





Abstract
Genetic factors are major determinants of thyroid function. Over the last two decades, multiple genetic variants have been associated with variations in normal range thyroid function tests. Most recently, a large-scale genome-wide association study (GWAS) doubled the number of known variants associated with normal range thyrotropin (TSH) and free thyroxine (FT4) levels.
This review summarizes the results of genetic association studies on normal range thyroid function and explores how these genetic variants can be used in future studies to improve our understanding of thyroid hormone regulation and disease.
Serum TSH and FT4 levels are determined by multiple genetic variants on virtually all levels of the hypothalamus-pituitary-thyroid (HPT) axis. Functional follow-up studies on top of GWAS hits has the potential to discover new key players in thyroid hormone regulation, as exemplified by the identification of the thyroid hormone transporter SLC17A4 and the metabolizing enzyme AADAT. Translational studies may use these genetic variants to investigate causal associations between thyroid function and various outcomes in Mendelian Randomization (MR) studies, to identify individuals with an increased risk of thyroid dysfunction, and to predict the individual HPT axis setpoint.
Recent genetic studies have greatly improved our understanding of the genetic basis of thyroid function, and have revealed novel pathways involved in its regulation. In addition, these findings have paved the way for various lines of research that can improve our understanding of thyroid hormone regulation and thyroid diseases, as well as the potential use of these markers in future clinical practice.
Thyroid diseases are common and have a negative impact on multiple health outcomes (1). Large observational and population-based studies showed that even variation in thyroid function within the reference range is associated with adverse clinical outcomes, such as atherosclerotic cardiovascular disease (2), stroke (3), atrial fibrillation (4), type 2 diabetes (5), metabolic syndrome (6), fracture risk (7), dementia (8), depression (9), increased risk of thyroid cancer in patients with thyroid nodules (10–12), and mortality (13). Therefore, it is essential to understand the mechanisms underlying these variations in thyroid function. In healthy persons, serum TSH and thyroid hormone (TH) levels show substantial interindividual variation, while the intraindividual variation lies within a much narrower range (14). The observed interindividual variation can be partially attributed to differences in individual characteristics (eg, age and BMI) and exposure to environmental factors (eg, iodine intake) (15). However, studies in mono- and dizygotic twins demonstrated that genetic factors are major determinants of thyroid function, responsible for 45–65% of the interindividual variation in TSH and TH levels (16). It has been suggested that besides rare variants with large effects (mutations), the overall effect of the genetic factors on an individual’s TSH and TH levels is determined by multiple common variants (polymorphisms) with small effect sizes (17, 18). Over the last two decades, several genetic variants have been associated with thyroid function in candidate gene studies, while more recently, many more variants were identified in genome-wide association studies (GWAS) (17, 18). The largest GWAS on thyroid function to date has been recently performed within the ThyroidOmics Consortium in 72 000 individuals and greatly increased the number of associated genetic variants (19). In the current review, we will discuss the knowns and unknowns of the genetic factors involved in the regulation of thyroid function, with a special emphasis on the most recently identified variants. Moreover, we will discuss several approaches in which these novel data can be used to improve our understanding of thyroid physiology and disease, as well as the potential use of these markers in future clinical practice.
Genetic Determinants of Thyroid Function
Early work on genetic determinants of thyroid function often involved candidate gene studies that analyzed a limited number of genetic variants located within specific genes selected based on their known function. Later on, GWAS allowed researchers to investigate the association of approximately 1 000 000 independent genetic variants across the entire genome with the trait of interest, using a hypothesis-free approach (20). To avoid false-positive results due to multiple testing, an adjusted P-value threshold (P < 5 × 10-8) is used to declare statistical significance in GWAS (21). Large sample sizes are therefore required to ensure sufficient power. The first GWAS on TSH levels, performed in 2008 by Arnaud-Lopez et al in 4300 Sardinians, identified one genome-wide significant locus (ie, Phosphodiesterase 8B[PDE8B]) (22). This list was further extended by several larger GWAS performed by Gudmundsson et al (23), Porcu et al (24), and others (25–31). As illustrated in Figure 1, the increasing number of subjects included in every next GWAS led to a steep increase in newly identified variants. The most recent GWAS performed by the ThyroidOmics Consortium (www.thyroidomics.com) identified 19 novel loci associated with normal range TSH levels and 16 novel loci associated with normal range free thyroxine (FT4) levels, leading to a total of 42 and 21 known and novel associated loci for these two traits (Figures 1 and 2) (19), which we briefly discuss in this section. As variants associated with hypothyroidism and hyperthyroidism are not in the scope of this review, we refer to previous comprehensive reviews (17, 32, 33).
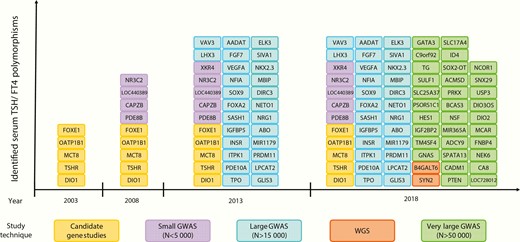
Identified serum TSH and/or FT4 associated loci over time, using different study techniques. Variants found to be significant in candidate gene studies were included when associations were replicated in at least one independent population (N > 500) or in case of in vitro evidence for functionality. Abbreviations: GWAS, genome-wide association study; N, number of subjects analyzed in the study; WGS, whole-genome sequencing.
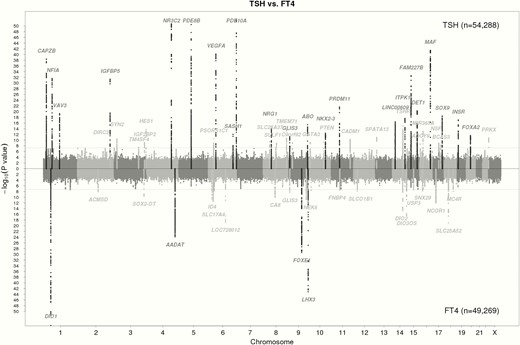
Manhattan plots of the GWAS meta-analysis results for TSH and FT4 contrasted with each other. Single nucleotide polymorphisms (SNPs) are plotted on the x-axis according to their position on each chromosome with -log10 (p-value) on the y-axis. The upper solid horizontal line indicates the threshold for genome-wide significance, ie, 5x10-8. Genomic loci previously known to contain trait-associated variants are colored in black, new loci in gray. Adapted from Teumer et al (19).
Established players
The results of genetic association studies demonstrate that TSH and TH levels are determined by variants in genes involved in the regulation of thyroid function on virtually all levels of the hypothalamus-pituitary-thyroid (HPT) axis and peripheral thyroid hormone regulation (17, 18). Based on their function, these genes can be classified into several groups, which often tend to predominantly determine either TSH or TH levels, as presented in Table 1. We highlight typical examples for each of these groups below.
Genetic loci associated with variation in normal range TSH and FT4 levels classified by their function in the hypothalamus–pituitary–thyroid axis and peripheral thyroid hormone regulation.
