





Abstract
The rapid pubertal height growth is unique to humans, but why do we have it? Although the spurt contributes 13% to 15% to the final adult height, we hypothesized that the biological significance of the high acromegalic levels of GH and IGF-I, which are behind the pubertal growth spurt, might primarily occur to stimulate the reproductive organs.
Animal data have demonstrated that adult Igf1 and Igf2 gene knockout mice that survive show a dramatic reduction in the size of the reproductive organs and are infertile. In humans, case reports of mutations in the genes affecting the GH–IGF axis and growth (GH, GHRH, GH-R, STAT5b, IGF-I, IGF-II, IGF-1R, PAPPA2) are also characterized by delayed pubertal onset and micropenis. Furthermore, GH treatment will tend to normalize the penile size in patients with GH deficiency. Thus, the endocrine effects of high IGF-I levels might be needed for the transition of the sexual organs, including the secondary sex characteristics, from the “dormant” stages of childhood into fully functioning reproductive systems. The peak IGF-I levels, on average, occur 2 years after the peak height growth velocity, suggesting reasons other than longitudinal growth for the high IGF-I levels, and remain high in the years after the height spurt, when the reproductive systems become fully functional.
We suggest that the serum levels of IGF-I should be monitored in children with poor development of sexual organs, although it remains to be investigated whether GH should be added to sex steroids in the management of hypogonadism for some pubertal children (e.g., boys with micropenis).
A rapid pubertal growth spurt is unique to humans. No other primates have such a dramatic increase in growth velocity (1). So why do we have it? A common assumption has been that it is needed to provide the individual with an appropriate adult height. This is plausible, considering that normal children grow much faster during the pubertal spurt. However, some children, who might entirely lack the pubertal growth spurt due to hypogonadism, can grow to be taller than their peers. Thus, the pubertal growth spurt contributes to the final height, but other factors such as the duration of the growth period might be even more important.
Human growth patterns vary throughout the neonatal and infancy periods, childhood, and puberty. After birth, a marked deceleration of height velocity is observed during the first years of life from the high speed seen during the fetal period (an average 52-cm gain in length in 9 months) to low speed growth during the later childhood years (4 to 6 cm annually), until growth acceleration begins at the onset of puberty at 10 to 12 years after birth. Postnatal growth has been divided into three phases according to the infancy-childhood-puberty growth model (2). Infant growth is primarily dependent on nutrition and insulin. In contrast, normal childhood growth depends on nutrition, thyroid hormones, and GH. The pubertal growth period depends on sex steroids increasing the activity of the GH–IGF axis and also exerting direct effects at the level of the growth plate. New mathematical models (Quadratic Exponential Puberty Stop) provide individualized estimates of growth during puberty, with accompanying SD-scores, which might enable more in-depth analysis of the complex human pubertal growth spurt (3).
A marked sex difference in the postnatal growth pattern exists, and the onset of growth acceleration in adolescence occurs 2 years earlier in girls than in boys. In addition, the increase in linear growth is a very early pubertal phenomenon in girls, occurring with the onset of puberty and sometimes even preceding other pubertal signs such as breast budding. In contrast, in boys, the onset of the pubertal growth spurt usually occurs 1 or 2 years after pubertal onset. The child’s age at pubertal onset affects the duration of puberty and the maximal height velocity (peak height velocity) (4). However, the final adult height will not differ significantly between subjects with early or late pubertal timing within the normal range of variation. In contrast, outside the normal range of pubertal timing, the adult height could depend on the child’s age at sexual maturation. The final height will be decreased in girls with overt precocious puberty (5). However, boys with markedly delayed puberty who do not show a pubertal growth spurt can continue to grow during adolescence without a spurt and often grow taller than their peers (6). Similarly, rare cases of estrogen receptor mutations (or in the aromatase gene), with the subsequent lack of estrogen action, have resulted in continuous growth without a spurt well into the 20s and increased adult height (7).
