





Abstract
Up to 20% of children with neurofibromatosis type 1 (NF1) develop low-grade optic pathway gliomas (OPGs) that can result in endocrine dysfunction. Data on prevalence and type of endocrine disorders in NF1-related OPGs are scarce.
The aim of the study was to determine the prevalence of endocrine dysfunctions in patients with NF1 and OPGs and to investigate predictive factors before oncological treatment.
Multicenter retrospective study.
Records were reviewed for 116 children (64 females, 52 males) with NF1 and OPGs followed at 4 Italian centers.
We evaluated endocrine function and reviewed brain imaging at the time of OPG diagnosis before radio- and chemotherapy and/or surgery. OPGs were classified according to the modified Dodge classification.
Thirty-two children (27.6%) with a median age of 7.8 years had endocrine dysfunctions including central precocious puberty in 23 (71.9%), growth hormone deficiency in 3 (9.4%), diencephalic syndrome in 4 (12.5%), and growth hormone hypersecretion in 2 (6.2%). In a multivariate cox regression analysis, hypothalamic involvement was the only independent predictor of endocrine dysfunctions (hazard ratio 5.02 [1.802-13.983]; P = .002).
Endocrine disorders were found in approximately one-third of patients with Neurofibromatosis type 1 and OPGs before any oncological treatment, central precocious puberty being the most prevalent. Sign of diencephalic syndrome and growth hormone hypersecretion, although rare, could be predictive of optic pathway gliomas in NF1. Tumor location was the most important predictor of endocrine disorders, particularly hypothalamic involvement.
Neurofibromatosis type 1 (NF1) is a multisystemic, autosomal dominant condition associated to neuro-oncological risk. It is due to heterozygous mutations of the NF1 tumor suppressor gene located on chromosome 17q11.2 which leads to secondary increased mTOR activation (1). Its product, neurofibromin, is highly expressed in Schwann and glial cells, explaining why up to 15% of NF1 patients develop a brain tumor (2), with optic pathway glioma (OPG) representing the commonest occurring in about 20% of patients within the first decade of life (3). OPG is considered a hallmark of NF1, and represents one of the diagnostic criteria together with café-au-lait macules, freckling, neurofibromas, Lisch nodules, bone dysplasia, and a first-degree relative affected.
Although OPG can have different histology (ie, pilocytic astrocytoma, diffusely infiltrating astrocytoma, pilomyxoid astrocytoma, and ganglioglioma), in almost all NF1 cases they present as a pilocytic grade 1 astrocytoma, usually tending to have more indolent behavior than in sporadic forms and to involve more frequently the anterior part of the optic pathway, bilaterally, with chances of spontaneous regression (4-12). On the other hand, about half of patients become symptomatic and need to be treated (surgically and/or with chemotherapy). In fact, OPG (13) may cause visual loss and endocrine disorders, and can exceptionally lead to death (14).
Although endocrine disorders have been reported in 1% to 3% of all NF1 patients (15-17), to our knowledge, data on prevalence, types, and outcomes of OPG-related pretreatment of endocrine dysfunctions are extremely scarce with just 2 papers published on this topic (15,17).
The aim of our study was to determine the prevalence of endocrine dysfunction in patients with NF1 related OPG before any treatment and to investigate their predictors, especially in relation to tumor location and hypothalamic involvement.
Patients and Methods
Study cohort
This retrospective, multicenter study included patients followed at 4 Italian tertiary pediatric referral centers for NF1: the Pediatric Referral Centre of Neurofibromatosis of the Università degli Studi della Campania in Naples, Istituto Giannina Gaslini in Genoa, ‘Meyer’ Children’s Hospital in Florence, and the Regina Margherita Children Hospital, in Turin (52 cases from Naples, 25 from Turin, 20 from Florence, and 19 from Genoa).
Patients with a clinically confirmed diagnosis of NF1 (18), and radiological diagnosis of OPG performed before the age of 16, were included. Gender, NF1 inheritance, age at diagnosis of NF1 and of OPG, indications for diagnostic magnetic resonance imaging (MRI) scan, and duration of follow-up were recorded.
OPGs were classified according to the modified Dodge classification (MDC), also known as the PLAN classification (10). MRI scans performed at the time of diagnosis were all reviewed by the same neuroradiologist (M.C.) with expertise in this field.
OPGs involving the optic nerves were recorded as MDC 1, chiasmatic OPGs as MDC 2, whereas tumors involving tracts and radiations were staged as MDC 3. The highest (most posterior) MDC stage was assigned to tumors involving multiple regions, in agreement with other studies (19). Primary treatment approaches were recorded as surgical resections (any tumor debulking or decompression procedures aimed at relieving raised intracranial pressure), radiotherapy, chemotherapy, and wait and see strategy. Chemotherapy was administered according to International Society of Paediatric Oncology (SIOP) trial protocols.