| Locus | Annotated gene | Gene full name | Function in the HPT axis | Associated trait | Direction (when affecting both TSH and FT4) | References |
|---|---|---|---|---|---|---|
| Genes encoding proteins implicated in development and function of the HPT axis | ||||||
| Transcription factors | ||||||
| 9q22 | FOXE1 | Forkhead box E1 | Main mediator of TSH-regulated expression of thyroid-specific genes | FT4/TSH | Same | (19, 23, 24, 28, 30, 34) |
| 9q34 | LHX3 | LIM homeobox 3 | Transcription factor required for pituitary development | FT4/(TSH) | Same | (19, 24, 34) |
| 9p24 | GLIS3 | GLIS family zinc finger 3 | Transcription factor implicated in thyroid development | TSH/FT4 | Opposite | (19, 24) |
| 1p31 | NFIA | Nuclear factor I A | Interacts with thyroid transcription factor 1, which is essential for the expression of thyroid-specific genes | TSH/(FT4) | Opposite | (19, 23, 24) |
| 14q13 | TTF1/MBIP | NK2 homeobox 1 (thyroid transcription factor 1)/ MAP3K12 binding inhibitory protein 1 | Transcription factor crucial for thyroglobulin expression and thyroid differentiation (TTF1) | TSH/(FT4) | Opposite | (19, 24, 34) |
| 17q23 | SOX9 | SRY-box 9 | Transcription factor expressed both in the pituitary and thyroid, which interacts with the TH receptor complex | TSH | NA | (19, 24) |
| 17p11-12 | NCOR1 | Nuclear receptor corepressor 1 | Corepressor involved in mediating TH action | FT4 | NA | (19) |
| Growth factors and their binding proteins | ||||||
| 6p12 | VEGFA | Vascular endothelial growth factor A | Growth factor implicated in angiogenesis and iodine supply to the thyroid | TSH/(FT4) | Opposite | (19, 23, 24, 28, 34) |
| 15q21 | FGF7 | Fibroblast growth factor 7 | Plays a role in the development of the thyroid | TSH/(FT4) | Opposite | (19, 23, 24) |
| 2q35 | IGFBP2/ IGFBP5 | Insulin like growth factor binding protein 2 / Insulin like growth factor binding protein 5 | Enhanced production of IGFBP5 is correlated with inhibition of thyroid function | TSH/(FT4) | Opposite | (19, 23, 24, 34) |
| 3q27 | IGF2BP2 | Insulin like growth factor 2 mRNA binding protein 2 | Regulates translation of IGF2, which may have proliferative effects on follicular thyroid cells | TSH | NA | (19) |
| 19p13 | INSR | Insulin receptor | Binds insulin, IGF1 and IGF2, which have proliferative effects on follicular thyroid cells | TSH | NA | (19, 23, 24) |
| 6q24 | SASH1 | SAM and SH3 domain containing 1 | Downstream target of the insulin/IGF1 signaling pathway | TSH | NA | (19, 24) |
| 20p11 | FOXA2 | Forkhead box A2 | Downstream target of the insulin/IGF1 signaling pathway | TSH | NA | (19, 23) |
| Genes encoding proteins involved in the TSH receptor signaling cascade | ||||||
| 14q31 | TSHR | Thyroid stimulating hormone receptor | Stimulates TH synthesis and release in response to TSH | TSH | NA | (19, 35, 36) |
| 5q13 | PDE8B | Phosphodiesterase 8B | Inactivates cAMP signaling activated by TSHR response to TSH | TSH/(FT4) | Opposite | (19, 22-24, 26, 27, 31, 34) |
| 6q27 | PDE10A | Phosphodiesterase 10A | Inactivates cAMP signaling activated by TSHR response to TSH | TSH | NA | (19, 23, 24, 28, 34) |
| 14q32 | ITPK1 | Inositol-tetrakisphosphate 1-kinase | Regulates the phosphatidyl inositol pathway activated by TSHR response to TSH | TSH/(FT4) | Opposite | (19, 23, 24) |
| 20q13 | GNAS | GNAS complex locus | Stimulates the activity of adenylate cyclase which directly regulates cAMP signaling | TSH | NA | (37) |
| Genes encoding proteins directly involved in thyroid hormone synthesis | ||||||
| 2p25 | TPO | Thyroid peroxidase | Catalyzes the iodination of tyrosine residues and their subsequent coupling to iodothyronines | TSH | NA | (23) |
| 8q24 | TG | Thyroglobulin | Acts as a substrate for the synthesis of T4 and T3 | TSH | NA | (19) |
| 1p36 | CAPZB | Capping actin protein of muscle Z-line subunit beta | Takes part in TSH-induced protrusion of microvilli and filopodia from the thyrocyte surface | TSH | NA | (19, 23, 24, 26, 30, 34) |
| Genes encoding thyroid hormone transporters | ||||||
| Xq13 | MCT8 (SLC16A2) | Monocarboxylate transporter 8 (Solute carrier family 16 member 2) | TH transporter | FT4 | NA | (38) |
| 12p12 | OATP1B1 (SLCO1B1) | Solute carrier organic anion transporter family member 1B1 | TH transporter | FT4 | NA | (19, 39) |
| 6p22 | SLC17A4 | Solute carrier family 17 member 4 | TH transporter | FT4 | NA | (19) |
| Genes encoding enzymes involved in thyroid hormone metabolism | ||||||
| 1p32 | DIO1 | Iodothyronine deiodinase 1 | Responsible for TH metabolism, mainly the conversion of T4 to T3 and rT3 to T2 | FT4 | NA | (19, 24, 34, 36, 40, 41) |
| 14q31 | DIO2 | Iodothyronine deiodinase 2 | Responsible for conversion of T4 to T3 and rT3 to T2 | FT4 | NA | (19) |
| 14q32 | DIO3OS | DIO3 opposite strand upstream RNA | Potential regulator of DIO3 expression | FT4 | NA | (19) |
| 4q33 | AADAT | Aminoadipate aminotransferase | TH metabolizing enzyme | FT4 | NA | (19, 24, 34) |
| Genes with unknown function in the HPT axis | ||||||
| NR3C2, XKR4, ELK3, SIVA1, NKX2.3, DIRC3, VAV3, NRG1, ABO, MIR1179, PRDM11, LPCAT2, NETO1, SYN2, B4GALT6 (SLC25A52), HES1, PSORS1C1, SLC25A37, SULF1, C9orf92, GATA3, PTEN, CADM1, SPATA13, ADCY9, MIR365A, NSF, BCAS3, PRKX, ACMSD, SOX2-OT, ID4, LOC728012, CA8, NEK6, FNBP4, USP3, SNX29, MC4R, TM4SF4, LOC440389 (MAF), CNTN5 | (19, 23, 24, 26, 28, 30, 34) |
| Locus | Annotated gene | Gene full name | Function in the HPT axis | Associated trait | Direction (when affecting both TSH and FT4) | References |
|---|---|---|---|---|---|---|
| Genes encoding proteins implicated in development and function of the HPT axis | ||||||
| Transcription factors | ||||||
| 9q22 | FOXE1 | Forkhead box E1 | Main mediator of TSH-regulated expression of thyroid-specific genes | FT4/TSH | Same | (19, 23, 24, 28, 30, 34) |
| 9q34 | LHX3 | LIM homeobox 3 | Transcription factor required for pituitary development | FT4/(TSH) | Same | (19, 24, 34) |
| 9p24 | GLIS3 | GLIS family zinc finger 3 | Transcription factor implicated in thyroid development | TSH/FT4 | Opposite | (19, 24) |
| 1p31 | NFIA | Nuclear factor I A | Interacts with thyroid transcription factor 1, which is essential for the expression of thyroid-specific genes | TSH/(FT4) | Opposite | (19, 23, 24) |
| 14q13 | TTF1/MBIP | NK2 homeobox 1 (thyroid transcription factor 1)/ MAP3K12 binding inhibitory protein 1 | Transcription factor crucial for thyroglobulin expression and thyroid differentiation (TTF1) | TSH/(FT4) | Opposite | (19, 24, 34) |
| 17q23 | SOX9 | SRY-box 9 | Transcription factor expressed both in the pituitary and thyroid, which interacts with the TH receptor complex | TSH | NA | (19, 24) |
| 17p11-12 | NCOR1 | Nuclear receptor corepressor 1 | Corepressor involved in mediating TH action | FT4 | NA | (19) |
| Growth factors and their binding proteins | ||||||
| 6p12 | VEGFA | Vascular endothelial growth factor A | Growth factor implicated in angiogenesis and iodine supply to the thyroid | TSH/(FT4) | Opposite | (19, 23, 24, 28, 34) |
| 15q21 | FGF7 | Fibroblast growth factor 7 | Plays a role in the development of the thyroid | TSH/(FT4) | Opposite | (19, 23, 24) |
| 2q35 | IGFBP2/ IGFBP5 | Insulin like growth factor binding protein 2 / Insulin like growth factor binding protein 5 | Enhanced production of IGFBP5 is correlated with inhibition of thyroid function | TSH/(FT4) | Opposite | (19, 23, 24, 34) |
