





Abstract
Malignant pheochromocytoma and paraganglioma (MPP) are characterized by prognostic heterogeneity. Our objective was to look for prognostic parameters of overall survival (OS) in MPP patients.
Retrospective multicenter study of MPP characterized by a neck-thoraco-abdomino-pelvic CT or MRI at the time of malignancy diagnosis in European centers between 1998 and 2010.
One hundred sixty-nine patients from 18 European centers were included. Main characteristics of patients with MPP were: primary pheochromocytoma in 53% of patients; tumor- or hormone-related symptoms in 57% or 58% of cases; positive plasma or urine hormones in 81% of patients; identification of a mutation in SDHB in 42% of cases. Metastatic sites included bone (64%), lymph node (40%), lung (29%), and liver (26%); mean time between initial and malignancy diagnosis was 43 months (range, 0 to 614). Median follow-up was 68 months and median survival 6.7 years. Using univariate analysis, better survival was associated with head and neck paraganglioma, age <40 years, metanephrines less than fivefold the upper limits of the normal range, and low proliferative index. In multivariate analysis, hypersecretion [hazard ratio 3.02 (1.65 to 5.55); P = 0.0004] was identified as an independent significant prognostic factor of worst OS.
Our results do not confirm SDHB mutations as a major prognostic parameter in MPP and suggest additional key molecular events involved in MPP tumor progression. Aside from SDHB mutation, the biology of aggressive MPP remains to be understood.
Pheochromocytoma and paraganglioma are rare neuroendocrine chromaffin tumors located at adrenal or extra-adrenal sites and defined as malignant by the World Health Organization by the occurrence of metastases in nonchromaffin organs (1). Malignant pheochromocytoma and paraganglioma (MPP) incidence is less than one case per million population per year (2). MPP are characterized by their heterogeneity including primary site location, genetic predisposition, hormonal secretion, and metastatic organs (3). In addition, we recently demonstrated that half of patients with MPP have stable disease at one year without any therapeutic intervention (4). Finally, heterogeneous survival has been described in the literature as ranging from 40% to 77% at five-year diagnosis (5–7) and heterogeneous progression-free survival ranged from 4 to 36 months in therapeutic trials (8–11).
Pheochromocytoma and paraganglioma occur as part of inherited syndrome in 40% of cases with up to 12 genes found to be related to this disease in the past two decades (12). Germline mutations in succinate dehydrogenase subunit B have been associated with higher rate of metastatic disease as well as rare mutations in FH, MAX, and SLC25A11 (13–17). Ten years ago, we reported that the presence or absence of SDHB mutation was associated with distinct median survival of 42 or 244 months in adults (5). The presence of the SDHB mutation was also associated with a low metastases-free survival in a pediatric series (18). However, these results have not been confirmed in more recent reports (19, 20). In addition, other prognostic factors have been been highlighted including age (7, 19, 21), extra-adrenal location (7, 22–24), primary tumor size (7, 19, 22, 24), or synchronous metastatic disease (7, 19–21).
In this study, we retrospectively searched for prognostic factors of overall survival (OS) in a large series of patients with MPP followed within the European Network for the Study of Adrenal Tumors (ENS@T).
Patients and Methods
Patients
The medical files of patients with MPP who were followed up between January 1998 and December 2010 were reviewed in 18 centers of ENS@T. Clinical data were entered in the ENS@T database by each center and then data were extracted and reviewed by one investigator (S.H.). Informed consent was obtained from all patients. Patients with the following criteria were included in the MetAstatic Pheochromocytoma and Paraganglioma PRONOstic (MAPP-Prono) study: (i) confirmed diagnosis of MPP (including pheochromocytoma, abdominal, thoracic, or head and neck paraganglioma) as defined by the presence of distant metastasis between 1998 and 2010; (ii) neck-thoracic-abdomen-pelvic CT (TAP CT) performed within four months of metastasis diagnosis; and (iii) entire follow-up in the center. Exclusion criteria were (i) benign pheochromocytoma and paraganglioma and (ii) patients with venous or loco-regional or proximal lymph node spread only.
The following parameters were recorded at the time of first TAP CT in the metastatic setting: gender, age, genetic status of dedicated genes including SDHB mutation analysis according to available guidelines at the time of patient management, location and size of the primary tumor, metastatic sites (bone, lymph nodes, lung, and liver), presence of hormone- or tumor-related symptoms, chromogranin A and total metanephrine levels, the mitotic count and Ki67, and the interval between the initial diagnosis and that of metastasis. Hormonal secretion was defined as elevated chromogranin A and/or metanephrines (i.e., twofold the upper limits of the normal range). Causes of death, related or not to MPP, and status at last follow-up were recorded. The description and cutoff values of each parameter as well as the number of patients evaluable for each parameter are provided in Table 1.
