | 6.5 ± 1.1 | 0.66 |
| Cholesterol, mg/dL | 227.9 ± 37.9 | 250.0 ± 41.8 | 0.25 |
| Triglycerides, mg/dL | 154.9 ± 83.9 | 170.9 ± 90.2 | 0.51 |
| LDL, mg/dL | 137.6 ± 31.8 | 161.1 ± 31.9 | 0.17 |
| HDL, mg/dL | 59.3 ± 14.8 | 54.6 ± 17.7 | 0.17 |
| VLDL, mg/dL | 31.0 ± 16.8 | 34.3 ± 17.8 | 0.49 |
Abbreviations: dBP, diastolic blood pressure; FBG, fasting blood glucose; HbA1c, hemoglobin A1c; HDL, high-density lipoprotein; LDL, low-density lipoprotein; sBP, systolic blood pressure; VLDL, very low-density lipoprotein.
Adverse Events
Table 5 summarizes AEs in the 9 patients treated with seliciclib in the pilot and multicenter studies. Elevated liver enzymes were seen in 3 patients; IRB and DSMB considered them all study drug–related as elevated liver enzymes have been seen in prior studies with seliciclib (18, 19). In all cases, signs and symptoms resolved uneventfully after dose interruption/reduction for grade ≤ 2 AEs or within 4 weeks after treatment discontinuation for grade 4 AEs. One patient developed asymptomatic grade 2 elevation of ALT and grade 1 elevation of AST and alkaline phosphatase after 2 weeks of drug therapy in the proof-of-concept study. Dose was held for 1 week then resumed at 75% dose for the remaining 2 weeks of treatment.
Adverse events
| Proof-of-concept single-center study | Pilot multicenter study | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| Liver-related | |||||||||
| ȃElevated enzymes (ALT, AST, Alk-phos, total bilirubin) | Grade 1-2 | Grade 1-4 | Grade 3-4 | ||||||
| ȃAbdominal pain | Yes | ||||||||
| ȃJaundice | Yes | ||||||||
| Anemia | Grade 1 | Grade 1 | |||||||
| Elevated creatinine | Grade 1 | ||||||||
| Proof-of-concept single-center study | Pilot multicenter study | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| Liver-related | |||||||||
| ȃElevated enzymes (ALT, AST, Alk-phos, total bilirubin) | Grade 1-2 | Grade 1-4 | Grade 3-4 | ||||||
| ȃAbdominal pain | Yes | ||||||||
| ȃJaundice | Yes | ||||||||
| Anemia | Grade 1 | Grade 1 | |||||||
| Elevated creatinine | Grade 1 | ||||||||
Grading per National Cancer Institute Common Toxicity Criteria for Adverse Events (NCI-CTC) version 4.0.
Abbreviations: Alk-phos, alkaline phosphatase; ALT, alanine transaminase; AST, aspartate transaminase.
Adverse events
| Proof-of-concept single-center study | Pilot multicenter study | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| Liver-related | |||||||||
| ȃElevated enzymes (ALT, AST, Alk-phos, total bilirubin) | Grade 1-2 | Grade 1-4 | Grade 3-4 | ||||||
| ȃAbdominal pain | Yes | ||||||||
| ȃJaundice | Yes | ||||||||
| Anemia | Grade 1 | Grade 1 | |||||||
| Elevated creatinine | Grade 1 | ||||||||
| Proof-of-concept single-center study | Pilot multicenter study | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| Liver-related | |||||||||
| ȃElevated enzymes (ALT, AST, Alk-phos, total bilirubin) | Grade 1-2 | Grade 1-4 | Grade 3-4 | ||||||
| ȃAbdominal pain | Yes | ||||||||
| ȃJaundice | Yes | ||||||||
| Anemia | Grade 1 | Grade 1 | |||||||
| Elevated creatinine | Grade 1 | ||||||||
Grading per National Cancer Institute Common Toxicity Criteria for Adverse Events (NCI-CTC) version 4.0.
Abbreviations: Alk-phos, alkaline phosphatase; ALT, alanine transaminase; AST, aspartate transaminase.
Two patients in the pilot multicenter study had serious AEs of grade 4 elevation of liver enzymes without prolongation of prothrombin time (INR) or encephalopathy and required hospitalization. One had abdominal pain and elevated liver enzymes after dosing on Day 1 Week 3 (ALT 14.6 × ULN, AST 21.6 × ULN, alkaline phosphatase 1.5 × ULN, total bilirubin 2.75 × ULN). Study drug was stopped, the patient was withdrawn from the study, and then admitted to the hospital for observation. She was discharged after symptoms resolved and liver enzymes normalized within 4 weeks. The other patient had a history of gallstones but normal liver function tests at baseline. She developed acute cholecystitis after Day 2 Week 1 with normal hepatic function tests including ALT, AST, alkaline phosphatase, total bilirubin, albumin, and total protein levels. Seliciclib dose was withheld per IRB and DSMB, and she was admitted to the hospital for a cholecystectomy. She was restarted on seliciclib 2.5 weeks after cholecystectomy when all test results had normalized. After the first week of seliciclib, she showed elevated liver enzymes (ALT 16.5 × ULN, AST 10.6 × ULN, alkaline phosphatase 1.9 × ULN, total bilirubin 3.4 × ULN) with jaundice requiring hospitalization, and she was withdrawn from the study per IRB and DSMB. Liver enzymes returned to near normal levels (< 1 × ULN) 4 weeks after seliciclib discontinuation (23).
