





Abstract
Cushing disease (CD) is a rare disorder with severe sequels and incompletely understood pathogenesis. The underlying corticotroph adenomas harbor frequently somatic mutations in the ubiquitin-specific peptidase 8 (USP8) gene. These mutations render USP8 hyperactive and prevent client proteins from degradation.
To investigate the impact of USP8 mutations on proteins deregulated in CD.
One hundred eight pituitary adenomas (75 corticotroph [58 USP8 wild type (WT) and 17 USP8 mutated], 14 somatotroph, and 19 nonfunctioning) were investigated by immunohistochemistry. All evaluated proteins [USP8, arginine vasopressin receptor 1b and 2, corticotropin-releasing hormone receptor, cAMP response element-binding protein (CREB), p27/kip1, cyclin E, heat shock protein 90 (HSP90), orphan nuclear receptor 4, epidermal growth factor receptor, histone deacetylase 2, glucocorticoid receptor, cyclin-dependent kinase 5 and Abelson murine leukemia viral oncogene homolog 1 enzyme substrate 1] were known to be deregulated in CD. Furthermore, AtT20 cells were transfected with USP8 to investigate the expression of possible downstream proteins by immunoblot.
Whereas most of the investigated proteins were not differentially expressed, the cell-cycle inhibitor p27 was significantly reduced in USP8 mutated corticotroph adenoma (H-score 2.0 ± 1.0 vs 1.1 ± 1.1 in WT adenomas; P = 0.004). In contrast, the chaperone HSP90 was expressed higher (0.5 ± 0.4 vs 0.2 ± 0.4; P = 0.29), and the phosphorylation of the transcription factor CREB was increased in USP8 mutated adenomas (1.30.5 ± 0.40.9 vs 0.70.5 ± 0.40.7; P = 0.014). Accordingly, AtT20 cells transfected with the USP8 P720R mutant had higher phosphorylated CREB (pCREB) levels than WT transfected cells (1.3 ± 0.14 vs 1 ± 0.23; P = 0.13).
We could demonstrate that USP8 mutations are associated with deregulation of p27/kip1, HSP90, and pCREB. These findings suggest that these proteins are direct or indirect clients of USP8 and could therefore be potential targets for therapeutic approaches in patients with CD.
Cushing disease (CD) is a rare [incidence: two to four per million (1)] but life-threatening endocrine disorder caused by adrenocorticotropic hormone (ACTH)-secreting (corticotroph) pituitary adenomas. It is associated with increased morbidity and mortality, resulting from chronic exposure to glucocorticoids, secreted by the adrenal cortex in response to ACTH stimulation. Patients with CD often suffer from hypertension, diabetes mellitus, proximal myopathy, and skin atrophy (2). Accordingly, the majority of deaths associated with CD are caused by cardiovascular, metabolic, and infectious complications. Treatment options include surgical resection of the tumor, irradiation of the sellar region, and medical therapy (3). However, the available drugs mainly combat the symptoms but not the cause, acting as blockers of the glucocorticoid receptor (GR; e.g., mifepristone) and inhibitors of either ACTH release (e.g., dopamine agonists, somatostatin analogs) or cortisol secretion (e.g., metyrapone, ketoconazole) (4, 5). These drugs may have several adverse effects and often also limited efficacy; thus, improved pharmacological options are needed (but require an improved understanding of the molecular events causing CD). The definitive treatment of CD is bilateral adrenalectomy, which is taken under consideration if other treatments or medication have failed (6). However, one complication of bilateral adrenalectomy is Nelson’s syndrome, an aggressive pituitary tumor that occurs in a small subset of patients (7). In the last years, important insights in the development of CD have been gained, including the major finding of somatic mutations in the deubiquitinase ubiquitin-specific peptidase 8 (USP8) gene in almost one-half of ACTH-secreting adenomas (8–10). These mutations were shown to render USP8 hyperactive, resulting in increased recycling of its substrates, such as the receptor tyrosine kinase epidermal growth factor receptor (EGFR), leading ultimately to increased proliferation and increased ACTH secretion (8), bypassing the negative feedback control exercised through the GR. In addition, other proteins are known to be deregulated in corticotroph tumors, such as the family of arginine vasopressin receptor 1b and 2 (AVPR1b and 2) and the corticotropin-releasing hormone receptor (CRHR), both not only involved in ACTH release of the anterior pituitary but also overexpressed in corticotroph tumors (11–15). Quite recently, also, the chaperone heat shock protein 90 (HSP90) and the orphan nuclear receptor 4 (TR4) were shown to be overexpressed in corticotropinomas compared with normal pituitaries (nP) (16, 17). Whereas HSP90 is involved in proper GR folding (16), TR4 induces pro-opiomelanocortin (POMC) promoter transcription (17). In addition, the histone deacetylase 2 (HDAC2; which deacetylates histone H4 at the POMC promoter, thereby leading to the repression of POMC transcription) was shown to have a reduced expression in a subset of corticotroph tumors (18). Apart from proteins obviously involved in ACTH hypersecretion, expression or activity of proteins contributing to cell proliferation and tumorigenesis was reported to be modified in corticotroph tumors. These include the upregulation of cyclin E (19, 20) and EGFR (21, 22) on the one hand and the downregulation of cyclin-dependent kinase (CDK) inhibitor p27/Kip1 (23–25) on the other. Furthermore, CDK5 and Abelson murine leukemia viral oncogene homolog 1 enzyme substrate 1 (CABLES1), a negative regulator of cell-cycle progression is activated in response to glucocorticoids, and its expression was shown to be lost in approximately one-half of corticotroph tumors (26). Additionally, mutations in the CABLES1 gene were described as a rare cause for CD (27). Although a direct link between EGFR signaling and USP8 mutations has been suggested by in vitro studies (8, 28), Hayashi and colleagues (29) did not observe activated EGFR signaling in several USP8 mutated tumors with increased levels of POMC mRNA. Thus, it is likely that activated EGFR signaling is not exclusively responsible for increased ACTH secretion in USP8 mutated corticotroph tumors. Other candidates might be the already-mentioned deregulated proteins but also proteins in the cAMP/protein kinase A (PKA) signaling pathway. The latter has been shown to promote ACTH secretion (30), and there is a possibility that increased PKA signaling could be a result of deubiquitination through USP8. The aim of this study was to find putative USP8 client proteins that could contribute to the ACTH oversecretion and/or tumorigenesis in CD. For this, we evaluated whether already-reported deregulated proteins associated with CD have a significantly altered expression when comparing corticotroph USP8 wild-type (WT) with mutated tumors.