Demographics and Baseline Characteristics of Patients at T0
| Characteristics | N Evaluable n (%) | Total n (%) |
|---|---|---|
| Number | 169 | |
| Gender ratio, male/female | 169 (100) | 93/76 (55/45) |
| Age at T0 | 169 (100) | 48 y ± 16 |
| Primary tumor site | 169 (100) | |
| Adrenal | 90 (53) | |
| Extra-adrenal | 79 (47) | |
| Tumor-related symptoms | 169 (100) | 94 (57) |
| Hormone-related symptoms | 169 (100) | 96 (58) |
| Hypersecretiona | 145 (86) | 117 (81) |
| Pathology | ||
| Size >5 cm | 144 (85) | 109 (76) |
| Mitotic count >3/10 HPF | 83 (49) | 22 (27) |
| Ki67 | ||
| 2%–10% | 19 (40) | |
| >10% | 47 (28) | 18 (38) |
| Time between initial diagnosis and malignancy | 169 (100) | |
| M1 within a year | 79 (47) | |
| M1 >5 y | 47 (28) | |
| Metastatic site | 169 (100) | |
| Bones | 108 (64) | |
| Lymph nodes | 67 (40) | |
| Lung | 49 (29) | |
| Liver | 44 (26) | |
| FDG-PET | 98 (58) | |
| Positive | 94 (96) | |
| Genetics | 151 (89) | |
| No mutation | 69 (46) | |
| NF1 clinical screening | 4 | |
| MEN2 | 3 | |
| VHL | 4 | |
| SDHB | 63 (42) | |
| SDHC | 1 | |
| SDHD | 6 | |
| MDH2 | 1 |
| Characteristics | N Evaluable n (%) | Total n (%) |
|---|---|---|
| Number | 169 | |
| Gender ratio, male/female | 169 (100) | 93/76 (55/45) |
| Age at T0 | 169 (100) | 48 y ± 16 |
| Primary tumor site | 169 (100) | |
| Adrenal | 90 (53) | |
| Extra-adrenal | 79 (47) | |
| Tumor-related symptoms | 169 (100) | 94 (57) |
| Hormone-related symptoms | 169 (100) | 96 (58) |
| Hypersecretiona | 145 (86) | 117 (81) |
| Pathology | ||
| Size >5 cm | 144 (85) | 109 (76) |
| Mitotic count >3/10 HPF | 83 (49) | 22 (27) |
| Ki67 | ||
| 2%–10% | 19 (40) | |
| >10% | 47 (28) | 18 (38) |
| Time between initial diagnosis and malignancy | 169 (100) | |
| M1 within a year | 79 (47) | |
| M1 >5 y | 47 (28) | |
| Metastatic site | 169 (100) | |
| Bones | 108 (64) | |
| Lymph nodes | 67 (40) | |
| Lung | 49 (29) | |
| Liver | 44 (26) | |
| FDG-PET | 98 (58) | |
| Positive | 94 (96) | |
| Genetics | 151 (89) | |
| No mutation | 69 (46) | |
| NF1 clinical screening | 4 | |
| MEN2 | 3 | |
| VHL | 4 | |
| SDHB | 63 (42) | |
| SDHC | 1 | |
| SDHD | 6 | |
| MDH2 | 1 |
Figures represent the number of evaluable patients (%) or means ± SD.
Abbreviation: FDG-PET, fludeoxyglucose–positron emission tomography.
Hypersecreting tumor is defined as chromogranin A and/or metanephrines > 2N.
Demographics and Baseline Characteristics of Patients at T0
| Characteristics | N Evaluable n (%) | Total n (%) |
|---|---|---|
| Number | 169 | |
| Gender ratio, male/female | 169 (100) | 93/76 (55/45) |
| Age at T0 | 169 (100) | 48 y ± 16 |
| Primary tumor site | 169 (100) | |
| Adrenal | 90 (53) | |
| Extra-adrenal | 79 (47) | |
| Tumor-related symptoms | 169 (100) | 94 (57) |
| Hormone-related symptoms | 169 (100) | 96 (58) |
| Hypersecretiona | 145 (86) | 117 (81) |
| Pathology | ||
| Size >5 cm | 144 (85) | 109 (76) |
| Mitotic count >3/10 HPF | 83 (49) | 22 (27) |
| Ki67 | ||
| 2%–10% | 19 (40) | |
| >10% | 47 (28) | 18 (38) |
| Time between initial diagnosis and malignancy | 169 (100) | |
| M1 within a year | 79 (47) | |
| M1 >5 y | 47 (28) | |
| Metastatic site | 169 (100) | |
| Bones | 108 (64) | |
| Lymph nodes | 67 (40) | |
| Lung | 49 (29) | |
| Liver | 44 (26) | |
| FDG-PET | 98 (58) | |
| Positive | 94 (96) | |
| Genetics | 151 (89) | |
| No mutation | 69 (46) | |
| NF1 clinical screening | 4 | |
| MEN2 | 3 | |
| VHL | 4 | |
| SDHB | 63 (42) | |
| SDHC | 1 | |
| SDHD | 6 | |
| MDH2 | 1 |
| Characteristics | N Evaluable n (%) | Total n (%) |
|---|---|---|
| Number | 169 | |
| Gender ratio, male/female | 169 (100) | 93/76 (55/45) |
| Age at T0 | 169 (100) | 48 y ± 16 |
| Primary tumor site | 169 (100) | |
| Adrenal | 90 (53) | |
| Extra-adrenal | 79 (47) | |
| Tumor-related symptoms | 169 (100) | 94 (57) |
| Hormone-related symptoms | 169 (100) | 96 (58) |
| Hypersecretiona | 145 (86) | 117 (81) |
| Pathology | ||
| Size >5 cm | 144 (85) | 109 (76) |
| Mitotic count >3/10 HPF | 83 (49) | 22 (27) |
| Ki67 | ||
| 2%–10% | 19 (40) | |
| >10% | 47 (28) | 18 (38) |
| Time between initial diagnosis and malignancy | 169 (100) | |
| M1 within a year | 79 (47) | |
| M1 >5 y | 47 (28) | |
| Metastatic site | 169 (100) | |
| Bones | 108 (64) | |
| Lymph nodes | 67 (40) | |
| Lung | 49 (29) | |
| Liver | 44 (26) | |
| FDG-PET | 98 (58) | |
| Positive | 94 (96) | |
| Genetics | 151 (89) | |
| No mutation | 69 (46) | |
| NF1 clinical screening | 4 | |
| MEN2 | 3 | |
| VHL | 4 | |
| SDHB | 63 (42) | |
| SDHC | 1 | |
| SDHD | 6 | |
| MDH2 | 1 |
Figures represent the number of evaluable patients (%) or means ± SD.