Two patients developed grade 1 anemia. In one patient, the anemia coincided with recent initiation of the antihypertensive hydralazine. Nevertheless, this was considered possibly related to study drug. Post-study follow-up demonstrated stable hemoglobin levels. The other patient had grade 1 anemia at the end of Week 3 to the beginning of Week 4, which coincided with onset of her regular menstrual period. By end of Week 4, the anemia had resolved.
One patient had grade 1 elevated serum creatinine (1.3 × ULN) at the beginning of Week 2. This coincided with addition of the antihypertensive spironolactone. Spironolactone was discontinued and seliciclib was held for 1.5 weeks, after which creatinine returned to baseline level. Nevertheless, this effect has also been reported in previous studies of seliciclib and may be study drug-related. In this case, seliciclib was restarted at 50% dose for Week 2 and the full dose was administered for Week 3 and Week 4 with no further AEs.
Discussion
We describe here the first clinical use of oral seliciclib in patients with de novo, recurrent, or persistent CD. In a proof-of-concept, single-center phase 2 trial and a multicenter pilot phase 2 trial, 3 of 9 (33%) patients achieved ≥ 50% reduction in 24-hour UFC and 2 (22%) exhibited 48% UFC reduction. Reduction in plasma ACTH levels was slightly greater among those who achieved near or ≥ 50% reduction in 24-hour UFC (range, 48%-57%), supporting the ACTH-targeting mechanism of action of seliciclib.
Pituitary ACTH and adrenal cortisol productions follow a circadian rhythm. Plasma ACTH and cortisol levels exhibit diurnal pattern in healthy individuals, and often fluctuate in patients with corticotroph tumors (24). We measured UFC levels as a mean of 3 separate 24-hour urine collections within 1 week of each clinic visit, which is consistent with current recommendations for hypercortisolemia (2). Currently, pasireotide is the only pituitary-targeting drug approved by FDA for treatment of CD. In a phase 2, 15-day study of pasireotide, 76% (22 of 29) showed a mean of 44.5% decrease of UFC levels from baseline, of whom 5 (17%) patients normalized UFC levels (10). In our two 4-week studies of seliciclib, all 9 patients demonstrated reduced mean 24-hour UFC, ranging from 11% to 75%, and mean UFC decreased by 42%. The rapid reduction in UFC in these short studies coupled with the reduction in plasma ACTH among those with a steeper decline in UFC supports the potential benefit of this pituitary-targeting treatment for patients with CD.
We selected a seliciclib dose regimen of 400 mg given twice daily for 4 days per week for 4 weeks based on published data from phase 1 and phase 2 clinical trials of seliciclib in patients with nasopharyngeal cancer, which showed antiproliferative activity and a manageable AE profile (20, 21). At this dose, we observed a 63% reduction of overall mean UFC levels after the first week of treatment. Mean UFC then rebounded slightly with the second week of treatment, resulting in an overall UFC reduction of 36% after 4 weeks. Given the fluctuations in ACTH secretion from corticotrophs (24) and the short half-life of seliciclib (19), it is possible that the “4-days on and 3-days off” regimen contributed to the observed rebound phenomenon. Of note, there was no increase in mean 24-hour UFC or mean ACTH levels in any patient between baseline and study end, and longitudinal analysis showed mean UFC for each week remained significantly reduced compared to baseline.
To avoid compromising standard of care in patients destined to undergo pituitary or adrenal surgery, our study protocols allowed a 4-week washout of cabergoline and pasireotide LAR, which may not be as complete as a 6- or 8-week washout period. Of the 9 study subjects, 1 subject discontinued pasireotide LAR more than 2 months before study enrollment due to gastrointestinal side effects and was switched to ketoconazole as well as cabergoline. UFC was 213.4 μg/dL at 33 days after drug washout. Baseline UFCs collected between 54 and 61 days after stopping ketoconazole and cabergoline continued to meet inclusion criteria. One subject received a short course of pasireotide LAR in a clinical trial 2 years before enrollment to our trial. Another patient had cabergoline washout for > 2 months before obtaining screening/baseline UFC. Therefore, a 4-week washout for cabergoline and pasireotide LAR had minimal impact on the primary outcome.