Definitions of endocrine diseases
Endocrine disorders that only occurred prior to any therapy (either surgery or chemotherapy) were included for analyses. In particular, central precocious puberty (CPP) was defined as breast budding before the age of 8 years in girls and testicular volume > 4 mL before the age of 9 years in boys, together with pubertal basal luteinizing hormone (LH) levels and/or gonadotropin-releasing hormone (GnRH)-stimulated (0.1 mg of Relefact LHRH, Sanofi-Aventis, Frankfurt am Main, Germany) LH levels > 5 IU/L. growth hormone hypersecretion (GHH) was diagnosed in the presence of linear growth acceleration, elevated insulin-like growth factor (IGF)-1 levels for age (adjusted for sex and pubertal stage), and lack of growth hormone (GH) suppression < 1.0 ng/mL during an oral 1.75 g/kg glucose tolerance test. GH deficiency (GHD) was defined as decreased height velocity over a period of at least 6 months in the absence of other causes, with a GH peak < 10 ng/mL until December 2014 and < 8 ng/mL on 2 different provocative tests (arginine, clonidine, insulin tolerance test, and glucagon test were used).
Thyrotropin deficiency was defined as low free-thyroxine levels with inappropriately low or normal thyrotropin levels based on age-appropriate reference ranges.
Adrenocorticotropin (ACTH) deficiency was defined as cortisol peak ≤ 500 mmol/L during the insulin-induced symptomatic hypoglycemia on the insulin tolerance test or low-dose (1 µg) ACTH test.
Diencephalic syndrome (DS) was defined as failure to thrive not explained by vomiting, diarrhea, decreased caloric intake, or other causes, and/or hypothalamic involvement confirmed at the time of diagnosis, and/or crossing down of the 2nd percentiles for weight with a normal growth rate, and/or body mass index (BMI) < –2 standard deviation (SD) and/or emaciation, euphoria, hyperactivity (20-22).
Age at onset and type of the endocrine disorder, auxological data, hormonal testing results, and specific treatments were recorded.
The study was approved by the local ethics committees and informed consent was obtained from patients whenever possible, or by their parents/guardians.
Statistics
Differences between groups were analyzed with the Mann–Whitney U-test for continuous nonparametric variables. Numbers and proportions were compared using the chi-square test. Kaplan–Meier analysis was run in order to determine endocrine event-free survival from diagnosis and the log-rank test was used to compare curves of patients with and without hypothalamic involvement. Only the first endocrine disorder occurred before any treatment was included in the analysis. Patients who did not develop any endocrine disorders were censored at treatment start time and at last follow-up for the ones who did not receive any treatment at all.
Univariate and multivariate Cox regression models were used to explore predictors of endocrine disorders before any treatment. Variables statistically significant in the univariate analysis were included in the multivariate one: MDC stage, hypothalamic involvement, and age at diagnosis of OPGs. P < .05 was considered statistically significant.
SPSS 23 software for Windows for Windows was used for all statistical analysis.
Results
Study population
We enrolled a total of 116 children (52 males) with NF1 (age at diagnosis 1.75 years, range 0-12.25 years) and OPG (age at diagnosis 4.17 years, range 0.42-13.75). Median NF1 follow-up was 9 years (range 0.2-35), while median duration of OPG follow-up was 7.9 years (0.5-24.5 range). Table 1 summarizes the patients’ demographic data, NF1 and OPG features, details about diagnosis, treatment, and follow-up. The data mentioned are also separately reported for patients with and without endocrine dysfunctions. OPGs were diagnosed earlier in the 31 (26.7%) children with endocrine disorders compared with those without endocrine dysfunctions (median age at diagnosis of 3.3 years [range 1.33-11.92] versus 4.71 years [range 0.42-13.75], P = .03); however, there was no significant difference in gender, age at NF1 diagnosis, NF1 inheritance, and indications to MRI. Indeed, almost half of the patients (n = 49, 42.7%) underwent brain MRI for screening within a diagnosis of NF1. Other indications for an MRI scan were visual symptoms (n = 40, 34.5%), headaches (n = 7, 2.5%), neurological signs (n = 6, 1.8%), plexiform neurofibromas (n = 2, 0.8%). Eight children (6.8%) underwent brain MRI because of clinical suspicion of endocrine dysfunction, and in 4 of them (3.4%) a diagnosis was confirmed (n = 2 CPP, n = 1 DS, n = 1 GHH) (Table 1).