Abbreviation: FDG-PET, fludeoxyglucose–positron emission tomography.
Hypersecreting tumor is defined as chromogranin A and/or metanephrines > 2N.
Statistical analysis
Quantitative values are reported as means ± SD or median interquartile range (IQR) and categorical variables as percentages. Differences according to patients’ characteristics were assessed using the χ2 test or nonparametric Mann-Whitney U tests. A P value < 0.05 was considered significant. The primary end point of the study was OS defined by time from diagnosis of MPP (first thorax-abdomen or head and neck CT in the metastatic setting) (time zero: T0) to death by any cause. Specific survival was also analyzed. OS was estimated using Kaplan-Meier curves. Prognostic factors for OS were evaluated using the log-rank test in univariate analysis. Factors validated in univariate analysis were further tested in multivariate analysis. Results are reported as hazard ratios with 95% CI. All analyses were conducted using SAS software (version 9.1; SAS Institute, Cary, NC).
Results
Population
Of the 222 patients identified as included in the MAPP-Prono study in the ENS@T database, reasons for exclusion were diagnosis of metastatic disease before 1998 or after 2010 in 6 or 38 cases, respectively, benign tumor in 1 case, and proximal metastatic lymph nodes only in 8 cases; 169 patients were included.
The median time between initial diagnosis and T0 was 43 months (range, 0 to 614). The metastases were diagnosed within the first year in 79 patients (47%). A delayed diagnosis was observed with a time since the initial diagnosis longer than 5 and 10 years in 47 (28%) and 26 (15%) of the patients, respectively.
At T0, a slight predominance of male gender was found (55%) and the mean age of the cohort was 48 ± 16 years (range, 10 to 80). The primary tumor was located in the adrenal in 90 patients (53%) and at extra-adrenal sites in 79 patients (47%) including abdomen/pelvis or thoracic/neck in 63 and 16 patients, respectively. Twenty-two patients (13%) had multiple primary tumors. Ninety-four patients (57%) had tumor-related symptoms at presentation and 96 had hormone-related symptoms (58%). Fifty-three patients (32%) presented with both tumor- and hormone-related symptoms at T0 whereas 29 (17%) had no symptoms. Hormonal secretion as defined by an excess of chromogranin A and or metanephrine/normetanephrine was reported in 117 of 145 evaluable patients (81%) and elevated metanephrine/normetanephrine was found in 100 patients (72%). The size of the primary tumor was available for 144 patients and was larger than 5 cm in 109 (76%). In patients with available pathological reports, the mitotic count exceeded 3 per high-power field (HPF) in 22 of 83 patients (27%) and Ki67 was measured as follows: between 2% and 10% in 19 patients (40%) and more than 10% in 18 patients (38%) of 47 patients. Bone was the most frequent site of metastases (64%) and 35 patients (21%) had isolated bone metastases at presentation. The other metastatic organs were distant lymph nodes (40%), lung (29%), and liver (26%). Seventy-three patients (43%) had metastases in both bone and soft tissues whereas 61 patients (36%) had no bone metastases. Fludeoxyglucose–positron emission tomography was positive for 94 of 98 patients (96%). Of the four patients with negative fludeoxyglucose–positron emission tomography, two had a neurofibromatosis type 1 (NF1), and two had an apparently sporadic disease. Characteristics of patients according to their primary location are reported in Table 2: patients with malignant pheochromocytoma were characterized by less frequent genetic disease, tumor-related symptoms, or bone metastasis compared with malignant paraganglioma.
Characterization of Patients According to the Primary Tumor
| Characteristics | Adrenal N (%) | AbdoP PGL N (%) | HN PGL N (%) |
|---|---|---|---|
| N | 90 | 63 | 16 |
| Male | 66 (73) | 35 (56) | 6 (37) |
| Age at T0, y | 48,2 | 48,3 | 47,2 |
| M1 <1 y | 39 (43) | 33 (52) | 7 (44) |
| Genetics | |||
| N evaluable | 76 | 59 | 16 |
| Sporadic | 52 (68) | 13 (21) | 4 (25) |
| SDHB | 12 (13) | 42 (67) | 9 (56) |
| SDHC / SDHD / MDH2 | 0 / 1 / 0 | 1 / 2 / 1 | 0 / 3 / 0 |
| VHL / MEN2 / NF1 | 4 / 3 / 4 | 0 | 0 |
| Tumor-related syndrome | 41/87 (47) | 40 (63) | 13 (81) |
| Hormone-related syndrome | 53/87 (61) | 39 (62) | 4 (25) |
| Hypersecretiona | 71/80 (89) | 42/53 (79) | 3/12 (25) |
| Metastases | |||
| Bone only | 14 (16) | 15 (24) | 6 (37) |
| Soft tissue only | 49 (54) | 19 (30) | 5 (31) |
| Characteristics | Adrenal N (%) | AbdoP PGL N (%) | HN PGL N (%) |
|---|---|---|---|
| N | 90 | 63 | 16 |
| Male | 66 (73) | 35 (56) | 6 (37) |
| Age at T0, y | 48,2 | 48,3 | 47,2 |
| M1 <1 y | 39 (43) | 33 (52) | 7 (44) |
| Genetics | |||
| N evaluable | 76 | 59 | 16 |
| Sporadic | 52 (68) | 13 (21) | 4 (25) |
| SDHB | 12 (13) | 42 (67) | 9 (56) |
| SDHC / SDHD / MDH2 | 0 / 1 / 0 | 1 / 2 / 1 | 0 / 3 / 0 |
| VHL / MEN2 / NF1 | 4 / 3 / 4 | 0 | 0 |
| Tumor-related syndrome | 41/87 (47) | 40 (63) | 13 (81) |
| Hormone-related syndrome | 53/87 (61) | 39 (62) | 4 (25) |
| Hypersecretiona | 71/80 (89) | 42/53 (79) | 3/12 (25) |
| Metastases | |||
| Bone only | 14 (16) | 15 (24) | 6 (37) |
| Soft tissue only | 49 (54) | 19 (30) | 5 (31) |
Figures represent the number of evaluable patients (%), means ± SD, or median (IQR).