It is not known which factors activate the onset of puberty. It has been hypothesized that a central growth-tracking mechanism responds to circulating peripheral growth-related factors such as leptin or IGF-I, which in turn increase GnRH release at puberty. Activation of the complex kisspeptin system, including KNDY neurons, results in activation of the hypothalamic GnRH pulse generator. Also, GnRH neurons express IGF-I receptors, and GnRH neuron-specific knockout animals have shown markedly delayed puberty. Furthermore, IGF-I administration advanced puberty in mice (8), rats (9), and monkeys (10), suggesting that IGF-I of peripheral origin might accelerate the central initiation of puberty. Although a strong genetic component exists regarding pubertal timing (11), prenatal hormones or exposure to endocrine disrupting chemicals (12), early infancy weight gain (13), prepubertal body size, and serum IGF-I levels at 8 years of age (14) all seem to be involved in the timing of puberty. Thus, overweight girls tend to mature early, and girls with undernutrition or anorexia will experience delayed sexual maturation.
The effect of the pubertal growth spurt on the actual achieved final adult height might be somewhat overrated. Pubertal growth contributes by only 13% to 15% to the final adult height, corresponding to 28 cm in males and 22 cm in females (3). Nevertheless, its biological significance has often been seen as a process toward securing an appropriate final adult height. However, we speculate that this might not be the primary reason for humans experiencing a pubertal growth spurt.
Although not denying the importance of the pubertal growth spurt for the final adult height, in the present study, we have proposed the hypothesis that a primary biological function of the high levels of GH and IGF-I during puberty might be to resume the rapid growth development and maturation of the sex organs, including the testes, seminal vesicles, penis, skin, prostate, and voice break of boys and ovaries, oviducts, uterus, and mammary glands of girls.
The fetal development of organs is associated with high levels of hormones and growth factors. For the somatic organs, the structures are essentially complete within the first year after birth. However, the development of the sex organs is disrupted early in the first year after mini-puberty, and they become dormant until puberty. Thus, the resumption of the development of the reproductive system in puberty might require high levels of GH and growth factors to secure the final development and maturation of the different organs of the reproductive system. Therefore, in puberty, tight signaling exists between the gonads and the GH–IGF-I axis, often resulting in acromegalic levels of IGF-I in the circulation. Thus, parallel accelerated linear growth continues for a short period in the early phases of puberty until the sex steroid-dependent closure of the growth plates has occurred.
Physiology of the GH–IGF Hormone Axis
The endogenous GH 22-kDa form is secreted in pulses from somatotroph cells in the anterior pituitary gland under the excitatory and inhibitory influence of the hypothalamic GHRH and somatostatin. GH exert its actions after binding to the GH receptor, which is localized in multiple target tissues, including liver, muscle, bone, growth plates, and gonads. Thus, GH stimulates intracellular signals through many intracellular pathways, including STAT5b. Activation of the GH receptor results in both mitogenic and metabolic pathways. The pulsatile GH secretion stimulates linear bone growth in postnatal life via the production of IGF-I and IGF-II, which subsequently interact with the ubiquitous IGF receptors. The biological activity of circulating IGFs is further modulated by six specific IGF-binding proteins (IGFBP-1 to -6). Of these, IGFBP-3 binds most circulating IGFs, together with the acid labile subunit (ALS), in a ternary complex. These large molecular weight complexes function as a reservoir and circulating buffer of IGF-I, which also prevents the glucose-lowering effects of free IGFs [for review, see Juul (15)]. Recently, pregnancy-associated plasma protein-A2 (PAPP-A2) and stanniocalcin-2 were identified as novel modulators of IGF-I bioavailability. PAPP-A2 is a protease that cleaves IGFBP-3 and -5, and stanniocalcin-2 inhibits PAPP-A and PAPP-A2 activity. Altogether, this complex network of growth-related factors influences linear growth. Consequently, mutations in individual genes resulting in insufficient production and/or function in any of these factors will result in a disturbed GH–IGF axis and impaired growth. Mutations in the GHRH, GHRH-R, or GH will result in extreme dwarfism due to lack of GH. Mutations in the GH-R (16) or STAT5b (17) cause complete GH insensitivity (Laron syndrome), resulting in severe dwarfism due to lack of GH action, despite high plasma levels. Mutations in genes encoding IGF-I (18), IGF-II (19), ALS (20), and IGF receptor (21) result in severe growth failure of the affected subjects. Recently, humans carrying mutations in the PAPPA2 gene who presented with short stature, showed elevated total IGF-I, IGFBP-3, IGFBP-5, and ALS but low free IGF-I (22). The role and mechanism of the GH–IGF axis in normal human growth have been unraveled during the past 4 decades, not only by exciting knock out animal studies, but, more importantly, by the detailed clinical, biochemical, and genetic characterization of unique and rare patients with mutations affecting GH action. A detailed review has been reported by David et al. (23).