Patients’ demographic, tumor- and treatment-related characteristics reported in total and separately for patients with and without endocrine dysfunctions.
| Patients with OPG (116) | Patients with OPG-related endocrine disorders (31) | Patients without OPG-related endocrine disorders (85) | P value | |
|---|---|---|---|---|
| Male gender, N (%) | 52 (44.8) | 15 (48.3) | 37 (43.5) | .2 |
| Median age at NF1 diagnosis, median (range) | 1.75 (0-12.25) | 1.3 (0-6.7) | 1.91 (0.1-12.2) | .7 |
| Inheritance, N (%) | ||||
| Sporadic | 77 (66.3) | 19 (61.3) | 58 (68.3) | .5 |
| Maternal | 19 (16.4) | 6 (22.5) | 13 (14.1) | .6 |
| Paternal | 20 (17.2) | 6 (19.4) | 14 (16.6) | .7 |
| Median duration of NF1 follow-up, years, median (range) | 9 (0.2-35) | 10 (1-25) | 9 (0.5-35) | .2 |
| Age at OPG diagnosis, years, median (range) | 4.2 (0.4-13.7) | 3.3 (1.3-11.9) | 4.7 (0.4-13.7) | .03 |
| Indication to MRI scan, N (%) | ||||
| Screening | 49 (42.2) | 14 (45) | 35 (41.1) | .7 |
| Visual features | 40 (34.5) | 9 (29) | 31(36.4) | .4 |
| Endocrinological problems | 8 (6.8) | 4 (12.9) | 4 (4.7) | |
| Headache | 7 (6) | 1 (3.2) | 6 (7) | |
| Macrocephaly | 3 (2.5) | 0 | 3 (3.5) | |
| Neurological signs | 6 (5.1) | 2 (6.4) | 4 (4.7) | |
| Plexiform neurofibromas | 2 (1.8) | 0 | 2 (2.3) | |
| Neuropsychiatric disorder | 1 (0.8) | 1 (3.2) | 0 | |
| PLAN classification N (%) | ||||
| MDC 1 | 39 (33.6) | 0 (0) | 39 (46.4) | |
| MDC 2 | 23 (19.8) | 6 (18.8) | 17 (20.2) | .9 |
| MDC 3/4 | 54 (46.6) | 26 (81.3) | 28 (33.3) | .0000 |
| Hypothalamic involvement N (%) | 39 (33.6) | 25 (78.1) | 14 (16.7) | .0000 |
| Treatment | ||||
| Observation | 69 (59.5) | 8 (25.8) | 61 (71.7) | 0.0006 |
| Chemotherapy | 39 (33.6) | 19 (32.2) | 20 (23.5) | 0.03 |
| Any surgery | 12 (10.3) | 7 (22.5) | 5 (5.9) | 0.40 |
| Surgical resection | 9 | 7 | 2 | |
| Decompression procedures | 6 | 3 | 3 | |
| Radiotherapy | 1 (0.9) | 1 (3.2) | 0 |
| Patients with OPG (116) | Patients with OPG-related endocrine disorders (31) | Patients without OPG-related endocrine disorders (85) | P value | |
|---|---|---|---|---|
| Male gender, N (%) | 52 (44.8) | 15 (48.3) | 37 (43.5) | .2 |
| Median age at NF1 diagnosis, median (range) | 1.75 (0-12.25) | 1.3 (0-6.7) | 1.91 (0.1-12.2) | .7 |
| Inheritance, N (%) | ||||
| Sporadic | 77 (66.3) | 19 (61.3) | 58 (68.3) | .5 |
| Maternal | 19 (16.4) | 6 (22.5) | 13 (14.1) | .6 |
| Paternal | 20 (17.2) | 6 (19.4) | 14 (16.6) | .7 |
| Median duration of NF1 follow-up, years, median (range) | 9 (0.2-35) | 10 (1-25) | 9 (0.5-35) | .2 |
| Age at OPG diagnosis, years, median (range) | 4.2 (0.4-13.7) | 3.3 (1.3-11.9) | 4.7 (0.4-13.7) | .03 |
| Indication to MRI scan, N (%) | ||||
| Screening | 49 (42.2) | 14 (45) | 35 (41.1) | .7 |
| Visual features | 40 (34.5) | 9 (29) | 31(36.4) | .4 |
| Endocrinological problems | 8 (6.8) | 4 (12.9) | 4 (4.7) | |
| Headache | 7 (6) | 1 (3.2) | 6 (7) | |
| Macrocephaly | 3 (2.5) | 0 | 3 (3.5) | |
| Neurological signs | 6 (5.1) | 2 (6.4) | 4 (4.7) | |
| Plexiform neurofibromas | 2 (1.8) | 0 | 2 (2.3) | |
| Neuropsychiatric disorder | 1 (0.8) | 1 (3.2) | 0 | |
| PLAN classification N (%) | ||||
| MDC 1 | 39 (33.6) | 0 (0) | 39 (46.4) | |
| MDC 2 | 23 (19.8) | 6 (18.8) | 17 (20.2) | .9 |
| MDC 3/4 | 54 (46.6) | 26 (81.3) | 28 (33.3) | .0000 |
| Hypothalamic involvement N (%) | 39 (33.6) | 25 (78.1) | 14 (16.7) | .0000 |
| Treatment | ||||
| Observation | 69 (59.5) | 8 (25.8) | 61 (71.7) | 0.0006 |
| Chemotherapy | 39 (33.6) | 19 (32.2) | 20 (23.5) | 0.03 |
| Any surgery | 12 (10.3) | 7 (22.5) | 5 (5.9) | 0.40 |
| Surgical resection | 9 | 7 | 2 | |
| Decompression procedures | 6 | 3 | 3 | |
| Radiotherapy | 1 (0.9) | 1 (3.2) | 0 |
Patient characteristics are reported as number and percentage. Continuous nonparametric variables are presented as median and range. Comparisons between patients with and without endocrine disorders are made with the chi-square or Fisher exact test for proportions and Mann–Whitney U-test for continuous nonparametric variables, where appropriate. A P value < .05 is considered statistically significant.