| 3q27 | IGF2BP2 | Insulin like growth factor 2 mRNA binding protein 2 | Regulates translation of IGF2, which may have proliferative effects on follicular thyroid cells | TSH | NA | (19) |
| 19p13 | INSR | Insulin receptor | Binds insulin, IGF1 and IGF2, which have proliferative effects on follicular thyroid cells | TSH | NA | (19, 23, 24) |
| 6q24 | SASH1 | SAM and SH3 domain containing 1 | Downstream target of the insulin/IGF1 signaling pathway | TSH | NA | (19, 24) |
| 20p11 | FOXA2 | Forkhead box A2 | Downstream target of the insulin/IGF1 signaling pathway | TSH | NA | (19, 23) |
| Genes encoding proteins involved in the TSH receptor signaling cascade | ||||||
| 14q31 | TSHR | Thyroid stimulating hormone receptor | Stimulates TH synthesis and release in response to TSH | TSH | NA | (19, 35, 36) |
| 5q13 | PDE8B | Phosphodiesterase 8B | Inactivates cAMP signaling activated by TSHR response to TSH | TSH/(FT4) | Opposite | (19, 22-24, 26, 27, 31, 34) |
| 6q27 | PDE10A | Phosphodiesterase 10A | Inactivates cAMP signaling activated by TSHR response to TSH | TSH | NA | (19, 23, 24, 28, 34) |
| 14q32 | ITPK1 | Inositol-tetrakisphosphate 1-kinase | Regulates the phosphatidyl inositol pathway activated by TSHR response to TSH | TSH/(FT4) | Opposite | (19, 23, 24) |
| 20q13 | GNAS | GNAS complex locus | Stimulates the activity of adenylate cyclase which directly regulates cAMP signaling | TSH | NA | (37) |
| Genes encoding proteins directly involved in thyroid hormone synthesis | ||||||
| 2p25 | TPO | Thyroid peroxidase | Catalyzes the iodination of tyrosine residues and their subsequent coupling to iodothyronines | TSH | NA | (23) |
| 8q24 | TG | Thyroglobulin | Acts as a substrate for the synthesis of T4 and T3 | TSH | NA | (19) |
| 1p36 | CAPZB | Capping actin protein of muscle Z-line subunit beta | Takes part in TSH-induced protrusion of microvilli and filopodia from the thyrocyte surface | TSH | NA | (19, 23, 24, 26, 30, 34) |
| Genes encoding thyroid hormone transporters | ||||||
| Xq13 | MCT8 (SLC16A2) | Monocarboxylate transporter 8 (Solute carrier family 16 member 2) | TH transporter | FT4 | NA | (38) |
| 12p12 | OATP1B1 (SLCO1B1) | Solute carrier organic anion transporter family member 1B1 | TH transporter | FT4 | NA | (19, 39) |
| 6p22 | SLC17A4 | Solute carrier family 17 member 4 | TH transporter | FT4 | NA | (19) |
| Genes encoding enzymes involved in thyroid hormone metabolism | ||||||
| 1p32 | DIO1 | Iodothyronine deiodinase 1 | Responsible for TH metabolism, mainly the conversion of T4 to T3 and rT3 to T2 | FT4 | NA | (19, 24, 34, 36, 40, 41) |
| 14q31 | DIO2 | Iodothyronine deiodinase 2 | Responsible for conversion of T4 to T3 and rT3 to T2 | FT4 | NA | (19) |
| 14q32 | DIO3OS | DIO3 opposite strand upstream RNA | Potential regulator of DIO3 expression | FT4 | NA | (19) |
| 4q33 | AADAT | Aminoadipate aminotransferase | TH metabolizing enzyme | FT4 | NA | (19, 24, 34) |
| Genes with unknown function in the HPT axis | ||||||
| NR3C2, XKR4, ELK3, SIVA1, NKX2.3, DIRC3, VAV3, NRG1, ABO, MIR1179, PRDM11, LPCAT2, NETO1, SYN2, B4GALT6 (SLC25A52), HES1, PSORS1C1, SLC25A37, SULF1, C9orf92, GATA3, PTEN, CADM1, SPATA13, ADCY9, MIR365A, NSF, BCAS3, PRKX, ACMSD, SOX2-OT, ID4, LOC728012, CA8, NEK6, FNBP4, USP3, SNX29, MC4R, TM4SF4, LOC440389 (MAF), CNTN5 | (19, 23, 24, 26, 28, 30, 34) |
Loci associated in candidate gene studies were included when associations were replicated in at least one independent population (N > 500) or in case of in vitro evidence for functionality. Effects on TSH and/or FT4 levels are shown between brackets when they were only significant in secondary analyses (and not in the main GWAS analysis).
Abbreviations: TH, thyroid hormone; NA, not applicable.
Genetic loci associated with variation in normal range TSH and FT4 levels classified by their function in the hypothalamus–pituitary–thyroid axis and peripheral thyroid hormone regulation.
| Locus | Annotated gene | Gene full name | Function in the HPT axis | Associated trait | Direction (when affecting both TSH and FT4) | References |
|---|---|---|---|---|---|---|
| Genes encoding proteins implicated in development and function of the HPT axis | ||||||
| Transcription factors | ||||||
| 9q22 | FOXE1 | Forkhead box E1 | Main mediator of TSH-regulated expression of thyroid-specific genes | FT4/TSH | Same | (19, 23, 24, 28, 30, 34) |
| 9q34 | LHX3 | LIM homeobox 3 | Transcription factor required for pituitary development | FT4/(TSH) | Same | (19, 24, 34) |
| 9p24 | GLIS3 | GLIS family zinc finger 3 | Transcription factor implicated in thyroid development | TSH/FT4 | Opposite | (19, 24) |
| 1p31 | NFIA | Nuclear factor I A | Interacts with thyroid transcription factor 1, which is essential for the expression of thyroid-specific genes | TSH/(FT4) | Opposite | (19, 23, 24) |
| 14q13 | TTF1/MBIP | NK2 homeobox 1 (thyroid transcription factor 1)/ MAP3K12 binding inhibitory protein 1 | Transcription factor crucial for thyroglobulin expression and thyroid differentiation (TTF1) | TSH/(FT4) | Opposite | (19, 24, 34) |
| 17q23 | SOX9 | SRY-box 9 | Transcription factor expressed both in the pituitary and thyroid, which interacts with the TH receptor complex | TSH | NA | (19, 24) |
| 17p11-12 | NCOR1 | Nuclear receptor corepressor 1 | Corepressor involved in mediating TH action | FT4 | NA | (19) |
| Growth factors and their binding proteins | ||||||
| 6p12 | VEGFA | Vascular endothelial growth factor A | Growth factor implicated in angiogenesis and iodine supply to the thyroid | TSH/(FT4) | Opposite | (19, 23, 24, 28, 34) |
| 15q21 | FGF7 | Fibroblast growth factor 7 | Plays a role in the development of the thyroid | TSH/(FT4) | Opposite | (19, 23, 24) |
| 2q35 | IGFBP2/ IGFBP5 | Insulin like growth factor binding protein 2 / Insulin like growth factor binding protein 5 | Enhanced production of IGFBP5 is correlated with inhibition of thyroid function | TSH/(FT4) | Opposite | (19, 23, 24, 34) |
| 3q27 | IGF2BP2 | Insulin like growth factor 2 mRNA binding protein 2 | Regulates translation of IGF2, which may have proliferative effects on follicular thyroid cells | TSH | NA | (19) |
| 19p13 | INSR | Insulin receptor | Binds insulin, IGF1 and IGF2, which have proliferative effects on follicular thyroid cells | TSH | NA | (19, 23, 24) |
| 6q24 | SASH1 | SAM and SH3 domain containing 1 | Downstream target of the insulin/IGF1 signaling pathway | TSH | NA | (19, 24) |
| 20p11 | FOXA2 | Forkhead box A2 | Downstream target of the insulin/IGF1 signaling pathway | TSH | NA | (19, 23) |
| Genes encoding proteins involved in the TSH receptor signaling cascade | ||||||
| 14q31 | TSHR | Thyroid stimulating hormone receptor | Stimulates TH synthesis and release in response to TSH | TSH | NA | (19, 35, 36) |
| 5q13 | PDE8B | Phosphodiesterase 8B | Inactivates cAMP signaling activated by TSHR response to TSH | TSH/(FT4) | Opposite | (19, 22-24, 26, 27, 31, 34) |
| 6q27 | PDE10A | Phosphodiesterase 10A | Inactivates cAMP signaling activated by TSHR response to TSH | TSH | NA | (19, 23, 24, 28, 34) |
| 14q32 | ITPK1 | Inositol-tetrakisphosphate 1-kinase | Regulates the phosphatidyl inositol pathway activated by TSHR response to TSH | TSH/(FT4) | Opposite | (19, 23, 24) |
| 20q13 | GNAS | GNAS complex locus | Stimulates the activity of adenylate cyclase which directly regulates cAMP signaling | TSH | NA | (37) |
| Genes encoding proteins directly involved in thyroid hormone synthesis | ||||||
| 2p25 | TPO | Thyroid peroxidase | Catalyzes the iodination of tyrosine residues and their subsequent coupling to iodothyronines | TSH | NA | (23) |