Abbreviations: AbdoP PGL, abdomino-pelvic paraganglioma; HN PGL, head and neck paraganglioma.
Hypersecreting tumor is defined as chromogranin A and/or metanephrines >2N.
Characterization of Patients According to the Primary Tumor
| Characteristics | Adrenal N (%) | AbdoP PGL N (%) | HN PGL N (%) |
|---|---|---|---|
| N | 90 | 63 | 16 |
| Male | 66 (73) | 35 (56) | 6 (37) |
| Age at T0, y | 48,2 | 48,3 | 47,2 |
| M1 <1 y | 39 (43) | 33 (52) | 7 (44) |
| Genetics | |||
| N evaluable | 76 | 59 | 16 |
| Sporadic | 52 (68) | 13 (21) | 4 (25) |
| SDHB | 12 (13) | 42 (67) | 9 (56) |
| SDHC / SDHD / MDH2 | 0 / 1 / 0 | 1 / 2 / 1 | 0 / 3 / 0 |
| VHL / MEN2 / NF1 | 4 / 3 / 4 | 0 | 0 |
| Tumor-related syndrome | 41/87 (47) | 40 (63) | 13 (81) |
| Hormone-related syndrome | 53/87 (61) | 39 (62) | 4 (25) |
| Hypersecretiona | 71/80 (89) | 42/53 (79) | 3/12 (25) |
| Metastases | |||
| Bone only | 14 (16) | 15 (24) | 6 (37) |
| Soft tissue only | 49 (54) | 19 (30) | 5 (31) |
| Characteristics | Adrenal N (%) | AbdoP PGL N (%) | HN PGL N (%) |
|---|---|---|---|
| N | 90 | 63 | 16 |
| Male | 66 (73) | 35 (56) | 6 (37) |
| Age at T0, y | 48,2 | 48,3 | 47,2 |
| M1 <1 y | 39 (43) | 33 (52) | 7 (44) |
| Genetics | |||
| N evaluable | 76 | 59 | 16 |
| Sporadic | 52 (68) | 13 (21) | 4 (25) |
| SDHB | 12 (13) | 42 (67) | 9 (56) |
| SDHC / SDHD / MDH2 | 0 / 1 / 0 | 1 / 2 / 1 | 0 / 3 / 0 |
| VHL / MEN2 / NF1 | 4 / 3 / 4 | 0 | 0 |
| Tumor-related syndrome | 41/87 (47) | 40 (63) | 13 (81) |
| Hormone-related syndrome | 53/87 (61) | 39 (62) | 4 (25) |
| Hypersecretiona | 71/80 (89) | 42/53 (79) | 3/12 (25) |
| Metastases | |||
| Bone only | 14 (16) | 15 (24) | 6 (37) |
| Soft tissue only | 49 (54) | 19 (30) | 5 (31) |
Figures represent the number of evaluable patients (%), means ± SD, or median (IQR).
Abbreviations: AbdoP PGL, abdomino-pelvic paraganglioma; HN PGL, head and neck paraganglioma.
Hypersecreting tumor is defined as chromogranin A and/or metanephrines >2N.
Genetic features of the cohort
A mutation of a gene encoding a succinate dehydrogenase subunit was found in 70 patients including 63 SDHB mutation carriers (42%), 6 SDHD mutation carriers, and 1 SDHC mutation carrier. An additional 69 patients (46%), defined as apparently sporadic, underwent a negative genetic screening including at least SDHB. Four patients had a clinical history of NF1, patients had von Hippel–Lindau disease, and three multiple endocrine neoplasia type 2 (MEN2). Thus, patients can be classified into different subgroups depending on the cluster according to the unsupervised classic transcriptomic classification described for paraganglioma with 75 patients belonging to cluster 1 (50%), 4% to cluster 2, and 46% with sporadic MPP (25). Main characteristics of patients according to their genetic status are reported in Table 3. Finally, the genetic status was unknown for 18 patients (11%). Patients with sporadic tumors were characterized by older age at T0, higher median time to T0, adrenal primary and soft tissue metastases only compared with cluster 1 patients.