Abbreviations: MDC, modified Dodge classification; NF-1, neurofibromatosis type 1; OPGs, optic pathway gliomas.
Patients’ demographic, tumor- and treatment-related characteristics reported in total and separately for patients with and without endocrine dysfunctions.
| Patients with OPG (116) | Patients with OPG-related endocrine disorders (31) | Patients without OPG-related endocrine disorders (85) | P value | |
|---|---|---|---|---|
| Male gender, N (%) | 52 (44.8) | 15 (48.3) | 37 (43.5) | .2 |
| Median age at NF1 diagnosis, median (range) | 1.75 (0-12.25) | 1.3 (0-6.7) | 1.91 (0.1-12.2) | .7 |
| Inheritance, N (%) | ||||
| Sporadic | 77 (66.3) | 19 (61.3) | 58 (68.3) | .5 |
| Maternal | 19 (16.4) | 6 (22.5) | 13 (14.1) | .6 |
| Paternal | 20 (17.2) | 6 (19.4) | 14 (16.6) | .7 |
| Median duration of NF1 follow-up, years, median (range) | 9 (0.2-35) | 10 (1-25) | 9 (0.5-35) | .2 |
| Age at OPG diagnosis, years, median (range) | 4.2 (0.4-13.7) | 3.3 (1.3-11.9) | 4.7 (0.4-13.7) | .03 |
| Indication to MRI scan, N (%) | ||||
| Screening | 49 (42.2) | 14 (45) | 35 (41.1) | .7 |
| Visual features | 40 (34.5) | 9 (29) | 31(36.4) | .4 |
| Endocrinological problems | 8 (6.8) | 4 (12.9) | 4 (4.7) | |
| Headache | 7 (6) | 1 (3.2) | 6 (7) | |
| Macrocephaly | 3 (2.5) | 0 | 3 (3.5) | |
| Neurological signs | 6 (5.1) | 2 (6.4) | 4 (4.7) | |
| Plexiform neurofibromas | 2 (1.8) | 0 | 2 (2.3) | |
| Neuropsychiatric disorder | 1 (0.8) | 1 (3.2) | 0 | |
| PLAN classification N (%) | ||||
| MDC 1 | 39 (33.6) | 0 (0) | 39 (46.4) | |
| MDC 2 | 23 (19.8) | 6 (18.8) | 17 (20.2) | .9 |
| MDC 3/4 | 54 (46.6) | 26 (81.3) | 28 (33.3) | .0000 |
| Hypothalamic involvement N (%) | 39 (33.6) | 25 (78.1) | 14 (16.7) | .0000 |
| Treatment | ||||
| Observation | 69 (59.5) | 8 (25.8) | 61 (71.7) | 0.0006 |
| Chemotherapy | 39 (33.6) | 19 (32.2) | 20 (23.5) | 0.03 |
| Any surgery | 12 (10.3) | 7 (22.5) | 5 (5.9) | 0.40 |
| Surgical resection | 9 | 7 | 2 | |
| Decompression procedures | 6 | 3 | 3 | |
| Radiotherapy | 1 (0.9) | 1 (3.2) | 0 |
| Patients with OPG (116) | Patients with OPG-related endocrine disorders (31) | Patients without OPG-related endocrine disorders (85) | P value | |
|---|---|---|---|---|
| Male gender, N (%) | 52 (44.8) | 15 (48.3) | 37 (43.5) | .2 |
| Median age at NF1 diagnosis, median (range) | 1.75 (0-12.25) | 1.3 (0-6.7) | 1.91 (0.1-12.2) | .7 |
| Inheritance, N (%) | ||||
| Sporadic | 77 (66.3) | 19 (61.3) | 58 (68.3) | .5 |
| Maternal | 19 (16.4) | 6 (22.5) | 13 (14.1) | .6 |
| Paternal | 20 (17.2) | 6 (19.4) | 14 (16.6) | .7 |
| Median duration of NF1 follow-up, years, median (range) | 9 (0.2-35) | 10 (1-25) | 9 (0.5-35) | .2 |
| Age at OPG diagnosis, years, median (range) | 4.2 (0.4-13.7) | 3.3 (1.3-11.9) | 4.7 (0.4-13.7) | .03 |
| Indication to MRI scan, N (%) | ||||
| Screening | 49 (42.2) | 14 (45) | 35 (41.1) | .7 |
| Visual features | 40 (34.5) | 9 (29) | 31(36.4) | .4 |
| Endocrinological problems | 8 (6.8) | 4 (12.9) | 4 (4.7) | |
| Headache | 7 (6) | 1 (3.2) | 6 (7) | |
| Macrocephaly | 3 (2.5) | 0 | 3 (3.5) | |
| Neurological signs | 6 (5.1) | 2 (6.4) | 4 (4.7) | |