Role of GH–IGF Axis for Organ Development and Organ Functions Throughout Life
Pregnancy—maternal circulation
High and increasing IGF-I levels in pregnant women are ensured by the placental secretion of a high tonic GH 20-kDa variant form (placental GH) into the maternal circulation (24). Placental GH is secreted constantly and completely suppresses the maternal pulsatile GH secretion from the pituitary gland from mid-gestation. These high constant GH levels in the maternal circulation during pregnancy resemble an acromegalic state and result in a doubling of maternal serum IGF-I concentrations in late pregnancy. Most likely, this fetal takeover of maternal endocrinology is to ensure appropriate transplacental nutrient supply to the fetus. Consequently, hypopituitary women with complete GH deficiency will—for the first time in their life—experience high-normal IGF-I serum levels during late pregnancy even without any exogenous GH treatment, stimulated by the fetus’ (placental) production of GH variant. Immediately after delivery of the child and placenta, the GH variant will be rapidly cleared from the maternal circulation (25), and normal nonpregnant endocrinology will slowly resume.
Fetal period, infancy, and childhood
IGF-I and IGF-II have an important role in fetoplacental growth throughout gestation. They have metabolic, mitogenic, and differentiation actions in a wide range of fetal tissues, including the placenta. Both IGF-I and IGF-II genes are expressed in fetal tissues. Expression of the IGF-II gene is more abundant than IGF-I gene expression during mid to late gestation. Fetal circulation contains 3- to 10-fold greater levels of IGF-II compared with IGF-I during late gestation (26). The embryonic role of IGF-I and IGF-II is mediated by the IGF1R and thought to be GH independent. The actions of IGF-I on fetal tissue growth appear to be more prominent compared with those of IGF-II and might be mediated by other fetal hormones. In contrast, IGF-II might have major role in placental growth and nutrient supply (26). Thus, Igf1 and Igf2 gene knockout mice will be born small, and most will die in the early neonatal stages (27). Postnatal growth curves have suggested that surviving Igf-1−/− mutants, which exhibit delayed bone development, will continue to grow with a lower growth rate and will be 30% shorter than wild-type mice (28). In adult mutants of both sexes, a dramatic reduction in the size of the reproductive organs was observed, and they were infertile (29).
In postnatal life, the serum levels of IGF-I increase in infancy shortly after birth (30) and depend on breast feeding. Furthermore, the testicular position is associated with the serum concentrations of IGF-I in healthy infants (31). Serum IGF-I levels are primarily dependent on nutrition and insulin in the first year of life.
Puberty
The drivers of the pubertal growth spurt are the substantial increases in the levels of the pulsatile GH and its circulating mediator of linear growth IGF-I, which for a couple of years during adolescence could reach acromegalic levels. The high IGF-I levels observed in normal or precocious puberty will coincide with increasing linear growth. IGF-I levels increase linearly during childhood and the prepubertal years. However, the increase is more pronounced in puberty, during which some interaction occurs with the pubertal stages: increasing IGF-I with increasing age in Tanner stages I to III and decreasing IGF-I with increasing age in Tanner stages IV and V (32). After maximal growth velocity in puberty, the serum IGF-I levels will remain elevated for some years, followed by a slow decline throughout adult life (Fig. 1).
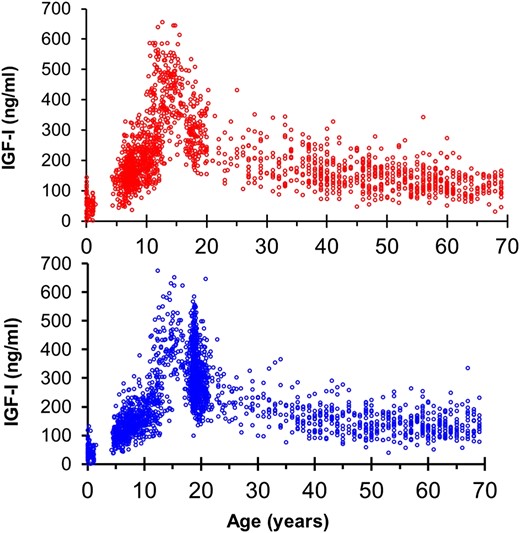
Serum concentrations of IGF-I in healthy infants, children, and adults according to age and sex. (Top) IGF-I levels in females and (bottom) males throughout life. Note the marked increased in IGF-I in the pubertal years in both sexes. Data redrawn from previous reported of healthy boys and girls and adults of both sexes (33–36).