Characterization of Patients According to Genetics
| Characteristics | Cluster 1 N (%) | Sporadic N (%) | P |
|---|---|---|---|
| Number | 75 | 69 | |
| Males | 38 (51) | 40 (58) | NS |
| Age at T0, y | 41 ± 16 | 53.8 ± 14 | <0.0001 |
| Median time to T0, months | 4.1 [0–613] | 26.9 [0–313] | 0.021 |
| M1 within a y | 43 (58) | 23 (33) | 0.004 |
| Primary tumor site | |||
| Adrenal | 17 (23) | 52 (75) | <0.0001 |
| HN PGL | 12 (16) | 4 (6) | 0.051 |
| AbdoP PGL | 46 (61) | 13 (19) | <0.0001 |
| Multiples | 15 (20) | 5 (7) | 0.027 |
| Tumor-related symptoms | 45 (60) | 34 (49) | NS |
| Hormone-related symptoms | 40 (53) | 40 (58) | NS |
| Hypersecretiona | 49 (65) | 48 (70) | NS |
| Metastatic site | |||
| Bone only | 20 (27) | 11 (16) | NS |
| Soft tissue only | 20 (27) | 43 (62) | <0.0001 |
| Characteristics | Cluster 1 N (%) | Sporadic N (%) | P |
|---|---|---|---|
| Number | 75 | 69 | |
| Males | 38 (51) | 40 (58) | NS |
| Age at T0, y | 41 ± 16 | 53.8 ± 14 | <0.0001 |
| Median time to T0, months | 4.1 [0–613] | 26.9 [0–313] | 0.021 |
| M1 within a y | 43 (58) | 23 (33) | 0.004 |
| Primary tumor site | |||
| Adrenal | 17 (23) | 52 (75) | <0.0001 |
| HN PGL | 12 (16) | 4 (6) | 0.051 |
| AbdoP PGL | 46 (61) | 13 (19) | <0.0001 |
| Multiples | 15 (20) | 5 (7) | 0.027 |
| Tumor-related symptoms | 45 (60) | 34 (49) | NS |
| Hormone-related symptoms | 40 (53) | 40 (58) | NS |
| Hypersecretiona | 49 (65) | 48 (70) | NS |
| Metastatic site | |||
| Bone only | 20 (27) | 11 (16) | NS |
| Soft tissue only | 20 (27) | 43 (62) | <0.0001 |
Figures represent the number of evaluable patients (%), means ± SD, or median (IQR).
Abbreviations: AbdoP PGL, abdomino-pelvic paraganglioma; HN PGL, head and neck paraganglioma; NS, not significant.
Hypersecreting tumor is defined as chromogranin A and/or metanephrines >2N
Characterization of Patients According to Genetics
| Characteristics | Cluster 1 N (%) | Sporadic N (%) | P |
|---|---|---|---|
| Number | 75 | 69 | |
| Males | 38 (51) | 40 (58) | NS |
| Age at T0, y | 41 ± 16 | 53.8 ± 14 | <0.0001 |
| Median time to T0, months | 4.1 [0–613] | 26.9 [0–313] | 0.021 |
| M1 within a y | 43 (58) | 23 (33) | 0.004 |
| Primary tumor site | |||
| Adrenal | 17 (23) | 52 (75) | <0.0001 |
| HN PGL | 12 (16) | 4 (6) | 0.051 |
| AbdoP PGL | 46 (61) | 13 (19) | <0.0001 |
| Multiples | 15 (20) | 5 (7) | 0.027 |
| Tumor-related symptoms | 45 (60) | 34 (49) | NS |
| Hormone-related symptoms | 40 (53) | 40 (58) | NS |
| Hypersecretiona | 49 (65) | 48 (70) | NS |
| Metastatic site | |||
| Bone only | 20 (27) | 11 (16) | NS |
| Soft tissue only | 20 (27) | 43 (62) | <0.0001 |
| Characteristics | Cluster 1 N (%) | Sporadic N (%) | P |
|---|---|---|---|
| Number | 75 | 69 | |
| Males | 38 (51) | 40 (58) | NS |
| Age at T0, y | 41 ± 16 | 53.8 ± 14 | <0.0001 |
| Median time to T0, months | 4.1 [0–613] | 26.9 [0–313] | 0.021 |
| M1 within a y | 43 (58) | 23 (33) | 0.004 |
| Primary tumor site | |||
| Adrenal | 17 (23) | 52 (75) | <0.0001 |
| HN PGL | 12 (16) | 4 (6) | 0.051 |
| AbdoP PGL | 46 (61) | 13 (19) | <0.0001 |
| Multiples | 15 (20) | 5 (7) | 0.027 |
| Tumor-related symptoms | 45 (60) | 34 (49) | NS |
| Hormone-related symptoms | 40 (53) | 40 (58) | NS |
| Hypersecretiona | 49 (65) | 48 (70) | NS |
| Metastatic site | |||
| Bone only | 20 (27) | 11 (16) | NS |
| Soft tissue only | 20 (27) | 43 (62) | <0.0001 |
Figures represent the number of evaluable patients (%), means ± SD, or median (IQR).
Abbreviations: AbdoP PGL, abdomino-pelvic paraganglioma; HN PGL, head and neck paraganglioma; NS, not significant.
Hypersecreting tumor is defined as chromogranin A and/or metanephrines >2N
Prognostic factors for survival in the cohort
The median follow-up from T0 was 64 months (range, 0.5 to 185). Median OS was 6.7 years and 5-year OS was 62%. Seventy-eight of the 92 deaths were reported as related to the MPP (85%). Because no difference between overall and specific survival analyses was found, parameters affecting OS only were reported as described in Table 4. At univariate analysis (Fig. 1), head and neck paraganglioma, younger age, absence of or low hormonal secretion (under five times upper normal range), and low proliferative index (as defined by mitosis ≤3/10 HPFand or Ki67 ≤2%) were associated with a better OS. Patients with MEN2 and NF1, but also proliferative index were excluded from the multivariate analysis because of their small number (Table 4). At multivariate analysis hypersecretion [hazard ratio 3.02 (1.65 to 5.55); P = 0.0004] was identified as independent prognostic factors of worst OS.