| Plexiform neurofibromas | 2 (1.8) | 0 | 2 (2.3) | |
| Neuropsychiatric disorder | 1 (0.8) | 1 (3.2) | 0 | |
| PLAN classification N (%) | ||||
| MDC 1 | 39 (33.6) | 0 (0) | 39 (46.4) | |
| MDC 2 | 23 (19.8) | 6 (18.8) | 17 (20.2) | .9 |
| MDC 3/4 | 54 (46.6) | 26 (81.3) | 28 (33.3) | .0000 |
| Hypothalamic involvement N (%) | 39 (33.6) | 25 (78.1) | 14 (16.7) | .0000 |
| Treatment | ||||
| Observation | 69 (59.5) | 8 (25.8) | 61 (71.7) | 0.0006 |
| Chemotherapy | 39 (33.6) | 19 (32.2) | 20 (23.5) | 0.03 |
| Any surgery | 12 (10.3) | 7 (22.5) | 5 (5.9) | 0.40 |
| Surgical resection | 9 | 7 | 2 | |
| Decompression procedures | 6 | 3 | 3 | |
| Radiotherapy | 1 (0.9) | 1 (3.2) | 0 |
Patient characteristics are reported as number and percentage. Continuous nonparametric variables are presented as median and range. Comparisons between patients with and without endocrine disorders are made with the chi-square or Fisher exact test for proportions and Mann–Whitney U-test for continuous nonparametric variables, where appropriate. A P value < .05 is considered statistically significant.
Abbreviations: MDC, modified Dodge classification; NF-1, neurofibromatosis type 1; OPGs, optic pathway gliomas.
Regarding tumor location, 33.6% of patients (n = 39) were staged as MDC 1, 19.8% as MDC 2 (n = 23), and 46.6% (n = 54) as MDC 3/4. In particular, the anterior part of the pathway was almost always involved (93.1%), followed by the chiasm (62.3%) and the posterior tracts (45.2%). No cases of leptomeningeal metastasis were observed. Thirty-nine patients presented tumors involving the hypothalamus, representing 33.6% of all OPGs: 6 classified as MDC 2 and 33 as MDC 3. Patients with endocrine dysfunctions presented with a significantly higher percentage of hypothalamic involvement than those without endocrine dysfunctions (78.1% vs 16.7%, P = .000) and none of the patients with nerve involvement only (MDC 1) presented endocrine dysfunctions (Table 1).
Finally, the number of patients with endocrine dysfunctions who later underwent treatment for OPGs (chemotherapy and/or surgery) was higher than those without endocrine disorders (75.6% vs 34.2%; P = .0001).
Endocrine dysfunctions
We identified 31 patients with endocrine dysfunction before oncological treatment. CPP was diagnosed in 23 children (72%). Among these patients, 12 were males with median age at diagnosis of 8.0 years range: 3.5-10.4) and median age at onset CPP signs of 7.9 years (range: 3.5-8.8), and 11 were females with median age at diagnosis of 7.8 years (range 4.7-8.6) and a median age at onset of CPP signs of 7.5 years (range: 4.7-7.5). Moreover GHD was diagnosed in 3 patients (9%; 3 males, median age at diagnosis 9.45 years); DS in 4 (12%; all females, median age at diagnosis 4.7 years) and GHH in 2 patients (6%; 2 females, median age at diagnosis 4 years). No patients with thyrotropin and ACTH deficiency were identified. The median time of follow-up between endocrine dysfunction and OPG diagnosis was 4.58 years (range 0.08-8.17). Age at endocrine dysfunction diagnosis was different according to type of disease, being lower in patients with DS and GHH (Table 2).