IGF-II levels will be relatively constant in pubertal years (37). In contrast, IGFBP-3 and ALS both increase in puberty (37, 38). Using a direct assay quantifying bioavailable IGF-I, we found increasing bioavailable IGF-I in puberty (39), which was verified in a small subpopulation of healthy children using a direct ultracentrifugation assay to quantify free IGF-I (40). Also, longitudinal studies of healthy children have detailed the pubertal increase in IGF-I compared with the height velocity and testosterone levels (41, 42). In mid- to late puberty, serum GH and IGF-I levels are clearly within the acromegalic range and are associated with transient insulin resistance (43) (Fig. 2).
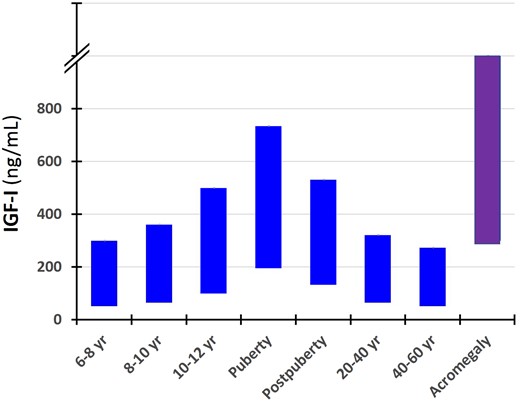
Serum concentrations of IGF-I (10th and 90th percentiles) in different age groups of healthy subjects and untreated patients with acromegaly at diagnoses. Note the overlapping of IGF-I values between the normal pubertal subjects and patients with acromegaly.
Girls with precocious puberty, who are characterized by increased linear growth, also have markedly elevated IGF-I levels compared with their prepubertal peers (44). Once linear growth has stopped owing to fusion of the epiphyseal growth plates in the later part of the pubertal transition, the GH and IGF-I levels will gradually return toward the levels seen before puberty, although with some years delay.
GH–IGF Axis and Reproductive Function: Lessons From Pathological Studies
Numerous examples from nature have suggested that the GH–IGF axis is, not only involved in longitudinal growth, but also in genital development and sexual maturation. Infants with isolated GH deficiency usually have a birthweight within the normal range, although impaired genital development (micropenis and cryptorchidism) seem to occur frequently in such children (45, 46). In support of the importance of GH on genital development is that the small penile size increased during GH treatment of male infants with congenital isolated GH deficiency. Thus, 13 of the 15 prepubertal boys with isolated GH deficiency (11 with a hereditary form) had a small penis, and 3 of these had a diagnosis of true micropenis. Treatment with GH improved penile length in all but 2 of the 13 boys (47–49). Likewise, IGF-I treatment appears to stimulate testicular growth in boys with GH insensitivity syndrome (50). Altogether, GH and IGF-I therapy seem to stimulate genital growth in male patients lacking these hormones. Also, GH treatment appeared to shorten the duration of puberty (1.5 years in both sexes) in a study of 134 children with idiopathic GH deficiency who had been treated with GH (51). Likewise, Stanhope et al. (52) evaluated the effects of GH in 52 children with GH deficiency. They reported that both boys and girls had a more rapid rate of pubertal maturation than normal and that these inappropriate effects on pubertal progression might counterbalance the improvement in growth prognosis during prepuberty (52). However, similar findings were not reported in a multicenter study of 239 children with idiopathic short stature, for whom GH treatment did not appear to accelerate pubertal onset, pace, or bone maturation (53). Despite these reported data, not much attention has been given to the possible biological role of the GH–IGF axis for genital development during puberty.