Methods
Tissue collection
In total, we investigated 108 formalin-fixed paraffin-embedded (FFPE) pituitary tumors [corticotroph (n = 75), somatotroph (growth hormone-secreting; n = 14), and nonfunctioning (n = 19)] from individual patients. At the University Hospital Hamburg, 51 corticotroph pituitary adenomas were assembled into one tissue microarray. To choose representative areas with well-preserved morphology of each adenoma, hematoxylin and eosin-stained tissues were re-evaluated by one experienced pathologist (W.S.), marking the corresponding area for tissue punching. Two cores with a diameter of 0.6 mm were punched from each tissue block and arrayed into a new block. Full FFPE slides were used in 24 corticotroph, 12 somatotrophs, and 19 nonfunctioning adenomas; these samples had been collected at the University of Wuerzburg. In addition, nP (n = 5) were available from autopsies performed at the University Hospital Wuerzburg.
Patients and clinical annotations
For patients with corticotroph adenomas, key clinical characteristics are listed in Table 1. Diagnosis of CD was performed according to established clinical, biochemical, and morphological criteria (31); the same was true for other pituitary tumor entities. All patients gave written, informed consent, and the study was approved by the Ethics Committees of the University Hospital Wuerzburg and the Chamber of Physicians in Hamburg.
Clinical Characteristics of Patients With Corticotroph Adenomas
| USP8 WT | USP8 Mutated | P Value | |
|---|---|---|---|
| Patients, n (%) | 58 (77) | 17 (23) | |
| Age, years | 54 (17–82) | 38 (17–77) | 0.02 |
| Sex, n (%) | |||
| Female | 36 (62) | 17 (100) | |
| Male | 22 (38) | 0 (0) | |
| Maximum adenoma size, mm | 10 (4–47) | 10 (3–22) | 0.14 |
| Body mass index, kg/m2 | 27 (20–46) | 29 (19–46) | 0.14 |
| Hormone levels before surgery | |||
| ACTH, pg/mL median (n) | 52 ± 106 (38) | 68 ± 7 (13) | 0.38 |
| Cortisol, µg/dL median (n) | 16 ± 2.4 (43) | 18 ± 2.6 (12) | 0.11 |
| USP8 WT | USP8 Mutated | P Value | |
|---|---|---|---|
| Patients, n (%) | 58 (77) | 17 (23) | |
| Age, years | 54 (17–82) | 38 (17–77) | 0.02 |
| Sex, n (%) | |||
| Female | 36 (62) | 17 (100) | |
| Male | 22 (38) | 0 (0) | |
| Maximum adenoma size, mm | 10 (4–47) | 10 (3–22) | 0.14 |
| Body mass index, kg/m2 | 27 (20–46) | 29 (19–46) | 0.14 |
| Hormone levels before surgery | |||
| ACTH, pg/mL median (n) | 52 ± 106 (38) | 68 ± 7 (13) | 0.38 |
| Cortisol, µg/dL median (n) | 16 ± 2.4 (43) | 18 ± 2.6 (12) | 0.11 |
Results are given as median (range).
Clinical Characteristics of Patients With Corticotroph Adenomas
| USP8 WT | USP8 Mutated | P Value | |
|---|---|---|---|
| Patients, n (%) | 58 (77) | 17 (23) | |
| Age, years | 54 (17–82) | 38 (17–77) | 0.02 |
| Sex, n (%) | |||
| Female | 36 (62) | 17 (100) | |
| Male | 22 (38) | 0 (0) | |
| Maximum adenoma size, mm | 10 (4–47) | 10 (3–22) | 0.14 |
| Body mass index, kg/m2 | 27 (20–46) | 29 (19–46) | 0.14 |
| Hormone levels before surgery | |||
| ACTH, pg/mL median (n) | 52 ± 106 (38) | 68 ± 7 (13) | 0.38 |
| Cortisol, µg/dL median (n) | 16 ± 2.4 (43) | 18 ± 2.6 (12) | 0.11 |
| USP8 WT | USP8 Mutated | P Value | |
|---|---|---|---|
| Patients, n (%) | 58 (77) | 17 (23) | |
| Age, years | 54 (17–82) | 38 (17–77) | 0.02 |
| Sex, n (%) | |||
| Female | 36 (62) | 17 (100) | |
| Male | 22 (38) | 0 (0) | |
| Maximum adenoma size, mm | 10 (4–47) | 10 (3–22) | 0.14 |
| Body mass index, kg/m2 | 27 (20–46) | 29 (19–46) | 0.14 |
| Hormone levels before surgery | |||
| ACTH, pg/mL median (n) | 52 ± 106 (38) | 68 ± 7 (13) | 0.38 |
| Cortisol, µg/dL median (n) | 16 ± 2.4 (43) | 18 ± 2.6 (12) | 0.11 |
Results are given as median (range).