Type 2 diabetes mellitus, dyslipidemia, hypertension, and other cardiovascular disease risk factors are common complications of CD (2). Four of 9 seliciclib-treated patients had a history of type 2 diabetes mellitus treated with daily insulin injections. Insulin dose was adjusted per standard care during the trial and there were no dose changes for metformin or other oral antihyperglycemic treatments, yet mean fasting blood glucose marginally decreased between baseline and end of study. As change in diabetes treatment was not a study endpoint, follow-up data were not collected, and the clinical relevance of this finding is unknown. Total cholesterol, triglyceride, and LDL levels increased mildly while HDL level decreased mildly from baseline to study end. The causes of these changes are unclear, although withholding of statin dosing due to concern for potential additive liver toxicity may have contributed to individual elevations in cholesterol levels. Of note, systolic blood pressure decreased significantly between baseline and study end (142.7 ± 18.3 mmHg vs 125.7 ± 8.4 mmHg, P = 0.027, n = 7). One patient with de novo CD required an addition of antihypertensive medication during the trial due to a high baseline blood pressure of 179/80 mmHg. Nevertheless, even after excluding this patient, mean systolic blood pressure remained significantly decreased (136.7 ± 9.9 mmHg vs 125.3 ± 9.1 mmHg, P = 0.0009, n = 6).
Transient and potentially dose-limiting elevated liver enzymes have been observed in patients with nasopharyngeal cancer and other solid cancers treated with seliciclib (19–21). In our proof-of-concept single-center study, 1 patient developed asymptomatic grade ≤ 2 elevated liver enzymes that resolved after dose interruption for 2 weeks. She tolerated introduction of reduced-dose seliciclib for the remainder of the study and liver enzymes were normal at study end. We therefore decided to use the same dose regimen for Cohort A of the pilot multicenter trial. However, of the 5 patients treated in this study, 2 developed serious AEs with symptomatic grade 4 liver enzyme elevation requiring hospitalization and permanent treatment discontinuation. All of these patients had normal liver enzyme levels at baseline. Investigation by study investigators and DSMB identified multiple factors that might have contributed to the elevated liver enzymes in these patients, including concomitant use of high-dose statins for hyperlipidemia, angiotensin receptor blocker for hypertension, and divalproex sodium for mood disorder; a history of biliary sludge disease; and underlying fatty liver due to chronic hypercortisolemia of CD. Larger studies with a broader patient population are needed to determine which factors might predispose patients to liver-related AEs.
Of note, preclinical data and results of other clinical trials with seliciclib indicate that tolerability is dose-dependent (data on file, Cyclacel Inc). However, there are as yet no data on a dose-response relationship (ED50) in patients with CD. Our preclinical studies showed that seliciclib inhibits proliferation, POMC transcription, and ACTH production in neoplastic corticotrophs (16, 17). Whether these mechanisms driving inhibition of ACTH production are dose-dependent will be addressed in a future clinical trial evaluating safety and efficacy of the FDA-recommended lower dose of seliciclib in patients with de novo, recurrent, or persistent CD.
Our investigations of seliciclib efficacy are limited by small sample size, and only females were studied. CD patients commonly exhibit multiple comorbidities and disease-related complications, precluding enrollment of high numbers of patients eligible to meet our rigorous inclusion criteria despite collaboration among 5 institutions with established pituitary centers. Beginning 1 year after initiating the multicenter study, state, county, and institutional COVID-19 restrictions further constrained patient recruitment efforts, and also affected our ability to collect complete data on secondary endpoints.
Continued study of seliciclib will further yield important safety and efficacy data of this pituitary-targeting treatment for patients with CD. The early results shown here offer evidence that oral seliciclib treatment may improve hypercortisolemia in patients with CD by directly suppressing pituitary ACTH secretion. The lowest effective dose of seliciclib remains to be determined.
Funding
This study was supported by R21DK103198 funded by NIDDK and R01FD006106 funded by the U.S. Food and Drug Administration. Study drug was provided by Cyclacel Inc. None had a role in study design, data analysis, or decision to publish.
Disclosures
There is no conflict of interest.
Data Availability
Restrictions apply to the availability of some or all data generated or analyzed during this study to preserve patient confidentiality. The corresponding author will on request detail the restrictions and any conditions under which access to some data may be provided.
References
Abbreviations
- ACTH
adrenocorticotropic hormone
- AE
adverse event
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- AUC
area under the concentration-time curve
- CD
Cushing disease
- DSMB
data and safety monitoring board
- FDA
U.S. Food and Drug Administration
- HbA1c
glycated hemoglobin
- HDL
high-density lipoprotein
- IRB
institutional review board
- LDL
low-density lipoprotein
- MRI
magnetic resonance imaging
- UFC
urinary free cortisol
- ULN
upper limit of normal

{kind=link}
{kind=link}