Recent advances in the genetics of the GH–IGF-I axis have provided results in line with the clinical evidence for a role of GH in the development of the reproductive system. Deleterious mutations in genes encoding the hormones and receptors involved in the activity of the GH–IGF axis have frequently been associated with impaired genital growth or delayed puberty. A study of 41 patients with GH insensitivity (GHIS) reported delayed and slow progression throughout puberty, especially in boys with GHIS, who started puberty, on average, at 15.6 years of age (54). Also, other cases of impaired penile growth in GHIS have been reported (55). A case of GHIS due to a mutation in the STAT5b gene was reported. At the age of 16.5 years, the girl had a height of 117.8 cm (−7.5 SD) and delayed secondary sex characteristics (breast stage 3) (17). A 15-year-old boy, born small for gestational age (birth weight, −3.9 SD for gestational age) with a verified mutation in the IGF-I gene, had also presented with markedly delayed puberty (genital stage 2; testicular volume, 4 mL) (56). Within a family affected by a mutation in the IGF-II gene, 2 children had had puberty occur later than average (19). One girl experienced menarche at 14 years of age, and one boy who had initiated puberty at 13 years of age (19). A 14.6-year-old boy, who had presented with short stature and pubertal delay, was found to carry an inactivating mutation of the ALS gene, resulting in markedly reduced IGF-I levels. At evaluation, he was prepubertal, and both testes were 3 mL in size, and he showed slow pubertal progression (20). A 14.5-year-old girl born with severe intrauterine growth retardation and postnatal growth failure with a mutation in the IGF-IR gene presented with breast stage 2 at the age of 13.5 years (57). Altogether, these findings suggest that an impaired GH–IGF axis results, not only in a short stature, but also in micropenis and/or insufficient or delayed pubertal development of sexual organs.
The mechanisms by which GH and/or IGF-I act on the reproductive system are not entirely clear. GH treatment increases human chorionic gonadotropin-induced T secretion in hypopituitary boys (58) and adult men (59). Both GH and IGF-I receptors are present in the human testis and appear to play a crucial role in testicular development, spermatogenesis, and steroidogenesis. Healthy men with a more sensitive GH receptor genotype (GHRd3) had a larger semen volume and greater serum inhibin B compared with men with a wild-type GH receptor genotype (33). Furthermore, GH treatment increased serum estradiol in normal men (60) and GH-deficient men (61).
Discussion and Perspectives
GH and IGF-I play key roles in the growth of all organs and in skeletal growth. However, the target for the acromegalic levels of IGF-I during puberty might not primarily be the bones but the organs of the reproductive system. Admittedly, in the clinic, the concern about children’s “growth” has most often been centered around longitudinal bone growth. However, the reproductive system must also grow quickly during puberty and is highly dependent on a normally functioning GH–IGF-I axis. GH and/or IGF-I receptors are widely distributed in the reproductive organs of both sexes. In the male, they are present in the Leydig, Sertoli, and germ cells in the testis and are also expressed in the prostate. In the female, they are found in the ovary (granulosa and germ cells), mammary glands, uterus, and oviducts (62). The development of gynecomastia is a well-described side effect during GH treatment in elderly men. In addition, serum IGF-I was associated with the presence of pubertal gynecomastia in healthy school boys (41), suggesting GH and/or IGF-I effects on breast glandular tissue. GH receptors are also present in the skin of both sexes.
In conclusion, we believe that GH and IGF-I play crucial roles in all phases of development and function of internal and external sex organs of both sexes. The increase in IGF-I early in normal puberty is especially conspicuous, because IGF-I levels will often reach levels comparable to those seen in adults with acromegaly. From a biological viewpoint, we find it tempting to hypothesize that these high IGF-I levels are needed to secure the resumption of growth and development of the “dormant” sexual organs, in concert with the increasing levels of sex steroids from the gonads. Also, despite the rapidly declining growth velocity in the years after the peak height velocity, circulating IGF-I levels remain significantly elevated for years, suggesting that GH and/or IGF-I have important physiological actions during this period, not related to the growth of the bones. Thus, the target for the high pubertal IGF-I levels stretching into adolescence might be the reproductive system.
Our hypothesis could be relevant to the treatment of children with GH deficiency and children and adolescents with malformations and delayed development of reproductive organs (e.g., management of micropenis). However, it remains to be investigated whether a combination of GH and sex hormone treatment would be preferable to treatment with sex hormones alone for children with severe hypogonadism and a poor response to sex steroids alone.
Acknowledgments
Disclosure Summary: The authors have nothing to disclose.
Abbreviations:
- ALS
acid labile subunit
- GHIS
GH insensitivity
- IGFBP
IGF-binding protein
- PAPP-A2
pregnancy-associated plasma protein-A2

{kind=link}
{kind=link}