DNA extraction from FFPE tissue
Full FFPE tissue slides of each 24 µm were used to extract DNA, using the QIAamp® DNA FFPE Tissue Kit (Qiagen), according to the manufacturer’s instructions. Elution was performed in 30 µL H2O. DNA quality and concentration were determined with the Nanodrop spectrophotometer (Thermo Fisher Scientific).
Sanger sequencing
Sanger sequencing was used to determine the USP8 mutation status. Two primers were used, spanning a length of 266 bp around the 14-3-3 binding domain of USP8: USP8 forward, ATATGTACCCACCGGAAATG, and USP8 reverse, CAGAGTAGAAACTTTGAAATACAGCA. For amplification of the PCR product in the following reaction, 30 to 60 ng DNA was used: 96°C for 2 minutes, 10 cycles with 96°C for 30 seconds, 58°C for 30 seconds, and 72°C for 30 seconds. Twenty cycles were followed with a reduced annealing temperature of 56°C. The same primers were used in the sequencing reaction. Results were analyzed with the Genome Genetic Analysis System (ABSciex).
Chromogenic immunohistochemistry
Slides were deparaffinized in xylol twice before being rehydrated, and antigen retrieval was performed in 10 mM citric acid monohydrate buffer (pH 6.5) within a pressure cooker for 13 minutes. Endogenous peroxidase was blocked with 3% H2O2 in methanol for 10 minutes, and unspecific binding sites were blocked in 20% human AB serum at room temperature for 1 hour. Primary antibodies were diluted in PBS and incubated at room temperature for 1 hour. The following antibodies were used: USP8 (#HPA004869, 1:500; Sigma), HSP90 (#EPR3953, 1:500; Abcam), TR4 (#HPA006313, 1:200; Sigma), cyclin E (#sc-481, 1:400; Santa Cruz Biotechnology), p27/kip1 (#610242, 1:1000; Becton Dickinson), cAMP response element-binding protein (CREB; #9197, 1:500; Cell Signaling Technology), phosphorylated CREB (pCREB; #9198, 1:500; Cell Signaling Technology), CRHR (#LS-A6687/41992, 1:250; LifeSpan BioSciences), AVPR1b (#LS-A3737/41899, 1:250; LifeSpan BioSciences), EGFR (SC-03, 1:500; Santa Cruz Biotechnology), AVPR2 (#LS-B8250/135767, 1:250; LifeSpan BioSciences), GR (#12041, 1:400; Cell Signaling Technology), HDAC2 (#ab32117, 1:1600; Abcam), CABLES1 (#ab75535, 1:2000; Abcam), and N-universal negative controls anti-rabbit or anti-mouse (Dako). Signal amplification was performed with the Advance horseradish peroxidase (HRP) link kit (Dako) for 40 minutes and development with the 3,3′-diaminobenzidine (DAB)+ liquid kit (Dako) for 10 minutes. Nuclei were counterstained with Mayer’s hematoxylin for 2 minutes, and bluing was performed for 5 minutes in running tap water. Stained slides were dehydrated in 100% ethyl alcohol and dried at 56°C for 30 minutes before mounting with Entellan.
Semiquantitative analysis of immunoreactivity
Staining intensities were always determined by two independent investigators (I.W. and L.K. or S.S.) and graded 0 (negative), 1 (low), 2 (medium), and 3 (high). The proportion of positive tumor cells was calculated for each specimen and scored 0 if 0% were positive, 0.1 if 0.1% to 9% were positive, 0.5 if 10% to 49% were positive, and 1 if ≥50% were positive. Cytoplasmic and/or membranous staining and nuclear staining were analyzed independently. A semiquantitative H-score was then calculated by the multiplication of the staining-intensity grading score with the proportion score, as described previously (32). In case of discrepant results, both investigators jointly assessed the respective stainings, and the final H-score was formed by consensus.
Cell culture and transfections
To investigate the expression of possible downstream proteins, the murine corticotroph adenoma cell line AtT20 was transfected, not only with different USP8 constructs (WT, P720R) but also with empty vector (mock). Cells were seeded into six-well plates (5 × 105/well) and transfected with 1 µg DNA, 24 hours later, using Effectene (Qiagen), according to the manufacturer’s instructions. USP8 WT and P720R plasmids were donated by Professor Marily Theodoropoulou (Ludwig Maximilian Universität, Munich, Germany) (8). Post-transfection (48 hours), cells were lysed in radioimmunoprecipitation assay buffer, containing protease and phosphatase inhibitors (Sigma).
Immunoblots
Protein lysates (10 µg) were loaded onto polyacrylamide gradient gels (4% to 12%; Bio-Rad) and transferred to a polyvinylidene difluoride membrane. Antibodies were incubated overnight in 5% blocking solution and developed on an X-ray film. The following primary antibodies were used: USP8 (#HPA004869, 1:1000; Sigma), pCREB (#9198, 1:500; Cell Signaling Technology), PKA catalytic subunit α (PKA C α; 5B, 1:1000; Becton Dickinson), α-tubulin (housekeeping gene; DM1A, 1:20,000; Sigma), and secondary antibodies—goat α-rabbit-HRP (1:10,000; Jackson ImmunoResearch) and goat α-mouse-HRP (1:10,000; Jackson ImmunoResearch). Protein-antibody complexes were visualized using the Amersham ECL Prime reagent (GE Healthcare).