Characteristics of endocrine disorders
| CPP | GHD | DS | GHH | |
|---|---|---|---|---|
| n (%) | 23 (71.9) | 3 (9.4) | 4 (12.5) | 2 (6.2) |
| M:F (n) | 12:11 | 3:0 | 0:4 | 0:2 |
| Median age at diagnosis, year (range) | 8.2 (3.5-10.4) | 9.75 (9.1-11.6) | 4.66 (1.4-5.8) | 4.0 (3.9-4.1) |
| Chemotherapy | 13 | 2 | 4 | 1 |
| Resection surgery | 3 | 0 | 3 | 1 |
| Decompression procedures | 0 | 0 | 1 | 1 |
| CPP | GHD | DS | GHH | |
|---|---|---|---|---|
| n (%) | 23 (71.9) | 3 (9.4) | 4 (12.5) | 2 (6.2) |
| M:F (n) | 12:11 | 3:0 | 0:4 | 0:2 |
| Median age at diagnosis, year (range) | 8.2 (3.5-10.4) | 9.75 (9.1-11.6) | 4.66 (1.4-5.8) | 4.0 (3.9-4.1) |
| Chemotherapy | 13 | 2 | 4 | 1 |
| Resection surgery | 3 | 0 | 3 | 1 |
| Decompression procedures | 0 | 0 | 1 | 1 |
Characteristics of endocrine disorders
| CPP | GHD | DS | GHH | |
|---|---|---|---|---|
| n (%) | 23 (71.9) | 3 (9.4) | 4 (12.5) | 2 (6.2) |
| M:F (n) | 12:11 | 3:0 | 0:4 | 0:2 |
| Median age at diagnosis, year (range) | 8.2 (3.5-10.4) | 9.75 (9.1-11.6) | 4.66 (1.4-5.8) | 4.0 (3.9-4.1) |
| Chemotherapy | 13 | 2 | 4 | 1 |
| Resection surgery | 3 | 0 | 3 | 1 |
| Decompression procedures | 0 | 0 | 1 | 1 |
| CPP | GHD | DS | GHH | |
|---|---|---|---|---|
| n (%) | 23 (71.9) | 3 (9.4) | 4 (12.5) | 2 (6.2) |
| M:F (n) | 12:11 | 3:0 | 0:4 | 0:2 |
| Median age at diagnosis, year (range) | 8.2 (3.5-10.4) | 9.75 (9.1-11.6) | 4.66 (1.4-5.8) | 4.0 (3.9-4.1) |
| Chemotherapy | 13 | 2 | 4 | 1 |
| Resection surgery | 3 | 0 | 3 | 1 |
| Decompression procedures | 0 | 0 | 1 | 1 |
All patients with CPP were treated with GnRH analogs; all 3 patients with GHD were treated with GH, 2 of them for a relatively short period of time while 1 patient is still on therapy. One of the 2 patients with GHH was treated with octreotide acetate for 1 year, while 1 did not receive any treatment, since GHH was a transient phenomenon.
Children with endocrine disorders collectively underwent 9 surgeries (n = 7 debulkings and n = 2 surgeries for hydrocephalus). Considering the entire population, the cumulative proportion of patients free from endocrine dysfunctions before any oncological treatment was 65.9% at 10 years of OPG follow-up. Endocrine event-free survival declined up to 8 years after OPG diagnosis (23) and patients with hypothalamic involvement showed a significant lower endocrine event-free survival (P < .0001; Fig. 1): indeed, almost 50% of this cohort presented an endocrine dysfunction 5 years after OPG diagnosis.
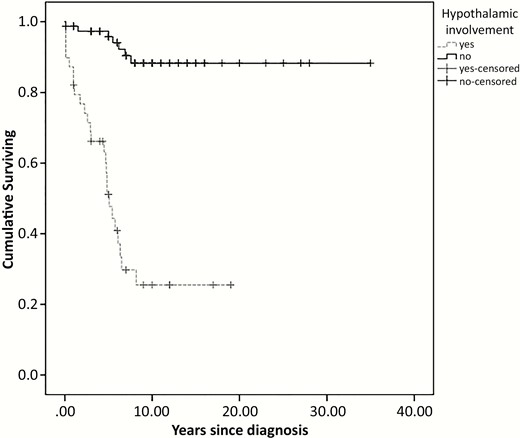
Kaplan–Meier survival curves for pretreatment endocrine event by hypothalamic involvement.
Predictive factors
Hypothalamic involvement, MDC stage, and age at OPG diagnosis less than 5 years were independent predictors of endocrine disorders (Table 3), whereas after adjusting for the same covariates, hypothalamic involvement was the only significant predictor of endocrine disorder before oncological treatment in multivariate analysis (hazard ratio 5.02 [95% confidence interval 1.802-13.983], P = .002) (Table 3).