Univariate and Multivariate Analysis of the Association of Prognostic Factors With OS
| Univariate Analysis | Multivariate Analysis | |||
|---|---|---|---|---|
| Parameter | HR (95% CI) | P | HR (95% CI) | P |
| Sex | 0.62 | |||
| Male | 1 | |||
| Female | 1.11 (0.73–1.68) | |||
| Primary | 0.023 | 0.097 | ||
| Adrenal | 1 | 1 | ||
| Head and neck | 0.15 (0.04–0.62) | 0.35 (0.07–1.62) | ||
| Abdomino-pelvic | 0.78 (0.51–1.2) | 1.44 (0.8–2.59) | ||
| Age at T0, y | 0.038 | 0.13 | ||
| <40 | 1 | 1 | ||
| 40–60 | 1.82 (1.07–3.09) | 1.57 (0.84–2.92) | ||
| >60 | 2.12 (1.15–3.91) | 2.08 (1.04–4.17) | ||
| Disease-free interval, y | 0.093 | 0.28 | ||
| <1 | 1 | 1 | ||
| 1–5 | 1.02 (0.63–1.65) | 0.77 (0.39–1.53) | ||
| >5 | 0.58 (0.34–0.98) | 0.58 (0.3–1.14) | ||
| Genetics | <0.0001 | 0.53 | ||
| Sporadic and cluster 2a | 1 | 1 | ||
| Cluster 1 | 0.71 (0.44–1.13) | 0.78 (0.38–1.62) | ||
| Metastatic site | 0.56 | |||
| Bone only | 1 | |||
| Soft tissue only | 0.81 (0.46–1.41) | |||
| Both | 1.02 (0.58–1.79) | |||
| Number of metastatic sites | 0.18 | |||
| 1 | 1 | |||
| 2 | 0.72 (0.44–1.17) | |||
| 3 | 1.30 (0.74–2.3) | |||
| 4 | 1.37 (0.64–2.95) | |||
| Tumor-related syndrome | 0.70 | |||
| No | 1 | |||
| Yes | 1.20 (0.79–1.82) | |||
| Unknown | 1.02 (0.24–4.23) | |||
| Hormone-related syndrome | 0.95 | |||
| No | 1 | |||
| Yes | 0.94 (0.62–1.44) | |||
| Unknown | 0.89 (0.21–3.68) | |||
| Hypersecretion | 0.0008 | 0.0004 | ||
| Absent or low | 1 | 1 | ||
| High | 2.42 (1.45–4.05) | 3.02 (1.65–5.55) | ||
| Proliferative index | 0.006 | 0.19 | ||
| Low | 1 | 1 | ||
| High | 2.39 (1.39–4.1) | 2.07 (0.94–4.53) | ||
| Univariate Analysis | Multivariate Analysis | |||
|---|---|---|---|---|
| Parameter | HR (95% CI) | P | HR (95% CI) | P |
| Sex | 0.62 | |||
| Male | 1 | |||
| Female | 1.11 (0.73–1.68) | |||
| Primary | 0.023 | 0.097 | ||
| Adrenal | 1 | 1 | ||
| Head and neck | 0.15 (0.04–0.62) | 0.35 (0.07–1.62) | ||
| Abdomino-pelvic | 0.78 (0.51–1.2) | 1.44 (0.8–2.59) | ||
| Age at T0, y | 0.038 | 0.13 | ||
| <40 | 1 | 1 | ||
| 40–60 | 1.82 (1.07–3.09) | 1.57 (0.84–2.92) | ||
| >60 | 2.12 (1.15–3.91) | 2.08 (1.04–4.17) | ||
| Disease-free interval, y | 0.093 | 0.28 | ||
| <1 | 1 | 1 | ||
| 1–5 | 1.02 (0.63–1.65) | 0.77 (0.39–1.53) | ||
| >5 | 0.58 (0.34–0.98) | 0.58 (0.3–1.14) | ||
| Genetics | <0.0001 | 0.53 | ||
| Sporadic and cluster 2a | 1 | 1 | ||
| Cluster 1 | 0.71 (0.44–1.13) | 0.78 (0.38–1.62) | ||
| Metastatic site | 0.56 | |||
| Bone only | 1 | |||
| Soft tissue only | 0.81 (0.46–1.41) | |||
| Both | 1.02 (0.58–1.79) | |||
| Number of metastatic sites | 0.18 | |||
| 1 | 1 | |||
| 2 | 0.72 (0.44–1.17) | |||
| 3 | 1.30 (0.74–2.3) | |||
| 4 | 1.37 (0.64–2.95) | |||
| Tumor-related syndrome | 0.70 | |||
| No | 1 | |||
| Yes | 1.20 (0.79–1.82) | |||
| Unknown | 1.02 (0.24–4.23) | |||
| Hormone-related syndrome | 0.95 | |||
| No | 1 | |||
| Yes | 0.94 (0.62–1.44) | |||
| Unknown | 0.89 (0.21–3.68) | |||
| Hypersecretion | 0.0008 | 0.0004 | ||
| Absent or low | 1 | 1 | ||
| High | 2.42 (1.45–4.05) | 3.02 (1.65–5.55) | ||
| Proliferative index | 0.006 | 0.19 | ||
| Low | 1 | 1 | ||
| High | 2.39 (1.39–4.1) | 2.07 (0.94–4.53) | ||
Hypersecretion is defined as high if chromogranin A and/or metanephrines > 5N; proliferative index is defined as high if mitosis count is >3/10 HPF and/or Ki67 >2%.
Abbreviation: HR, hazard ratio.
Cluster 2 patients were excluded for the multivariate analysis because of their small number.