Statistical analysis
The Mann-Whitney U test was performed to determine statistically significant differences between two nonparametric datasets. One-way ANOVA test with Tukey post hoc test was done to determine significant differences between different groups of samples. For statistical analyses, GraphPad Prism (version 6.0) was used. P < 0.05 was considered statistically significant.
Results
Protein expression pattern in USP8 WT vs USP8 mutated corticotroph adenomas
As already described in the original report (8), cytoplasmatic expression of USP8 was unchanged between USP8 WT and mutated (mut) tumors [H-score 1.1 ± 0.6 (WT) vs 1.2 ± 0.5 (mut); P = 0.49], but nuclear USP8 was predominant in USP8 mutated tumors [0.7 ± 0.6 (WT) vs 1.6 ± 0.8 (mut); P = 0.0001; Fig. 1(a) and 1(b)]. EGFR levels were similar in the nucleus [0.6 ± 0.6 (WT) vs 0.6 ± 0.8 (mut); P = 0.76], whereas significantly lower in the membrane in USP8 mutated compared with USP8 WT adenomas [1.3 ± 0.7 (WT) vs 0.9 ± 0.3 (mut); P = 0.04; Fig. 1(a) and 1(c)].
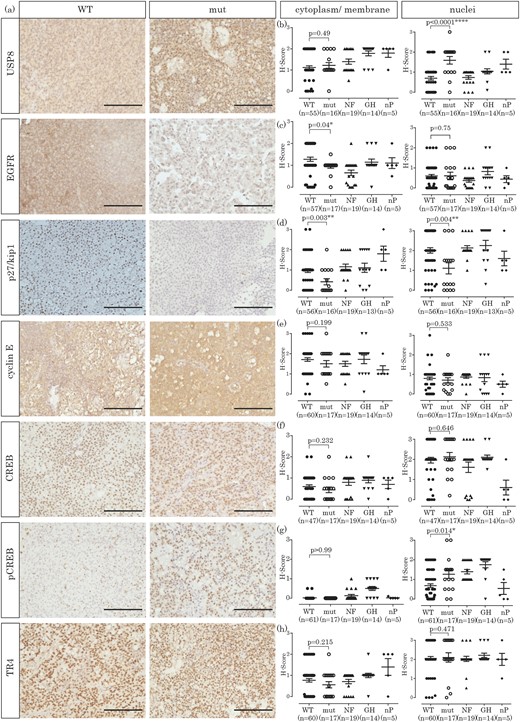
Immunohistochemical investigation of pituitary adenomas (corticotroph, somatotroph, and nonfunctioning) and nP. (a) Exemplary staining of a USP8 WT and a mutated (mut) corticotroph adenoma for each protein investigated (original scale bars, 100 µm). (b) H-Score quantification of USP8 expression, either in cytoplasm and membrane or in the nucleus. (c) H-Score quantification of EGFR expression, either in cytoplasm and membrane or in the nucleus. (d) H-Score quantification of p27/kip1 expression, either in cytoplasm and membrane or in the nucleus. (e) H-Score quantification of cyclin E expression, either in cytoplasm and membrane or in the nucleus. (f) H-Score quantification of CREB expression, either in cytoplasm and membrane or in the nucleus. (g) H-Score quantification of pCREB expression, either in cytoplasm and membrane or in the nucleus. (h) H-Score quantification of TR4 expression, either in cytoplasm and membrane or in the nucleus. GH, growth hormone; NF, nonfunctioning adenomas.
The cell-cycle inhibitor p27/kip1 is significantly decreased in USP8 mutated corticotroph tumors
The CDK inhibitor p27/kip1 has been shown to be downregulated in corticotroph tumors (21, 23–25). With the analysis of USP8 WT and mutated adenomas independently, a reduction in p27/kip1 expression was observed in USP8 mutated adenomas in both the cytoplasm (1 ± 0.7 vs 0.4 ± 0.6; P = 0.003) and the nuclei [2 ± 1 vs 1.1 ± 1.1; P = 0.004; Fig. 1(a) and 1(d)]. Compared with the other tumor entities, p27/kip1 expression was also only reduced in USP8 mutated but not in USP8 WT tumors [Fig. 1(d)]. Another cell-cycle regulator (cyclin E) has been described as overexpressed in corticotroph tumors (19, 20). In our cohort, cyclin E was indeed expressed stronger in all pituitary tumors, independent of their specific hormone secretion compared with the nP. However, its expression in corticotroph tumors was independent of USP8 mutation status [WT vs mut, cytoplasm: 1.7 ± 0.7 vs 1.5 ± 0.7, P = 0.199; nuclei: 0.8 ± 0.6 vs 0.7 ± 0.5, P = 0.533; Fig. 1(a) and 1(e)].