| 8q24 | TG | Thyroglobulin | Acts as a substrate for the synthesis of T4 and T3 | TSH | NA | (19) |
| 1p36 | CAPZB | Capping actin protein of muscle Z-line subunit beta | Takes part in TSH-induced protrusion of microvilli and filopodia from the thyrocyte surface | TSH | NA | (19, 23, 24, 26, 30, 34) |
| Genes encoding thyroid hormone transporters | ||||||
| Xq13 | MCT8 (SLC16A2) | Monocarboxylate transporter 8 (Solute carrier family 16 member 2) | TH transporter | FT4 | NA | (38) |
| 12p12 | OATP1B1 (SLCO1B1) | Solute carrier organic anion transporter family member 1B1 | TH transporter | FT4 | NA | (19, 39) |
| 6p22 | SLC17A4 | Solute carrier family 17 member 4 | TH transporter | FT4 | NA | (19) |
| Genes encoding enzymes involved in thyroid hormone metabolism | ||||||
| 1p32 | DIO1 | Iodothyronine deiodinase 1 | Responsible for TH metabolism, mainly the conversion of T4 to T3 and rT3 to T2 | FT4 | NA | (19, 24, 34, 36, 40, 41) |
| 14q31 | DIO2 | Iodothyronine deiodinase 2 | Responsible for conversion of T4 to T3 and rT3 to T2 | FT4 | NA | (19) |
| 14q32 | DIO3OS | DIO3 opposite strand upstream RNA | Potential regulator of DIO3 expression | FT4 | NA | (19) |
| 4q33 | AADAT | Aminoadipate aminotransferase | TH metabolizing enzyme | FT4 | NA | (19, 24, 34) |
| Genes with unknown function in the HPT axis | ||||||
| NR3C2, XKR4, ELK3, SIVA1, NKX2.3, DIRC3, VAV3, NRG1, ABO, MIR1179, PRDM11, LPCAT2, NETO1, SYN2, B4GALT6 (SLC25A52), HES1, PSORS1C1, SLC25A37, SULF1, C9orf92, GATA3, PTEN, CADM1, SPATA13, ADCY9, MIR365A, NSF, BCAS3, PRKX, ACMSD, SOX2-OT, ID4, LOC728012, CA8, NEK6, FNBP4, USP3, SNX29, MC4R, TM4SF4, LOC440389 (MAF), CNTN5 | (19, 23, 24, 26, 28, 30, 34) |
| Locus | Annotated gene | Gene full name | Function in the HPT axis | Associated trait | Direction (when affecting both TSH and FT4) | References |
|---|---|---|---|---|---|---|
| Genes encoding proteins implicated in development and function of the HPT axis | ||||||
| Transcription factors | ||||||
| 9q22 | FOXE1 | Forkhead box E1 | Main mediator of TSH-regulated expression of thyroid-specific genes | FT4/TSH | Same | (19, 23, 24, 28, 30, 34) |
| 9q34 | LHX3 | LIM homeobox 3 | Transcription factor required for pituitary development | FT4/(TSH) | Same | (19, 24, 34) |
| 9p24 | GLIS3 | GLIS family zinc finger 3 | Transcription factor implicated in thyroid development | TSH/FT4 | Opposite | (19, 24) |
| 1p31 | NFIA | Nuclear factor I A | Interacts with thyroid transcription factor 1, which is essential for the expression of thyroid-specific genes | TSH/(FT4) | Opposite | (19, 23, 24) |
| 14q13 | TTF1/MBIP | NK2 homeobox 1 (thyroid transcription factor 1)/ MAP3K12 binding inhibitory protein 1 | Transcription factor crucial for thyroglobulin expression and thyroid differentiation (TTF1) | TSH/(FT4) | Opposite | (19, 24, 34) |
| 17q23 | SOX9 | SRY-box 9 | Transcription factor expressed both in the pituitary and thyroid, which interacts with the TH receptor complex | TSH | NA | (19, 24) |
| 17p11-12 | NCOR1 | Nuclear receptor corepressor 1 | Corepressor involved in mediating TH action | FT4 | NA | (19) |
| Growth factors and their binding proteins | ||||||
| 6p12 | VEGFA | Vascular endothelial growth factor A | Growth factor implicated in angiogenesis and iodine supply to the thyroid | TSH/(FT4) | Opposite | (19, 23, 24, 28, 34) |
| 15q21 | FGF7 | Fibroblast growth factor 7 | Plays a role in the development of the thyroid | TSH/(FT4) | Opposite | (19, 23, 24) |
| 2q35 | IGFBP2/ IGFBP5 | Insulin like growth factor binding protein 2 / Insulin like growth factor binding protein 5 | Enhanced production of IGFBP5 is correlated with inhibition of thyroid function | TSH/(FT4) | Opposite | (19, 23, 24, 34) |
| 3q27 | IGF2BP2 | Insulin like growth factor 2 mRNA binding protein 2 | Regulates translation of IGF2, which may have proliferative effects on follicular thyroid cells | TSH | NA | (19) |
| 19p13 | INSR | Insulin receptor | Binds insulin, IGF1 and IGF2, which have proliferative effects on follicular thyroid cells | TSH | NA | (19, 23, 24) |
| 6q24 | SASH1 | SAM and SH3 domain containing 1 | Downstream target of the insulin/IGF1 signaling pathway | TSH | NA | (19, 24) |
| 20p11 | FOXA2 | Forkhead box A2 | Downstream target of the insulin/IGF1 signaling pathway | TSH | NA | (19, 23) |
| Genes encoding proteins involved in the TSH receptor signaling cascade | ||||||
| 14q31 | TSHR | Thyroid stimulating hormone receptor | Stimulates TH synthesis and release in response to TSH | TSH | NA | (19, 35, 36) |
| 5q13 | PDE8B | Phosphodiesterase 8B | Inactivates cAMP signaling activated by TSHR response to TSH | TSH/(FT4) | Opposite | (19, 22-24, 26, 27, 31, 34) |
| 6q27 | PDE10A | Phosphodiesterase 10A | Inactivates cAMP signaling activated by TSHR response to TSH | TSH | NA | (19, 23, 24, 28, 34) |
| 14q32 | ITPK1 | Inositol-tetrakisphosphate 1-kinase | Regulates the phosphatidyl inositol pathway activated by TSHR response to TSH | TSH/(FT4) | Opposite | (19, 23, 24) |
| 20q13 | GNAS | GNAS complex locus | Stimulates the activity of adenylate cyclase which directly regulates cAMP signaling | TSH | NA | (37) |
| Genes encoding proteins directly involved in thyroid hormone synthesis | ||||||
| 2p25 | TPO | Thyroid peroxidase | Catalyzes the iodination of tyrosine residues and their subsequent coupling to iodothyronines | TSH | NA | (23) |
| 8q24 | TG | Thyroglobulin | Acts as a substrate for the synthesis of T4 and T3 | TSH | NA | (19) |
| 1p36 | CAPZB | Capping actin protein of muscle Z-line subunit beta | Takes part in TSH-induced protrusion of microvilli and filopodia from the thyrocyte surface | TSH | NA | (19, 23, 24, 26, 30, 34) |
| Genes encoding thyroid hormone transporters | ||||||
| Xq13 | MCT8 (SLC16A2) | Monocarboxylate transporter 8 (Solute carrier family 16 member 2) | TH transporter | FT4 | NA | (38) |
| 12p12 | OATP1B1 (SLCO1B1) | Solute carrier organic anion transporter family member 1B1 | TH transporter | FT4 | NA | (19, 39) |
| 6p22 | SLC17A4 | Solute carrier family 17 member 4 | TH transporter | FT4 | NA | (19) |
| Genes encoding enzymes involved in thyroid hormone metabolism | ||||||
| 1p32 | DIO1 | Iodothyronine deiodinase 1 | Responsible for TH metabolism, mainly the conversion of T4 to T3 and rT3 to T2 | FT4 | NA | (19, 24, 34, 36, 40, 41) |
| 14q31 | DIO2 | Iodothyronine deiodinase 2 | Responsible for conversion of T4 to T3 and rT3 to T2 | FT4 | NA | (19) |
| 14q32 | DIO3OS | DIO3 opposite strand upstream RNA | Potential regulator of DIO3 expression | FT4 | NA | (19) |
| 4q33 | AADAT | Aminoadipate aminotransferase | TH metabolizing enzyme | FT4 | NA | (19, 24, 34) |
| Genes with unknown function in the HPT axis | ||||||
| NR3C2, XKR4, ELK3, SIVA1, NKX2.3, DIRC3, VAV3, NRG1, ABO, MIR1179, PRDM11, LPCAT2, NETO1, SYN2, B4GALT6 (SLC25A52), HES1, PSORS1C1, SLC25A37, SULF1, C9orf92, GATA3, PTEN, CADM1, SPATA13, ADCY9, MIR365A, NSF, BCAS3, PRKX, ACMSD, SOX2-OT, ID4, LOC728012, CA8, NEK6, FNBP4, USP3, SNX29, MC4R, TM4SF4, LOC440389 (MAF), CNTN5 | (19, 23, 24, 26, 28, 30, 34) |
Loci associated in candidate gene studies were included when associations were replicated in at least one independent population (N > 500) or in case of in vitro evidence for functionality. Effects on TSH and/or FT4 levels are shown between brackets when they were only significant in secondary analyses (and not in the main GWAS analysis).