Univariate and Multivariate Analysis of the Association of Prognostic Factors With OS
| Univariate Analysis | Multivariate Analysis | |||
|---|---|---|---|---|
| Parameter | HR (95% CI) | P | HR (95% CI) | P |
| Sex | 0.62 | |||
| Male | 1 | |||
| Female | 1.11 (0.73–1.68) | |||
| Primary | 0.023 | 0.097 | ||
| Adrenal | 1 | 1 | ||
| Head and neck | 0.15 (0.04–0.62) | 0.35 (0.07–1.62) | ||
| Abdomino-pelvic | 0.78 (0.51–1.2) | 1.44 (0.8–2.59) | ||
| Age at T0, y | 0.038 | 0.13 | ||
| <40 | 1 | 1 | ||
| 40–60 | 1.82 (1.07–3.09) | 1.57 (0.84–2.92) | ||
| >60 | 2.12 (1.15–3.91) | 2.08 (1.04–4.17) | ||
| Disease-free interval, y | 0.093 | 0.28 | ||
| <1 | 1 | 1 | ||
| 1–5 | 1.02 (0.63–1.65) | 0.77 (0.39–1.53) | ||
| >5 | 0.58 (0.34–0.98) | 0.58 (0.3–1.14) | ||
| Genetics | <0.0001 | 0.53 | ||
| Sporadic and cluster 2a | 1 | 1 | ||
| Cluster 1 | 0.71 (0.44–1.13) | 0.78 (0.38–1.62) | ||
| Metastatic site | 0.56 | |||
| Bone only | 1 | |||
| Soft tissue only | 0.81 (0.46–1.41) | |||
| Both | 1.02 (0.58–1.79) | |||
| Number of metastatic sites | 0.18 | |||
| 1 | 1 | |||
| 2 | 0.72 (0.44–1.17) | |||
| 3 | 1.30 (0.74–2.3) | |||
| 4 | 1.37 (0.64–2.95) | |||
| Tumor-related syndrome | 0.70 | |||
| No | 1 | |||
| Yes | 1.20 (0.79–1.82) | |||
| Unknown | 1.02 (0.24–4.23) | |||
| Hormone-related syndrome | 0.95 | |||
| No | 1 | |||
| Yes | 0.94 (0.62–1.44) | |||
| Unknown | 0.89 (0.21–3.68) | |||
| Hypersecretion | 0.0008 | 0.0004 | ||
| Absent or low | 1 | 1 | ||
| High | 2.42 (1.45–4.05) | 3.02 (1.65–5.55) | ||
| Proliferative index | 0.006 | 0.19 | ||
| Low | 1 | 1 | ||
| High | 2.39 (1.39–4.1) | 2.07 (0.94–4.53) | ||
| Univariate Analysis | Multivariate Analysis | |||
|---|---|---|---|---|
| Parameter | HR (95% CI) | P | HR (95% CI) | P |
| Sex | 0.62 | |||
| Male | 1 | |||
| Female | 1.11 (0.73–1.68) | |||
| Primary | 0.023 | 0.097 | ||
| Adrenal | 1 | 1 | ||
| Head and neck | 0.15 (0.04–0.62) | 0.35 (0.07–1.62) | ||
| Abdomino-pelvic | 0.78 (0.51–1.2) | 1.44 (0.8–2.59) | ||
| Age at T0, y | 0.038 | 0.13 | ||
| <40 | 1 | 1 | ||
| 40–60 | 1.82 (1.07–3.09) | 1.57 (0.84–2.92) | ||
| >60 | 2.12 (1.15–3.91) | 2.08 (1.04–4.17) | ||
| Disease-free interval, y | 0.093 | 0.28 | ||
| <1 | 1 | 1 | ||
| 1–5 | 1.02 (0.63–1.65) | 0.77 (0.39–1.53) | ||
| >5 | 0.58 (0.34–0.98) | 0.58 (0.3–1.14) | ||
| Genetics | <0.0001 | 0.53 | ||
| Sporadic and cluster 2a | 1 | 1 | ||
| Cluster 1 | 0.71 (0.44–1.13) | 0.78 (0.38–1.62) | ||
| Metastatic site | 0.56 | |||
| Bone only | 1 | |||
| Soft tissue only | 0.81 (0.46–1.41) | |||
| Both | 1.02 (0.58–1.79) | |||
| Number of metastatic sites | 0.18 | |||
| 1 | 1 | |||
| 2 | 0.72 (0.44–1.17) | |||
| 3 | 1.30 (0.74–2.3) | |||
| 4 | 1.37 (0.64–2.95) | |||
| Tumor-related syndrome | 0.70 | |||
| No | 1 | |||
| Yes | 1.20 (0.79–1.82) | |||
| Unknown | 1.02 (0.24–4.23) | |||
| Hormone-related syndrome | 0.95 | |||
| No | 1 | |||
| Yes | 0.94 (0.62–1.44) | |||
| Unknown | 0.89 (0.21–3.68) | |||
| Hypersecretion | 0.0008 | 0.0004 | ||
| Absent or low | 1 | 1 | ||
| High | 2.42 (1.45–4.05) | 3.02 (1.65–5.55) | ||
| Proliferative index | 0.006 | 0.19 | ||
| Low | 1 | 1 | ||
| High | 2.39 (1.39–4.1) | 2.07 (0.94–4.53) | ||
Hypersecretion is defined as high if chromogranin A and/or metanephrines > 5N; proliferative index is defined as high if mitosis count is >3/10 HPF and/or Ki67 >2%.
Abbreviation: HR, hazard ratio.
Cluster 2 patients were excluded for the multivariate analysis because of their small number.