CREB is significantly stronger phosphorylated in USP8 mutated than in USP8 WT corticotroph adenomas
It has been demonstrated that the cAMP/PKA signaling pathway (including CREB) is involved in POMC promoter activation (33) and ACTH release from cells of the anterior pituitary gland (30). Therefore, we investigated the expression of CREB and its activated form, pCREB, both in USP8 WT and USP8 mutated corticotroph tumors. As CREB is a transcription factor, both forms were predominantly localized to the nucleus. However, CREB was also weakly expressed in the cytoplasm [Fig. 1(a) and 1(f)], whereas pCREB was exclusively nuclear [Fig. 1(a) and 1(g)]. Whereas CREB protein levels did not differ between USP8 WT and USP8 mutated tumors [neither in the cytoplasm (0.6 ± 0.5 vs 0.4 ± 0.6; P = 0.23) nor in the nucleus (2 ± 1 vs 2.1 ± 0.9; P = 0.65; Fig. 1(a) and 1(f)], nuclear levels of pCREB were significantly higher in USP8 mutated adenomas (0.7 ± 0.7 vs 1.3 ± 0.9; P = 0.014). Compared with nP, nuclear CREB protein expression was higher in all pituitary tumor entities [Fig. 1(f); P < 0.05], with similar results for pCREB, even if to a lesser extent [Fig. 1(g)]. As CREB is phosphorylated by PKA Cα, we analyzed the expression of the PKA C α in a subset of these tumors (34) and found a stronger expression of this subunit in USP8 mutated tumors compared with USP8 WT tumors. Nevertheless, this difference was not statistically significant.
Expression of TR4 is not changed between corticotroph USP8 WT and mutated tumors
More recently, orphan transcription factor TR4 (also known as NR2C2) was demonstrated to be significantly overexpressed in corticotroph tumors compared with nP glands (16, 17). In our cohort, the expression of TR4 was unchanged between USP8 WT and mutated tumors, both in the cytoplasm (0.8 ± 0.6 vs 0.6 ± 0.6; P = 0.215) and in the nuclei [2.0 ± 0.8 vs 2.1 ± 1; P = 0.471; Fig. 1(a) and 1(h)] or when compared with other pituitary tumors [Fig. 1(h)].
Further differential protein expression analyses in corticotroph USP8 WT and mutated tumors
We also examined the expression of several markers closely related to corticotroph deregulation in corticotroph adenomas and nP. AVPR1b protein levels were extremely low in corticotroph adenomas but were comparable between USP8 WT and USP8 mutated cells [membranous: 0.07 ± 0.2 (WT) vs 0.08 ± 0.1 (mut); P = 0.166 and completely absent expression within the nucleus; Fig. 2(a) and 2(b)]. In contrast, the five nP had significantly higher protein levels of AVPR1b compared with corticotroph adenomas independent of their mutation status (cytoplasmatic/membranous: 1.8 ± 0.4 vs 0.07 ± 0.2; P < 0.0001) (34). Expression of AVPR2 was, in general, relatively high: its expression varied from H-score 0 to 3 in both USP8 WT and mutated corticotroph tumors [Fig. 2(a) and 2(c)]. There was no significant difference in AVPR2 expression between USP8 WT and mutated corticotroph adenomas in either cytoplasm [1.5 ± 1 (WT) vs 1.5 ± 1.2 (mut); P = 0.94] or nucleus [0.1 ± 0.3 (WT) vs 0.1 ± 0.3 (mut); P = 0.66].
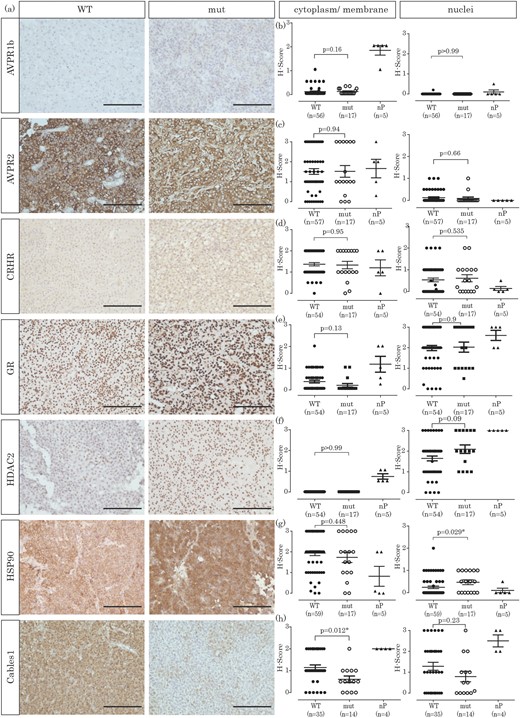
Immunohistochemical investigation of corticotroph pituitary adenomas and nP. (a) Exemplary staining of a USP8 WT and a mutated corticotroph adenoma for each protein investigated (original scale bars, 100 µm). (b) H-Score quantification of AVPR1b expression, either in cytoplasm and membrane or in the nucleus. (c) H-Score quantification of AVPR2 expression, either in cytoplasm and membrane or in the nucleus. (d) H-Score quantification of CRHR expression, either in cytoplasm and membrane or in the nucleus. (e) H-Score quantification of GR expression, either in cytoplasm and membrane or in the nucleus. (f) H-Score quantification of HDAC2 expression, either in cytoplasm and membrane or in the nucleus. (g) H-Score quantification of HSP90 expression, either in cytoplasm and membrane or in the nucleus. (h) H-Score quantification of CABLES1 expression, either in cytoplasm and membrane or in the nucleus.
H-Scores for CRHR expression were also largely unaffected by the USP8 mutation status, with similar expression levels between USP8 WT and USP8 mutated corticotroph adenomas [membranous: 1.4 ± 0.6 (WT) vs 1.3 ± 0.7 (mut); P = 0.95; Fig. 2(a) and 2(d)].