Abbreviations: TH, thyroid hormone; NA, not applicable.
Genes encoding proteins implicated in development and function of the HPT axis.
Several variants associated with TSH and/or TH levels localize within genes encoding proteins implicated in the development and function of the HPT axis. These include important transcription factors as well as growth factors and their binding proteins (Table 1).
Transcription factors.
The first group includes variants in genes encoding transcription factors expressed in the HPT axis, which are often associated with both TSH and TH levels (Table 1). The associated biochemical fingerprint depends on whether these factors primarily contribute to the development and function of the thyroid or pituitary. For example, LIM homeobox 3 (LHX3) encodes a transcription factor involved in pituitary development. LHX3 variants have been predominantly associated with lower FT4 levels, with either no effects on TSH levels (16) or lower TSH levels (21), which is indeed the hallmark of a central hypothyroidism-like effect. This is supported by the observation that inactivating mutations of the LHX3 gene can lead to central congenital hypothyroidism accompanied by growth hormone and gonadotropin deficiency (42).
On the other hand, variants of genes encoding transcription factors primarily involved in thyroid development and function tend to be associated with FT4 and TSH levels in the opposite direction, as illustrated by variants in the GLIS family zinc finger 3 (GLIS3) gene. GLIS3 encodes a zinc finger transcription factor, acting as both repressor and activator of gene transcription in the thyroid (43). Animal models and in vitro studies showed that GLIS3 is essential for proliferation of thyroid follicular cells and synthesis of TH, as it is involved in transcription of the iodide transporters, sodium iodide symporter (NIS/SLC5A5) and pendrin (PDS/SLC26A4) (44). Mutations in the GLIS3 gene have been associated with primary congenital hypothyroidism, accompanied by neonatal diabetes (45). Of note, GLIS3 was the only locus associated with both TSH and FT4 levels at the genome-wide significant level in the most recent GWAS (19), and the same GLIS3-rs10814915 allele was associated with higher TSH and lower FT4 levels.
Growth factors and their binding proteins.
Variants in genes encoding growth factors expressed in the thyroid are also predominantly associated with TSH levels and, to a lesser extent, with FT4 levels in the opposite direction (Table 1). Such biochemical fingerprints were indeed noted for variants in VEGFA (encoding vascular endothelial growth factor A) and FGF7 (encoding fibroblast growth factor 7). VEGFA promotes angiogenesis and proper microvasculature development, which is essential for iodine and TSH supply to the thyrocytes (46,47). In turn, FGF7 plays an important role in thyroid development and promotes thyrocyte proliferation (48). Of note, FGF7 was also one of the significant hits in the goiter GWAS by Teumer et al (49). Interestingly, the same FGF7-rs4338740 allele was associated with both increased thyroid volume in the study by Teumer et al (49) and lower TSH levels in several GWAS (19, 23, 24). These findings are in line with the results of observational studies showing that higher thyroid volume is associated with lower TSH levels in euthyroid adults (50–52) and children (53, 54). These results suggest that variants associated with increased FGF7 activity result in increased thyroid cell mass and volume, and it appears that for these thyroids lower TSH levels are required to maintain normal FT4 levels.
Genes encoding proteins involved in the TSH receptor signaling cascade.
Another important group of genetic determinants of thyroid function includes variants in genes encoding the TSH receptor (TSHR) and other proteins involved in the cascade, which follows after TSH is bound to the TSHR. As can be physiologically expected, such variants primarily affect TSH levels (Table 1), with some also having minor effects on TH levels in the opposite direction. This can be illustrated by variants in the PDE8B gene. PDE8B encodes a phosphodiesterase, which is highly expressed in the thyroid and responsible for the inactivation of cyclic adenosine monophosphate (cAMP), which is produced after TSHR activation. All large GWAS have identified that several PDE8B variants are associated with serum TSH levels, while their association with FT4 levels is much weaker (19,23,24). It is speculated that the identified genetic variants increase PDE8B activity, leading to lower cAMP levels. Consequently, a higher TSH level is required to maintain normal FT4 levels.
Genes encoding TH transporters and enzymes involved in TH metabolism.
In contrast, variants in genes encoding proteins involved in TH transport and metabolism result in altered TH levels without any evident change in TSH levels (Table 1). This can be illustrated by DIO1 encoding type 1 iodothyronine deiodinase. Several independent DIO1 variants have been associated with TH levels but not TSH levels in GWAS and candidate gene studies (19, 24, 36, 40, 41). DIO1 plays an important role in the peripheral conversion of thyroxine (T4) to triiodothyronine (T3) and the degradation of reverse T3 (rT3). Therefore, variants associated with lower DIO1 activity lead to higher T4 levels and lower T3 levels, resulting in a net euthyroid status, as reflected by the absence of an association with TSH.
The examples above illustrate that the identified effects of many variants fit nicely with the gene’s role in TH regulation. In turn, the biochemical fingerprint of variants in genes with a yet unknown role in TH regulation can therefore also be used to provide clues about the role of the associated gene in the HPT axis or peripheral TH regulation. This is illustrated by the recent identification of the roles of aminoadipate aminotransferase (AADAT) and solute carrier family 17 member 4 (SLC17A4) in TH regulation, which we discuss more extensively below. The exact location of the gene in the HPT axis and their associated effects on TSH and TH levels is also important when considering the use of these variants in Mendelian Randomization and prediction studies, as discussed later in this review.
Novel players
The most recent GWAS on thyroid function doubled the number of loci associated with TSH and FT4 levels (19). This included novel variants within known thyroid-related genes or related pathways, but also a number of variants within genes without an established role in the regulation of TSH and FT4 levels (Table 1). Functional follow-up studies on two of these genes (AADAT and SLC17A4) confirmed their role in TH regulation, as discussed below.
AADAT – a novel thyroid hormone metabolizing enzyme.