Hazard ratios and 95% confidence interval for endocrinological disorders onset
| Univariate | Multivariate | |||
|---|---|---|---|---|
| Hazard ratio (95% CI) | P | Hazard ratio (95% CI) | P | |
| Male sex | 0.99 (0.467-1.875) | .851 | - | - |
| Hypothalamic involvement | 11.55 (4.944 - 26.983) | .00000002 | 5.02 (1.802-13.983) | .002 |
| MDC | 3.985 (2.008 - 7.909) | .00008 | 2.140 (0.970 - 4.922) | .06 |
| Inheritance | 1.331 (0.657 - 2.696) | .427 | – | – |
| Age at OPG diagnosis < 5 years | 2.503 (1.124 - 5.575) | .025 | 1.959 (0.863-4.451) | .108 |
| Univariate | Multivariate | |||
|---|---|---|---|---|
| Hazard ratio (95% CI) | P | Hazard ratio (95% CI) | P | |
| Male sex | 0.99 (0.467-1.875) | .851 | - | - |
| Hypothalamic involvement | 11.55 (4.944 - 26.983) | .00000002 | 5.02 (1.802-13.983) | .002 |
| MDC | 3.985 (2.008 - 7.909) | .00008 | 2.140 (0.970 - 4.922) | .06 |
| Inheritance | 1.331 (0.657 - 2.696) | .427 | – | – |
| Age at OPG diagnosis < 5 years | 2.503 (1.124 - 5.575) | .025 | 1.959 (0.863-4.451) | .108 |
Hazard ratios and 95% confidence interval for endocrinological disorders onset
| Univariate | Multivariate | |||
|---|---|---|---|---|
| Hazard ratio (95% CI) | P | Hazard ratio (95% CI) | P | |
| Male sex | 0.99 (0.467-1.875) | .851 | - | - |
| Hypothalamic involvement | 11.55 (4.944 - 26.983) | .00000002 | 5.02 (1.802-13.983) | .002 |
| MDC | 3.985 (2.008 - 7.909) | .00008 | 2.140 (0.970 - 4.922) | .06 |
| Inheritance | 1.331 (0.657 - 2.696) | .427 | – | – |
| Age at OPG diagnosis < 5 years | 2.503 (1.124 - 5.575) | .025 | 1.959 (0.863-4.451) | .108 |
| Univariate | Multivariate | |||
|---|---|---|---|---|
| Hazard ratio (95% CI) | P | Hazard ratio (95% CI) | P | |
| Male sex | 0.99 (0.467-1.875) | .851 | - | - |
| Hypothalamic involvement | 11.55 (4.944 - 26.983) | .00000002 | 5.02 (1.802-13.983) | .002 |
| MDC | 3.985 (2.008 - 7.909) | .00008 | 2.140 (0.970 - 4.922) | .06 |
| Inheritance | 1.331 (0.657 - 2.696) | .427 | – | – |
| Age at OPG diagnosis < 5 years | 2.503 (1.124 - 5.575) | .025 | 1.959 (0.863-4.451) | .108 |
Discussion
We found a considerable prevalence of endocrine dysfunctions in almost one-third of children with NF1 and OPGs before oncological treatment. To our knowledge the number of patients enrolled in the study represents the largest cohort ever reported with this condition. In particular 72% of the OPG patients with endocrine disorders presented central precocious puberty, 12.5% had DS, 9.4% had GH deficiency, and 6% had GH oversecretion. Our data differ from the literature based on the report by Cnossen et al., who studied 122 children with NF1 among which 15 with OPG: the authors found a 5% prevalence of endocrine dysfunction in the entire cohort that increased up to 13% considering those with OPG, 1 of the patients being GHD and another 1 with CPP (15). In addition, Habiby et al. found CPP in 3% of 219 children with NF1, including 33 with associated OPG. The prevalence of CPP increased up to 21% in those with OPG (7 patients), all with optic chiasm involvement and, intriguingly, it increased up to 38% in subjects with optic chiasm involvement. However, the authors did not analyze the impact of hypothalamic involvement or prevalence of other endocrine dysfunctions (24).
Most studies investigating the prevalence of endocrine disorders among patients with optic gliomas tended to include both patients with and without NF1 (19,25,26), making it difficult to extrapolate the specific prevalence of endocrine comorbidities in NF1 with OPG. Moreover, they focused mostly on treatment-related endocrine sequelae without analyzing patients naïve to oncological treatment. To the best of our knowledge, Sani and Albanese investigated the prevalence of endocrine disorders in children with NF1 and OPGs before any treatment (17). They retrospectively reviewed a small cohort of 36 patients, and found a 55.6% prevalence of endocrine dysfunction in 20 patients. This prevalence was very high, even compared with studies on optic gliomas in a mixed population (19).
Our data showed that the most prevalent endocrine disorder in NF1 patients with OPGs before any treatment was CPP, in accordance with other studies on brain tumors involving the hypothalamic region (20). We confirmed, moreover, that males are more affected than in idiopathic CPP with a similar gender ratio (12:11) (14). Indeed, GHD in our population was an uncommon finding (3.4%), in contrast with the report by Sani and Albanesi (17) where GHD was the most common hypothalamic–pituitary defect in 36.1% of their cohort. The high prevalence of GHD may be due to the adopted criteria of GDH diagnosis based on a single stimulation test and/or to the different selection strategy of patients to submit to a GH stimulation test. In other studies including patients with optic gliomas, GHD was mostly diagnosed after treatment, particularly related to radiotherapy (27). Our data together with other reports suggest that GHD might be secondary to surgery and/or radiotherapy rather than related to OPG itself (19).
In our cohort, rare endocrine disorders like DS and GHH were diagnosed in younger patients. In particular, DS has been exceptionally reported in NF1-related OPGs and, unlike in sporadic cases of OPGs where DS tends to occur in the first year of life, the median age of our children with DS was 4.66 years. This might be related to the natural history of OPGs, which tend to grow more slowly in patients with NF1 than in sporadic cases (19). Cavicchiolo et al. reported a good outcome of DS in a 3-year-old boy with NF1 and hypothalamic-chiasmatic OPG after chemotherapy (28). However, analyses of the chemotherapy protocol SIOP for low grade gliomas show that patients with DS as the indication for treatment have a significantly worse outcome (29).