When we investigated the expression of the GR itself in corticotroph adenomas and nP, we did not observe significant differences between USP8 WT and mutated adenomas [Fig. 2(a) and 2(e)]. Cytoplasmatic staining was very low in corticotroph tumors [0.3 ± 0.5 (WT) vs 0.1 ± 0.3 (mut); P = 0.13], whereas nuclei staining was high in most samples [2 ± 0.9 (WT) vs 2 ± 1 (mut); P = 0.9].
The expression of HDAC2 in corticotroph adenomas was previously shown to be reduced, resulting in glucocorticoid resistance and promotion of CD (18). In our cohort, HDAC2 expression was exclusively nuclear [Fig. 2(a) and 2(f)], with only slightly higher expression in USP8 mutated adenomas compared with USP8 WT adenomas [1.6 ± 0.9 (WT) vs 2.1 ± 0.8 (mut); P = 0.09].
Expression of HSP90 is increased in USP8 mutated tumors
More recently, HSP90, a chaperone involved in proper GR folding, was found to be overexpressed in corticotroph adenomas (16).
To investigate whether this could be a result of mutation-induced USP8 hyperactivity, we analyzed HSP90 expression. HSP90 was not differentially expressed between USP8 WT and mutated adenomas in the cytoplasm [1.9 ± 0.9 (WT) vs 1.7 ± 1.0 (mut); P = 0.448], however its expression was significantly increased in the nucleus of mutated adenomas [0.2 ± 0.4 (WT) vs 0.5 ± 0.4 (mut); P = 0.029; Fig. 2(a) and 2(g)]. As observed by Riebold et al. (16), HSP90 expression was higher in corticotroph tumors than in nP.
Expression of CABLES1 is reduced in USP8 mutated tumors
It has been reported that expression of the negative cell-cycle regulator CABLES1 is absent in approximately one-half of corticotroph tumors and that CABLES1 expression correlates with p27/kip1 expression (26). When we analyzed the expression of CABLES1 in corticotroph tumors with known USP8 mutation status, we observed a reduction in CABLES1 expression in mutated adenomas compared with USP8 WT adenomas, both in the cytoplasm and in the nucleus. However, this reduction was only significant in the cytoplasm [1.1 ± 0.7 (WT) vs 0.6 ± 0.6 (mut); P = 0.012] but not in the nucleus [1.3 ± 1.1 (WT) vs 0.8 ± 0.9 (mut); P = 0.23; Fig. 2(a) and 2(h)].
USP8 mutation increases the levels of pCREB in the corticotroph adenoma cell line AtT20
In USP8 mutated adenomas, we did not only observe increased levels of pCREB [Fig. 1(g)] but also observed a tendency for increased nuclear expression of PKA C α in USP8 mutated adenomas (34). To investigate a direct link with USP8 mutations, we transiently transfected the murine corticotroph adenoma cell line AtT20 with USP8 WT, USP8 P720R mutant, and empty vector (mock). Cells transfected with both USP8 plasmids showed an increased expression of USP8 [Fig. 3(a) and 3(b)]. Furthermore, cells transfected with the mutated form of USP8 showed increased levels of pCREB (1.3 ± 0.14 vs 1 ± 0.23 vs 1.27 ± 0.12) and PKA C α (1.3 ± 0.12 vs 1 ± 0.38 vs 1.2 ± 0.34) compared with cells transfected with USP8 WT or empty vector [Fig. 3(a), 3(c), and 3(d)].
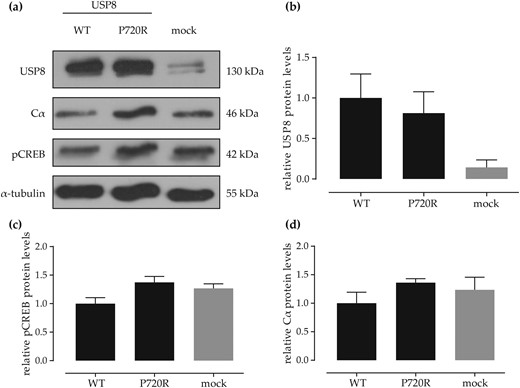
Western blot (WB) results of USP8 WT and USP8 P720R mut introduction in AtT20 cells. (a) Exemplary WB of transfected AtT20 cells. (b) Densitometric analysis of WB USP8 bands normalized to loading control (α-tubulin) compared with mock transfected cells. (c) Densitometric analysis of WB pCREB bands normalized to loading control compared with mock transfected cells. (d) Densitometric analysis of WB PKA Cα bands normalized to loading control compared with mock transfected cells. Analysis of three independent replicates. Overall P = not significant.
Discussion
In recent years, two types of molecular alterations have been reported in the context of CD: altered expression of different proteins (2, 35) on one hand and hotspot mutations in the deubiquitinase gene USP8 on the other (with the latter occurring in approximately one-half of the cases) (8, 10, 28). The underlying molecular causes for the alterations in protein expression, however, have often remained elusive.
As a starting point of our study, we hypothesized that activating mutations in USP8 might be responsible for some of these changes. To test this hypothesis, the most relevant of the previously reported deregulated proteins were investigated immunohistochemically. With the observation of proteins that are differentially expressed between USP8 WT and USP8 mutated adenomas, which is possibly related to an increased recycling induced by USP8 activating mutations, we aimed to identify potential USP8 client proteins.