GWAS have found that genetic variation in the AADAT locus is associated with variation in FT4 levels (19, 24). AADAT is highly expressed in multiple tissues, including the liver, gastrointestinal tract, and kidney (55), but until recently its role in TH regulation remained completely unknown. Given the observed associations with TH levels and the fact that AADAT encodes a mitochondrial aminotransferase with broad substrate specificity, Dr Theo Visser postulated that AADAT could encode a TH metabolizing enzyme. This was indeed confirmed by in vitro studies, in which cell lysates of human AADAT transfected cells were incubated with THs. When compared to empty vector transfected cells, this led to an increased conversion of THs (amongst others, T4 and T3) to their pyruvic acid metabolites (19). Further studies showed that the identified AADAT gene variant (rs6854291) was also associated with lower serum T3 levels and a lower T3/T4 ratio (19). These results show that AADAT is a TH metabolizing enzyme, which is likely to play a critical role in the rate of TH metabolism.
SLC17A4 – a novel thyroid hormone transporter.
The most recent GWAS on FT4 levels identified two independent variants within the SLC17A4 gene (rs9356988 and rs137964359) to be associated with FT4 levels (19). The SLC17A4 gene encodes an organic anion transporter that is particularly expressed in the liver, kidney, and gastrointestinal tract (56,57). Given its association with TH levels, it was postulated that SLC17A4 could encode a TH transporter. In vitro studies indeed confirmed that SLC17A4 is a high-affinity T3 and T4 transporter, with similar properties as monocarboxylate transporter 8 (MCT8), the most specific TH transporter identified to date (19).
DIO3OS – a potential regulator of DIO3 expression.
Type 3 deiodinase (DIO3) is responsible for the degradation of TH (T4 to rT3 and T3 to T2). In mice, DIO3 is critical for the maturation and function of the HPT axis, as mice lacking DIO3 activity develop perinatal thyrotoxicosis followed by mild central hypothyroidism in adulthood (58, 59). Whereas genetic variation in DIO3 was found not to be associated with TSH and/or TH levels in humans in candidate gene studies (36, 40, 41) or previous GWAS (22–31), two independent variants within the DIO3 opposite strand upstream RNA (DIO3OS) gene have been associated with FT4 levels in the most recent GWAS on thyroid function (19). DIO3 and DIO3OS are overlapping genes transcribed in opposite directions (60), and Kester et al demonstrated that DIO3 and DIO3OS are co-expressed in various human cell lines (61). Whereas DIO3 is an imprinted gene, DIO3OS may be involved in maintaining monoallelic expression of DIO3 (62, 63), which could explain the observed associations with TH levels. Future studies should clarify whether these variants indeed affect DIO3 expression.
Novel variants within thyroid-related genes and known pathways.
Type 2 iodothyronine deiodinase (DIO2) is involved in the conversion of T4 to T3 and rT3 to T2. The most frequently tested DIO2 variants are rs225014 (also known as p.92T > A) and rs12885300 (also known as ORFa-Gly3Asp). DIO2-rs12885300 is located in a short open reading frame within the 5’UTR of the DIO2 gene. This part of the gene is thought to be responsible for the inhibitory effect of the 5’UTR on DIO2 transcription, which is supported by in vitro analyses showing that the Asp variant is associated with increased gene transcription and DIO2 activity (64). In line with this finding, Peeters et al showed that this variant is associated with an increased T3/T4 ratio (65). However, this could not be replicated by others (66, 67), while this variant was neither associated with T4 levels in the most recent GWAS (19), suggesting that this variant does not have any substantial effects on circulating T3 and T4 levels, or their ratio.
DIO2-rs225014 is a non-synonymous variant resulting in the Thr92Ala substitution, which has been investigated in several in vitro studies (36, 68–70). Initial studies did not find effects of the Ala variant on T4 to T3 conversion in transfected human kidney (HEK) cells and fibroblast-like cells derived from monkey kidney tissue (COS cells) (36, 68). However, Canani et al did show decreased DIO2 activity in muscle and thyroid samples from patients with type 2 diabetes mellitus who were homozygous for the Ala variant (68). More recently, Castagna et al performed a series of experiments on DIO2-rs225014 functionality in mice, amongst others, demonstrating lower T4 to T3 conversion in DIO2-92Ala-transfected myoblasts and pituitary cells compared with DIO2-92Thr-transfected myoblasts and pituitary cells in DIO2-null mice (69). Collectively, these studies provide strong evidence for the functional effects of the DIO2-rs225014 variant in multiple cell lines. Despite this, DIO2-rs225014 was not associated with serum TSH and/or TH levels in several candidate gene studies (36, 67, 71, 72) and previous GWAS (24, 28, 29). However, the most recent GWAS proved that rs225014 and two other DIO2 variants (rs150816132 and rs978055, but not rs12885300) are indeed associated with normal-range FT4 levels (19), which clearly demonstrates the advantage of well-powered large-scale studies in identifying variants associated with thyroid function. Multiple studies investigated the potential clinical implications of the DIO2-rs225014 variant, which we will discuss later in this review. Of note, the recent GWAS also provided evidence for other thyroid-related genes (eg, TSHR, TG, OATP1B1) and identified several novel independent variants within the established loci (eg, PDE8B, CAPZB, DIO1) (Table 1).
Genes of unknown function in thyroid hormone regulation
Finally, a large part of the newly and previously identified loci (eg, MAF, PRDM11, DIRC3, CA8, PRKX, XKR4, and SNX29; see Table 1) do not have any known role in TH regulation. These loci form a particularly interesting group for further research, possibly elucidating new pathways in thyroid physiology as illustrated by the successful in vitro studies on AADAT and SLC17A4. Importantly, GWAS hits do not necessarily always pinpoint causal variants and genes, as the observed association might in some cases result from linkage disequilibrium with other (rare) variants yielding the causal effect (73). For example, the beta-1,4-galactosyltransferase 6 (B4GALT6) variant (rs113107469) identified by Taylor et al (34) is tagging a much rarer and potentially functional variant (rs28933981, minor allele frequency of 0.4%) in the gene encoding transthyretin (TTR), resulting in the Thr139Met substitution, which was associated with an increased thyroxine-binding affinity (34). Therefore, follow-up studies for these loci are a key next step to understand the mechanisms underlying the observed associations.
Implications for Clinical Practice
As reviewed above, many genetic determinants of thyroid function have been identified in the last few years. Here we discuss ways in which these data could serve future clinical practice. In particular, we will discuss how identified genetic variants could be used to: (1) investigate causal associations between thyroid function and adverse outcomes, (2) predict the individual risk of thyroid dysfunction and its response to treatment, and (3) define the individual HPT axis setpoint.
Mendelian randomization studies
In the last decade, multiple observational studies have reported associations between minor variations in thyroid function and adverse clinical outcomes (2-9, 13). This has opened up a discussion whether reference ranges for thyroid function tests should be adapted (74, 75). However, observational studies are prone to biases in study design, residual confounding, and reverse causality (76). In most cases, it is therefore unclear whether causal relationships underlie the observed associations. One possible and currently popular approach is to perform a Mendelian Randomization (MR) study. In MR, the effect of an exposure (eg, thyroid function) on an outcome (eg, cardiovascular disease) is evaluated using genetic variants associated with the exposure as the instruments (Figure 3). This concept draws from the fact that genetic variants segregate randomly from parents to offspring, which can be compared to randomization used in clinical trials. As genetic variants can affect an outcome (via exposure) but not the other way around, an association between the genetically estimated exposure and outcome can confirm the causality of the observed association. However, this approach requires several assumptions (77, 78). Most importantly, the instruments have to be truly associated with the exposure, and the effect of the instruments on the outcome has to be mediated solely by the exposure. This means that weak and pleiotropic instruments should be avoided since they can strongly bias the causal estimates (79, 80). Another important prerequisite is that a conclusive MR requires appropriate power, which depends on several parameters, including study sample size, magnitude of the causal association between the exposure and outcome, and proportion of variance in the exposure explained by the genetic instruments used (81).