GHH has been mainly described as a transient condition (30,31), and also in our cohort this endocrine feature spontaneously regressed in 1 patient, while targeted treatment was stopped after 1 year in the other patient. Of note, both children were very young (3.9 and 4.1 years respectively).
The reason why GHH may be transient remains to be elucidated. Our results together with already published data suggest that lesions located into the hypothalamus may interfere with its physiological control of the pituitary GH secretion. We may hypothesize that inconstant radiological tumor behavior may reflect a transient endocrinological disruption. In fact, OPGs in NF1 may show periods of quiescence and of partial spontaneous regression (3).
The prevalence GH oversecretion in our cohort is remarkably lower than in a previous study by Cambiaso et al. (1.7 vs 10.9%) (32), but this latter study was specifically designed to investigate GHH in patients with NF1 and OPGs. We believe that this condition may very likely be underdiagnosed due to its intriguing self-limited behavior; hence, a careful auxological follow-up is needed to recognize it. Whether all patients with NFI and OPG should be treated for their GH oversecretion or whether a personalized approach based on GH systemic effects should be adopted remain questionable.
We wondered whether endocrine dysfunctions caused by OPGs in our cohort were associated to lesions contiguous with or directly involving the hypothalamus. MDC (also known as PLAN classification), which better describes the involvement of each segment of the optic pathway and allows the independent involvement of the hypothalamus to be highlighted, suggests that tumor location was associated with an increased risk of endocrine disorders. Multivariate analysis, in fact, showed that hypothalamic involvement was the only predictor, with a hazard ratio of 5.02 (95% confidence interval 1.80-13.98); P = .002. This finding supports that a thorough auxological and biochemical follow-up is mandatory for patients with OPGs involving the hypothalamus at diagnosis, regardless of which segment of the optic pathway is involved, and emphasizes that auxological evaluation is imperative in all NF1 children since clinical signs of endocrine disorders might lead to a diagnosis of OPG (7,33). We also found that patients with hypothalamic involvement received more treatment than patients without: surgery in 7 and chemotherapy in 11 cases. This might be because hypothalamic and chiasmatic OPGs often require surgical debulking (34) although the role of surgery in OPGs involving the hypothalamus is still debated.
In particular, in our cohort, surgery was offered to 6 patients who had involvement of the hypothalamic region and whose OPGs were extended to all pathways (Dodge 3); this was also in order to achieve control of tumor growth prior to chemotherapy in 5 individuals, and to treat DS in 1 child, in line with literature recommendations (35). Moreover, 3 patients also needed a consensual decompression of hydrocephalus secondary to hypothalamic involvement.
Another hypothesis, which should be confirmed in future, is that tumors involving the chiasmatic region (MDC 2) might be associated with worse OPG outcomes in terms of wider tumor extension and secondary visual deterioration. It is worth pointing out that age less than 5 years at OPG diagnosis represented a further predictor for endocrine dysfunction univariate analysis. This might be linked to the worse prognosis of OPGs in very young children as age at presentation of OPG has been associated with an increased risk of clinical progression (34).
In conclusion, endocrine disorders are common in patients with NF1 and OPGs before any treatment and might be a sign of suspicion of an OPG.
Endocrine symptoms in NF1 patients should lead to MRI execution for early detection of OPG with likely hypothalamic involvement. It has also been suggested that chiasmatic and postchiasmatic OPG in children with NF1 have the highest risk for progression and vision loss and that early identification of this type of OPG may lead to improved visual outcomes (36). However, it is still unclear if early detection of OPG in patients with NF1 might improve patients’ prognosis as there is a lack of conclusive prospective studies (37).
The high prevalence of CPP could represent a hallmark of OPG in children with NF1 while GHD does not appear to be as common as it has been previously reported. DS and GHH, although rare, deserve careful attention as they can occur in young patients with NF1 and OPGs.
All endocrine disorders, except DS for which specific tumor treatment is required, respond to specific medical blocking or replacement treatment. In this context early diagnosis prevents the development of related complications such as, for example, short stature.
Because hypothalamic involvement by the tumor was the most important predictor of endocrine disorders, physicians should be aware that patients with OPGs encompassing the hypothalamic region need a more careful auxological follow-up.
Abbreviations
- ACTH
adrenocorticotropin
- BMI
body mass index
- CPP
central precocious puberty
- DS
diencephalic syndrome
- GH
growth hormone
- GHD
growth hormone deficiency
- GHH
growth hormone hypersecretion
- GnRH
gonadotropin-releasing hormone
- IGF
insulin-like growth factor
- LH
luteinizing hormone
- MDC
modified Dodge classification
- NF1
neurofibromatosis type 1
- OPG
optic pathway glioma
- SD
standard deviation
Acknowledgments
M.M., N.D. and A.G. belong to the DINOGMI (Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health) University of Genova, Department of Excellence of MIUR 2018-2022 (legge 232 del 2016).
Financial Support: Nothing to declare.
Additional Information
Disclosure Summary: nothing to declare.
Data Availability: The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References

{kind=link}