The CDK inhibitor p27/kip1, which is involved in cell-cycle arrest (36), has often been demonstrated to be downregulated in corticotroph adenomas (23–25). In our series, we observed reduced protein expression of p27/kip1 predominantly in USP8 mutated adenomas, which was significantly reduced when compared with USP8 WT tumors. A downregulation of p27/kip1 is likely to result in increased cell-cycle progression and probably contributes to cell proliferation. Furthermore, reduced levels of p27/kip1 in corticotroph tumors have been linked to phosphorylated EGFR at the amino acid residue Tyr992 (21), which is able to bind phospholipase Cγ (37). The latter though has been shown to facilitate the export of p27/kip1 from the nucleus into the cytoplasm, where it is then degraded (38). In addition, reduced expression of p27/kip1 was shown to correlate with reduced expression of CABLES1, a negative regulator of cell-cycle progression that is likely a direct glucocorticoid target (26). In our cohort, we could confirm this correlation, especially with mutated corticotroph adenomas, showing significantly lower CABLES1 expression contributing to glucocorticoid resistance (Fig. 2). Although the phosphorylation status of EGFR in USP8 mutated cells has not been assessed so far, its expression in USP8 mutated tumors was shown to be increased at least by some groups (8, 28, 29). In our cohort, EGFR expression was even reduced in USP8 mutated tumors compared with WT, making the theory that USP8 hyperactivity and EGFR recycling directly correlate even more questionable. USP8 mutations seem to be accompanied by reduced levels of p27/kip1; however, the reason for this change remains elusive, as there is no indication that p27/kip is regulated through ubiquitination. One might speculate that another protein is deubiquitinated by USP8, thereby leading to an increased abundance and reduced p27/kip1 levels. To test if USP8 is in close proximity to p27/kip1, we performed a proximity ligation assay in a small subset of these tumors. Only in USP8, mutated adenomas USP8 and p27/kip1 are located in close proximity, possibly in the same complex but not in USP8 WT adenomas (34). A correlation between reduced levels of p27/kip1 and USP8 mutations is therefore likely.
It has been repeatedly demonstrated that mutated USP8 is predominantly located within the nucleus. Consequently, the transcription factor CREB appeared to be a good candidate for direct regulation by USP8. CREB is not only localized in the nucleus but also phosphorylated by PKA; furthermore, it is involved in POMC promoter activation (33). Thus, we investigated CREB in both its unphosphorylated (i.e., inactive) and its phosphorylated (i.e., active) form [Fig. 1(a), 1(f), and 1(g)]. With respect to the nucleus, a substantial increase of pCREB was observed in USP8 mutated tumors compared with USP8 WT tumors (Fig. 1). It has been outlined that only pCREB at serine 133 is able to bind cAMP-response elements for induction of POMC promoter activation (39). As the expression of unphosphorylated CREB was not altered between USP8 WT and mutated adenomas, it can be hypothesized that PKA, which phosphorylates CREB (40), is deregulated. With the investigation of the expression of PKA C α (PRKACA) in a subset of pituitary adenomas, we found a trend toward increased levels of this protein in USP8 mutated adenomas in the nucleus. However, possibly as a result of the small sample size (n = 21), this discrepancy was statistically not significant. Introduction of the P720R USP8 mutation into AtT20 corticotroph cells increased the levels of pCREB [Fig. 3(a) and 3(b)], suggesting that either pCREB or PKA is a direct client protein of USP8. The increased levels of pCREB could lead, in turn, to enhanced ACTH secretion by activation of the transcription of the POMC promoter (Fig. 4).
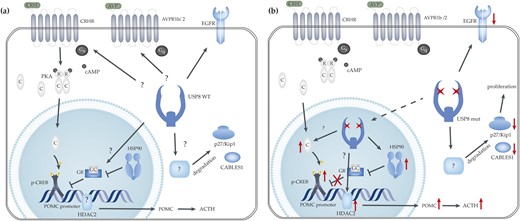
Proposed action of USP8 in corticotroph pituitary adenomas. (a) USP8 WT conditions. To date, it has not been ascertained if the three receptors AVPR1b /2 and CRHR are deubiquitinated by USP8 or not. The POMC promoter is normally activated by several transcription factors (including pCREB) and is inhibited through binding of the GR. HDAC2 deacetylates histone H4 within the POMC promoter, leading to reduced transcription of the POMC gene. (b) Mutated USP8 is predominantly located in the nucleus, possibly allowing for targets that are also located within the nucleus. Here, HSP90 or pCREB expression is possibly higher as a result of USP8 hyperactivity. An increase in pCREB could lead to increased POMC expression and ACTH secretion. Increased HSP90 in the nucleus might be a result of increased USP8 activity in the nucleus. This leads to increased HSP90-GR binding and reduced inhibition of the POMC promoter, resulting in increased ACTH secretion. Strongly reduced levels of p27/kip1 are associated with USP8 mutations. This observation is also true for CABLES1, however neither p27/kip1 nor CABLES1 seems to be direct USP8 client proteins. Black, solid arrow-headed lines, without further specification, symbolize USP8 deubiquitination action; black bar-headed lines demonstrate a blockage of normal protein activity; dashed arrow-headed lines illustrate protein translocation; red arrow-headed lines symbolize protein upregulation; and red crosses symbolize a block in activity.
The chaperone HSP90 was recently demonstrated to be overexpressed in corticotroph adenomas (16), resulting in increased inhibition of GR activity through HSP90-GR binding. Accordingly, GR is no longer able to inhibit efficiently POMC promoter activity and ACTH secretion (41). In the cytoplasm of corticotroph adenomas, we did not observe differences in HSP90 protein abundance; however, we found significantly higher protein expression of HSP90 in the nuclei of USP8 mutated adenomas [Fig. 2(a) and 2(g)]. As mutated USP8 also shows a nuclear localization, one could speculate that HSP90 is a nuclear client protein of USP8, as HSP90 has already been demonstrated to bind GR and is localized in the nucleus (42). Interestingly, the possible inhibition of GR action by increased HSP90 protein levels in the context of USP8 mutations could be therapeutically relevant. Riebold and colleagues (16) showed that inhibition of HSP90 with silibinin reduced ACTH secretion of AtT20 cells, both in vitro and in vivo, making this inhibitor particularly interesting for patients with CD tumors carrying USP8 mutations.