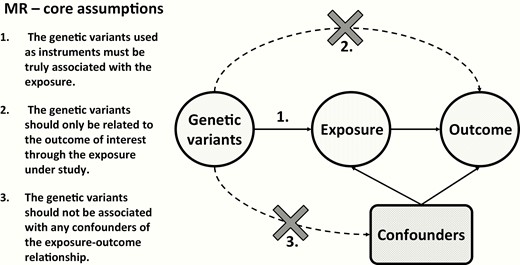
Core assumptions for Mendelian Randomization (MR) studies. Arrows indicate the direct effect of one variable on the other, while dashed arrows with a cross indicate that there should be no direct effect of one variable on the other.
Several MR studies have attempted to clarify the causal relationships between minor variation in thyroid function and various outcomes, including cardiovascular disease (82, 83), type 2 diabetes (84), bone mineral density and fracture risk (85), and kidney function (86). However, none of them found evidence for a causal association between thyroid function and the tested outcome. More recently, two MR studies (87, 88) provided evidence for causal effects of minor variation in thyroid function on atrial fibrillation (AF) risk, as they found that genetically-predicted decreased TSH levels, as well as increased FT3:FT4 ratios (but not FT4 levels), were associated with an increased risk of AF (87, 88). While negative results of MR studies could indicate lack of causal effects of thyroid function on tested outcomes, they might also result from insufficient power to detect existing effects. The identification of many novel genetic variants associated with serum TSH and FT4 levels in the most recent GWAS by the ThyroidOmics Consortium will significantly increase the power of future MR studies. However, there are more requirements to ensure a high quality of future MR studies (89). First of all, a good understanding of mechanisms underlying the association between thyroid function and genetic variants used as instruments in MR studies is essential. For example, several DIO1 variants have been associated with FT4 levels in GWAS. Since DIO1 is responsible for peripheral conversion of T4 to T3, the higher T4 levels coincide with lower T3 levels, leading to a net euthyroid status, as reflected by the absence of an association with TSH. Consequently, while these variants can be used as instruments for variation in normal-range FT4 levels, they should not be interpreted as being instruments for increased thyroid function, despite their relation with higher FT4 levels. Therefore, a good understanding of the role of identified loci in the HPT axis and their resulting associations with thyroid function tests (Table 1) is a key for an appropriate interpretation of MR study results. Moreover, it is important to remember that thyroid dysfunction is mostly caused by autoimmunity and that autoimmune diseases frequently coincide. For this reason, it is not always clear whether the causal effect of hypothyroidism or hyperthyroidism on an outcome observed in a MR study is attributed solely to thyroid dysfunction or rather to concomitant autoimmune disorders. Furthermore, even the associations with normal range TSH and FT4 levels can reflect either alterations in the HPT axis setpoint or mild (early stage) thyroid disease. Data from GWAS on (subclinical) hypo/hyperthyroidism (19) and thyroid peroxidase antibody positivity (90) available at the ThyroidOmics Consortium website (www.thyroidomics.com; genetic associations section) can be used to distinguish between these two groups.
Predicting the individual risk of thyroid disease and treatment response
It is an intriguing question as to whether data from large-scale genetic association studies can eventually serve the management of individual patients. In the following sections we discuss the potential role of genetic variations in DIO2, as well as the use of genetic risk scores (GRS).
Type 2 deiodinase.
Approximately 70% of circulating T3 derives from peripheral T4 deiodination catalyzed by DIO2 (91). As discussed above, DIO2-rs225014 has been shown to affect DIO2 activity in several cell lines (68–70). Several small-scale epidemiological studies have shown associations between this variant and various clinical endpoints, including type 2 diabetes, hypertension, osteoarthritis, bipolar disorder, and others (17, 18, 92). Additionally, it has been hypothesized that, because of impaired T4/T3 conversion, hypothyroid patients harboring the Ala variant may experience a benefit from LT4/LT3 combination therapy. For these reasons, three clinical studies have assessed the potential role of this variant in determining LT4/LT3 combination therapy efficacy. The first study was performed by Appelhof et al (71) in 2005, who did not find effects of the DIO2-rs225014 genotype on baseline well-being or appreciation of LT4/LT3 combination therapy in 141 hypothyroid patients (P > 0.05). In 2009, Panicker et al (93) performed a retrospective analysis in 552 hypothyroid patients from the Weston Area T4 /T3 Study (WATTS), in which patients on a stable dose of LT4 therapy were previously randomized to either LT4/LT3 combination therapy or the original LT4 monotherapy dose (94). In this study, patients homozygous for the Ala variant had an impaired psychosocial well-being at baseline as assessed by the General Health Questionnaire (GHQ) and showed greater improvement in the GHQ score on LT4/LT3 combination therapy when compared with LT4 monotherapy (by 2.3 GHQ points at 3 months and 1.4 points at 12 months, P = 0.03) (93). However, these results were not statistically significant when correcting for multiple testing, whereas significant heterogeneity in baseline psychosocial well-being and improvement on LT4/LT3 combination therapy was observed within the groups of patients stratified by the DIO2-rs225014 genotype (93). No association between the DIO2-rs225014 variant and health-related quality of life measured using the RAND36-Item Health Survey (95) was either found in 364 patients on LT4 treatment by Wouters et al (72). More recently, Carle et al (96) performed a small (N = 45) prospective double-blind randomized clinical trial in which they found that hypothyroid patients harboring combined MCT10-rs17606253 and DIO2-rs225014 variants (P = 0.009), but not DIO2-rs225014 alone (P > 0.05), may prefer LT4/LT3 combination treatment. While these results need to be interpreted with caution given the small sample size of the study (96), they may suggest that a combination of multiple variants, rather DIO2-rs225014 alone, may contribute to differences in response to LT4/LT3 combination therapy observed between patients. In summary, while it has been proven that the DIO2-rs225014 variant affects DIO2 function as well as circulating serum T4 levels, it is highly unlikely that this single variant alone is of substantial predictive value in the individual patient. The interindividual differences in response to LT4 monotherapy, as well as LT4/LT3 combination therapy, are more likely to be explained by a combination of multiple variants (common or rare with large individual effects sizes), which should be the scope of future genetic and clinical studies.
Genetic risk scores.
The most prominent research using GRS comes from the cardiovascular field. For example, using a GRS that combined 50 single nucleotide polymorphisms (SNPs) associated with coronary artery disease (CAD) in GWAS, Khera et al (97) showed that participants with a GRS in the highest quintile had a 91% higher relative risk of incident coronary events compared to those with a GRS in the lowest quintile. A more robust polygenic score was able to identify 8% of population at a greater than threefold increased risk of CAD (98). Given the ongoing identification of novel genetic variants associated with CAD and the continuously decreasing costs of genotyping arrays, implementation of such GRS into everyday clinical practice starts to be considered as a cost-effective method for improving the effectiveness of prevention and treatment for CAD (99, 100).
As GWAS on thyroid function have identified many variants in the last decade (Figure 1), a number of studies have also investigated the use of these variants to identify individuals with increased risk of thyroid dysfunction. In a follow-up analysis for their GWAS on serum TSH and FT4 levels using GRS based on 20 variants associated with normal range TSH levels, Porcu et al (24) showed that the odds of increased TSH levels were 6.65 times greater in individuals with a GRS in the top quartile compared to individuals in the bottom quartile (P = 3.4x10-20). In another study, Schultheiss et al (101) showed that a GRS based on nine variants associated with thyroid peroxidase antibodies (TPOAbs) in a dedicated GWAS (90) can be used to identify individuals with an increased risk of both overt and subclinical hypothyroidism (interquartile odds ratio of 1.89 and 1.80, respectively). Furthermore, the most recent GWAS on thyroid function also showed that a GRS based on variants associated with TSH levels within the normal range strongly correlates with the risk of both (subclinical) hypothyroidism and hyperthyroidism (Figure 4) (19). However, it should be noted that while showing impressive effects at the extremes, the number of subjects carrying extremely low or high GRS was limited, and these markers still have insufficient discriminatory power to be used in clinical practice. Nevertheless, these results illustrate in which ways they could be used to help identify individuals with an increased risk of thyroid dysfunction or personalize the treatment of patients with thyroid disease in the future.