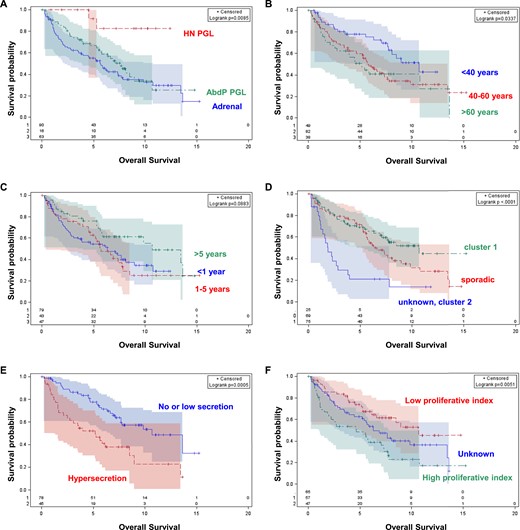
OS of patients with MPP according to (A) primary location, (B) age at T0, (C) disease-free interval, (D) genetic status, (E) hormonal secretion, and (F) proliferative index. Cluster 1 genetic status includes patients with a SDHx or MDH2 mutation and von Hippel–Lindau disease; cluster 2 genetic status includes patients with MEN2 and NF1. Hypersecretion is defined as high if chromogranin A and/or metanephrines >5N; Proliferative index is defined as high if mitosis count is >3/10 HPF and/or Ki67 >2%. AbdP PGL, abdomino-pelvic paraganglioma; HN PGL, head and neck paraganglioma.
Discussion
To the best of our knowledge, this series is the largest cohort of adult patients with MPP. Our aim was to improve the prognostic stratification of this heterogeneous group of neuroendocrine tumors. Our results do not confirm SDHB mutations as a major prognostic parameter in MPP and suggest additional key molecular events involved in MPP tumor progression. Primary tumor location and genetics were found highly involved with MPP characterization.
Our series confirms several critical characteristics of MPP patients that include a prolonged disease-free interval justifying lifelong follow-up especially in asymptomatic patients. We found SDHB mutation in 42% of cases and well-balanced pheochromocytoma and paraganglioma origin as expected because of the lower rate of malignancy but higher frequency of pheochromocytoma. We also found frequent bone locations isolated in 22% of cases prompting specific bone site screening. Furthermore, in our cohort, the primary size was larger than 5 cm in 76% of cases.
The five-year survival was 62% in the range of published series (4–7). Interestingly, the majority of deaths were classified as MPP related and no difference was found between OS and disease-free survival analysis, suggesting that OS constitutes a valuable primary end point in this group of tumors. Highlighted prognostic parameters deserve further comments. First, in contrast to other paraganglioma locations, patients with head and neck paraganglioma were associated with improved prognosis in univariate analysis but also a trend was found in multivariate analysis. Our result suggests that primary location of MPP could be considered at the time of prognostic stratification in two separate categories: head and neck paraganglioma and pheochromocytoma/abdomen-pelvic paraganglioma. We cannot confirm the prognostic role of paraganglioma site compared with pheochromocytoma in MPP and suggest that paraganglioma location may cover different entities with various behaviors. Larger series are required to validate this hypothesis definitively. Second, as for other well-differentiated neuroendocrine tumors, hypersecretion but not hormonal or tumor symptoms were found pejorative. These biomarkers may be considered as reliable surrogates of both the secretory activity of the tumor and also the tumor burden. The efficacy of systemic and also local therapeutic options including surgery in reducing these secretions remains to be explored further. In line with this, the fact that the tumor burden as indicated by the number or type of metastasized organs did not emerge as a prognostic parameter in our study is intriguing and may suggest insufficient tumor characterization in centers. Indeed, all patients underwent a thoracic, abdominal, and pelvic imaging but information on TEP imaging is missing in 58%. Third, genetics were found prognostic as defined by a worse prognosis of sporadic MPP. SDHB status was analyzed in 151 of 169 patients (89%). Half of the patients with MPP harbor a mutation belonging to cluster 1. That SDHB mutation rate conformsto other published series. However, in line with recent publications, we do not confirm a prognostic role of SDHB mutation, which remains questionable. Indeed, SDHB was initially considered a risk factor for malignancy and secondly hypothesized as potentially carrying a prognostic value. Our results suggest that these two roles could be dissociated, and additional molecular markers of tumor aggressiveness are involved beyond SDHB mutation. However, confounding factors should be kept in mind: SDHB patient characteristics were associated with other favorable prognostic factors such as younger age, earlier diagnosis, lower tumor burden at diagnosis but also more frequent head and neck paraganglioma, and absence of hypersecretion. In addition, SDHB patients may respond better to systemic therapy and benefit from a better surveillance screening. We also observe a trend for a better prognosis for patients with prolonged disease-free interval and low Ki67. Both parameters constitute markers of tumor growth velocity that may constitute useful additional factors at the time of the therapeutic decision. As proposed in an earlier study, we therefore propose to evaluate the spontaneous radiological tumor rate of progression, as a surrogate of disease-free interval, but also proliferative index in future prognostic studies (4).
Limitations of our study include the absence of functioning imaging in all patients, absence of consistent analysis of proliferative index, and methoxytyramine evaluation. In addition, the scarcity and retrospective nature of our study constitute well-known limitations when evaluating rare cancers.
To conclude, patients with MPP with sporadic disease and hypersecretion experienced worst OS. Head and neck paraganglioma may constitute a new favorable prognostic category. We recommend that treatments should aim at reducing the secretory burden. The prognostic relevance of SDHB mutation is challenged. Efforts to implement international cohorts of patients with well-characterized MPP should be performed to improvethe prognostic stratification.
Abbreviations:
- ENS@T
European Network for the Study of Adrenal Tumors
- IQR
interquartile range
- MAPP-Prono
Metastatic Pheochromocytoma and Paraganglioma Prognostic study, MEN2, multiple endocrine neoplasia type 2
- MPP
malignant pheochromocytoma and paraganglioma
- NF
neurofibromatosis
- OS
overall survival
- TAP-CT
neck-thoracic-abdomen-pelvic CT
Acknowledgments
We thank Antony Stell for managing the ENS@T database.
Disclosure Summary: The authors have nothing to disclose.

{kind=link}