One of the proteins where we observed differences between nP and corticotroph adenomas was the AVPR1b, which in normal conditions, is activated by vasopressin, thereby potentiating the stimulating effects of CRH on ACTH secretion (43). AVPR1b is the vasopressin receptor predominantly expressed on corticotroph cells of nP and has been shown to be overexpressed in corticotroph adenomas (11–13). In contrast, we observed a complete loss of the AVPR1b in corticotroph adenomas compared with nP tissue in our series. This might be a result of either the small reference sample size (only five nP) or (probably more likely) the fact that our study investigated AVPR1b expression at the protein level, whereas all other studies analyzed mRNA expression only (11–13). The decrease of AVPR1b might be a result of increased vasopressin stimulation in corticotroph adenomas compared with nP and somatotroph adenomas. We are well aware that this is not in line with the observed responsiveness of corticotroph adenomas (but not of other pituitary tumors) to desmopressin, a synthetic AVP analog that binds all three AVP receptors (13). Nevertheless, it is well suitable to the observation that AVPR1b may be expressed in primary corticotropinoma cell cultures, although the patients did not respond to desmopressin (44). Of note, both USP8 WT and mutated samples had similarly low levels, indicating that this receptor might not be a direct substrate for USP8. Interestingly, the AVPR2 was much stronger and differentially expressed between different corticotroph tumors, with tumors being characterized by either high or low AVPR2 expression, however, independent of the USP8 mutation status [Fig. 2(a) and 2(c)]. As desmopressin also binds this AVP receptor subtype, its high expression, at least in some corticotroph tumors, could possibly explain the responsiveness of corticotroph tumors to desmopressin. In addition, we observed reduced expression of HDAC2 in corticotroph tumors compared with nP [Fig. 2(a) and 2(f)], as already described elsewhere (18). This reduction was not as pronounced in USP8 mutated as in WT tumors, however we can only speculate that this might be a result of USP8 hyperactivity in mutated adenomas.
However, all other proteins that have been demonstrated to be overexpressed in corticotroph adenomas compared with nP (including GR, CRHR, cyclin E, and TR4) were unchanged between USP8 WT and mutated adenomas (Figs. 1 and 2). Although the activating role of CRHR and TR4 in ACTH secretion is well known, both are probably no client proteins of USP8; their overexpression in all corticotroph adenomas may have a different causality instead. For TR4, we also could not corroborate the results generated by Du et al. (17), who described an overexpression in corticotroph adenomas.
Strengths and limitations
To our knowledge, this is the largest study of corticotroph tumors with known USP8 mutation status in which 14 different corticotroph-relevant proteins were investigated. As a result of the rarity of the tumor entity, samples had been collected multicentrically over the past decades. However, clinical follow-up of patients was not standardized and potentially interesting clinical data (e.g., on remission rates or responsiveness to desmopressin) were unfortunately not available.
Conclusion
To summarize, our results demonstrate a possible role of USP8 mutations in the regulation of protein levels for several markers associated with corticotroph adenomas, most notably, p27/kip1, CABLES1, HSP90, and activated CREB (Fig. 4), all of which have already been described as contributors to either ACTH hypersecretion or increased cell proliferation and CD (16). These findings might have clinical relevance, as an array of existing inhibitors could be used to tailor the best therapy for CD patients with USP8 mutations, especially the HSP90 inhibitor silibinin, which is in clinical use and has been shown to have an excellent safety record (45).
Acknowledgments
We thank Professor Marily Theodoropoulou (Ludwig Maximilian Universität, Munich, Germany) for generously donating the USP8 WT and P720R plasmids.
Financial Support: This work has been supported by grants from the German Research Foundation (DGE) [Grant SB 52/1-1 (to S.S.), Grant FA 466/5-1 (to M.F.), Grant DE 2657/1-1 (to T.D.), and SFB Transregio CRC/TRR 205/1 (to M.F.)].
Disclosure Summary: The authors have nothing to disclose.
Abbreviations:
- ACTH
adrenocorticotropic hormone
- AVPR1b
arginine vasopressin receptor 1b
- AVPR2
arginine vasopressin receptor 2
- CABLES1
cyclin-dependent kinase 5 and Abelson murine leukemia viral oncogene homolog 1 enzyme substrate 1
- CD
Cushing disease
- CDK
cyclin-dependent kinase
- CREB
cAMP response element-binding protein
- CRHR
corticotropin-releasing hormone receptor
- EGFR
epidermal growth factor receptor
- FFPE
formalin-fixed paraffin-embedded
- GR
glucocorticoid receptor
- HDAC2
histone deacetylase 2
- HRP
horseradish peroxidase
- HSP90
heat shock protein 90
- mut
mutated
- nP
normal pituitaries
- pCREB
phosphorylated cAMP response element-binding protein
- PKA
protein kinase A
- PKA C α
PKA catalytic subunit α
- POMC
pro-opiomelanocortin
- TR4
orphan nuclear receptor 4
- USP8
ubiquitin-specific peptidase 8
- WT
wild type

{kind=link}
{kind=link}
{kind=link}
{kind=link}