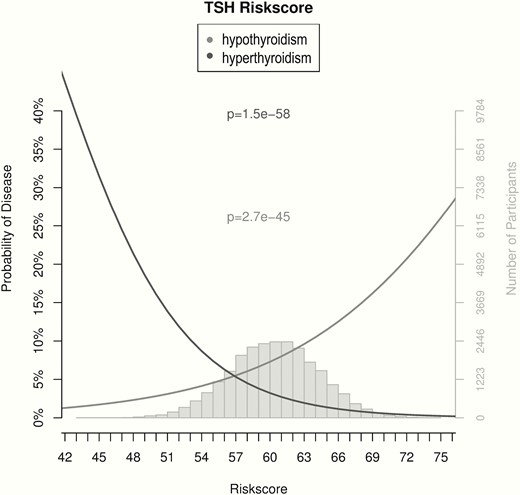
Associations of genetic risk scores with hypothyroidism and hyperthyroidism. The probability of hypothyroidism (gray) or hyperthyroidism (black), as defined by an increased or decreased TSH, respectively, based on a weighted genetic risk score (GRS) built using the 61 SNPs associated with TSH levels in the most recent GWAS by Teumer et al (19) is shown with respect to the percent of total risk alleles (x-axis). The gray histogram shows the distribution of the GRS in the study sample. Adapted from Teumer et al (19).
Defining the individual HPT axis setpoint
Serum TSH and TH levels in healthy persons show substantial interindividual variation, while the intraindividual variation is much smaller (14). This suggests that every person has a unique individual TSH and TH setpoint. Therefore, normalization of TSH and TH levels in thyroid disease patients after the initiation of treatment to levels within the reference ranges cannot guarantee the euthyroid status of that specific individual, as the achieved levels can still deviate from the individual setpoint. In addition, even TSH and TH levels within the reference range have been associated with an increased risk of cardiovascular events, including atherosclerotic cardiovascular disease and stroke (2, 3). Unfortunately, we seldom have TSH and TH levels before the onset of the thyroid disease, which could be used as a proxy of the individual setpoint. Mathematical models based on repeated measurements of TSH and TH levels of an individual on LT4 therapy have been proposed to estimate the individual HPT axis setpoint (102, 103), but this approach still requires clinical validation. While we currently cannot predict an individual’s HPT axis setpoint, we do know that genetic factors are a major determinant of TSH and FT4 levels, next to individual and environmental factors such as age, gender, BMI, and iodine status (15). Future studies should therefore investigate if these factors can be used to predict the individual TSH and TH setpoint, which is a first step towards the personalized treatment of thyroid disease patients. This is important, as ~10% of hypothyroid patients on T4 treatment have persisting hypothyroid complaints (104, 105), which could be partially explained by the fact that their TSH and FT4 levels, despite being in the reference range, still deviate from their individual setpoint.
Moreover, knowing the individual TSH setpoint could be particularly useful in individuals with TSH levels close to (above/below) the upper limit of the reference range. This likely concerns a heterogeneous group, including individuals with normal thyroid function (ie, a setpoint at the extremes of the normal distribution) as well as individuals with (mild, early stage) thyroid disease. This is expected to be important as observational studies showed that subclinical hypothyroidism but also minor alterations in thyroid function are associated with adverse cardiovascular outcomes (2–4). Knowing the individual TSH setpoint could therefore further direct personalized treatment decisions. However, a better insight into the genetic determinants of thyroid function is still required in order to fully address this research question, as discussed below.
Knowledge Gaps and Further Research Directions
While recent GWAS have led to an impressive increase in the identification of variants associated with thyroid function, these variants so far only explain 33% and 21% of the genetic variance in TSH and FT4 levels, respectively (19). This is still a major limitation for all clinical applications discussed above and identifying this “missing heritability” (106) will be a key task for this field.
Further GWAS, WES, and WGS studies
The results of genetic association studies in complex traits and disorders, such as thyroid function and cardiovascular disease, have demonstrated that a large part of their genetic susceptibility is defined by multiple common variants with modest individual effect sizes. Therefore some of the “missing heritability” may be explained by common polymorphisms that contribute to the genetic susceptibility with even smaller effect sizes than those already identified in previous GWAS. For this, larger GWAS are needed. Moreover, while most of GWAS performed so far included mainly Caucasians (19, 22-28), future studies should also investigate the genetic basis of thyroid function in other ethnic groups. This is important since interethnic variations in allele frequencies may result in substantial differences in effect sizes and overall contribution for the identified variants across populations. Although requiring large sample sizes, gene–gene and gene–environment interactions, such as with iodine status, should also be investigated in these future studies, as these are potentially significant contributors to the genetic architecture of complex traits and disorders (107, 108).
While GWAS on thyroid function managed to identify multiple variants associated with TSH and FT4 levels, still little is known on the genetic factors determining FT3 levels, as they were not assessed in well-powered studies. Triiodothyronine (T3) is the active form of TH responsible for most of its metabolic effects (109). In 2010, a GWAS performed by Panicker et al in a cohort of 2014 female twins from the British population failed to identify any variants associated with FT3 levels at the genome-wide significant level (25). Most recently, another small (N = 1731) GWAS in the Croatian population identified the EPH receptor B2 (EPHB2) gene variant (rs67142165) as a new locus for FT3 levels (110). However, this finding requires confirmation in an independent study. Therefore, similar to TSH and FT4, well-powered GWAS meta-analyses are needed to effectively investigate the genetic determinants of FT3 levels.
Of note, there may be also rare variants conferring larger effects that can be identified in whole genome sequencing (WGS) or whole exome sequencing (WES) studies (106). The potential success of such studies is, for example, illustrated by a WGS study in 21 620 unrelated individuals of European ancestry, which showed that rare variants can be responsible for approximately 54% and 51% of heritability for height and BMI, respectively (111). While the first WGS study on thyroid function performed by Taylor et al managed to identify only two novel loci associated with TSH and TH levels (B4GALT6 and SYN2) (34), further WES or WGS studies with larger sample size are expected to identify more rare variants that could explain a substantial proportion of the “missing heritability” (112).
Epigenetic determinants of thyroid function and other omics in consortia
Epigenetic modifications such as DNA methylation, acetylation, phosphorylation, ubiquitination, and methylation of histones can regulate gene expression by alternations in DNA configuration. These nonpermanent changes are involved in cell proliferation and differentiation, but some of them can also be heritable or modified by environmental factors (113). Therefore, epigenetic modifications can be responsible for an important part of the “missing heritability” (114). While other fields in medicine have made significant progress in the understanding of the role of methylation (115, 116), its role in the regulation of thyroid function is still poorly understood. Therefore, further studies, including epigenome-wide association studies (EWAS), are warranted.
Finally, population-based studies employing other omics, such as transcriptomics, proteomics, and metabolomics, are outside the scope of this review, but they may also lead to new insights in thyroid physiology and pathophysiology, as recently discussed elsewhere (117).
Functional studies
As mentioned before, an important part of the loci identified in GWAS on thyroid function has no known role yet in TH regulation. As illustrated by the AADAT and the SLC17A4 examples (19), further in vitro studies are a key step to discover new players involved in the regulation of thyroid function and investigate their potential role in thyroid disease. Similarly, in vitro studies are also needed for the newly associated variants in known TH pathway genes, which will further increase our understanding of TH regulation.
Conclusions
In recent years, GWAS have identified many novel genetic variants, which determine individual TSH and FT4 levels. These findings have not only led to the discovery of new genes that play a role in regulation of thyroid function, but have also paved the way for various other applications. Mendelian Randomization studies are expected to improve our understanding of the relationships between thyroid (dys)function and various adverse health outcomes. Furthermore, with the identified genetic variants, we can start exploring ways to use these in future clinical practice, including thyroid disease risk prediction and personalization of its treatment. However, as their discriminative power is expected to still be too limited to be used in clinical practice yet, future studies in large-scale consortia are needed to decipher the unexplained genetic variance in thyroid function.
Acknowledgments
Funding: A.K. is supported by the Exchange in Endocrinology Expertise (3E) program of the European Union of Medical Specialists (UEMS), Section and Board of Endocrinology. M.M. is supported by research grants from the American Thyroid Association and European Thyroid Association, and an Erasmus University EUR fellowship grant.
Additional Information
Disclosure Summary: The authors have nothing to disclose.
Data Availability: Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}