





Abstract
Delta-like homolog 1 (DLK1), also called preadipocyte factor 1, prevents adipocyte differentiation and has been considered a molecular gatekeeper of adipogenesis. A DLK1 complex genomic defect was identified in five women from a single family with central precocious puberty (CPP) and increased body fat percentage.
We studied 60 female patients with a diagnosis of CPP or history of precocious menarche. Thirty-one of them reported a family history of precocious puberty. DLK1 DNA sequencing was performed in all patients. Serum DLK1 concentrations were measured using an ELISA assay in selected cases. Metabolic and reproductive profiles of adult women with CPP caused by DLK1 defects were compared with those of 20 women with idiopathic CPP.
We identified three frameshift mutations of DLK1 (p.Gly199Alafs*11, p.Val271Cysfs*14, and p.Pro160Leufs*50) in five women from three families with CPP. Segregation analysis was consistent with the maternal imprinting of DLK1. Serum DLK1 concentrations were undetectable in three affected women. Metabolic abnormalities, such as overweight/obesity, early-onset glucose intolerance/type 2 diabetes mellitus, and hyperlipidemia, were more prevalent in women with the DLK1 mutation than in the idiopathic CPP group. Notably, the human metabolic alterations were similar to the previously described dlk1-null mice phenotype. Two sisters who carried the p.Gly199Alafs*11 mutation also exhibited polycystic ovary syndrome and infertility.
Loss-of-function mutations of DLK1 are a definitive cause of familial CPP. The high prevalence of metabolic alterations in adult women who experienced CPP due to DLK1 defects suggests that this antiadipogenic factor represents a link between reproduction and metabolism.
Early menarche (≤11 years) has been considered a risk factor for metabolic syndrome, cardiovascular disease, and cancer (1–4). In girls with central precocious puberty (CPP), a pediatric disorder characterized by the appearance of secondary sexual features before 8 years of age due to early activation of the hypothalamic-pituitary-ovarian axis, menarche may occur prematurely, usually before 9 years, and health implications in adulthood may be aggravated in this group (5).
Timing of menarche is a highly polygenic trait (6). More than 300 independent signals were identified for age at menarche in a large genome-wide association study of ∼370,000 women (7, 8). Notably, common variants near the imprinted genes makorin ring finger 3 (MKRN3) and delta-like homolog 1 (DLK1) were identified, exhibiting major effects only when paternally inherited (7, 8). In addition, loss-of-function mutations of MKRN3 are a prevalent cause of familial CPP (up to 46%) (9–11).
DLK1, or preadipocyte factor 1 (Pref-1), is a transmembrane protein containing epidermal growth factor (EGF)‒like repeats homologous to the Notch/Delta/Serrate family (12, 13). Human DLK1 is encoded by a paternally expressed gene located on the long arm of chromosome 14 (14q32.2) within a locus associated with Temple syndrome, an imprinting disorder caused mainly by maternal uniparental disomy (14–16). Clinical findings of Temple syndrome include prenatal and postnatal growth failure, relative macrocephaly, truncal obesity, hypotonia, small hands and feet, and CPP (14–16). A neuroendocrine function of DLK1 was suggested by evidence of postnatal DLK1 expression in several hypothalamic nuclei as well as in kisspeptin neuron‒derived cell lines (17, 18). In 2017, whole-genome sequencing revealed a complex genomic defect of DLK1 in five female members of an Afro-descendent Brazilian family with CPP (18). The affected patients did not demonstrate additional clinical Temple syndrome features except for increased fat tissue (18).
Animal and in vitro studies have demonstrated that DLK1 acts as an adipogenesis gatekeeper by preventing adipocyte differentiation (12, 19, 20). Indeed, Dlk1-null mice display accelerated weight gain and hyperlipidemia at adulthood, supporting the antiadipogenic actions (19). Here, we demonstrate DLK1 defects in adult women who experienced CPP. Interestingly, metabolic alterations were detected in the majority of these women, suggesting that DLK1 plays an important role in pubertal development and metabolic homeostasis.
Methods
Sixty unrelated pediatric or adult patients with a clear history of CPP or precocious menarche were selected for DLK1 genetic analysis. The idiopathic CPP diagnosis was defined as Tanner stage 2 before 8 years of age in girls, basal and/or GnRH-stimulated LH levels within the pubertal range, advanced bone age (Greulich and Pyle method), and normal findings on MRI of the brain. Patients were classified as having familial CPP if they had at least one first- or second-degree relative with documented CPP or on the basis of self-reported age at menarche for female relatives (≤9 years) or premature age at onset of pubertal changes in male relatives. Among the 60 patients with CPP, 31 reported a family history of premature sexual development. Two sisters who belonged to a cohort of patients with polycystic ovary syndrome (PCOS) from Sao Paulo Medical School were also studied because they had a history of precocious menarche. Loss-of-function mutations of MKRN3 were excluded in all of these patients.
Long-term follow-up of anthropometric, metabolic, and reproductive parameters of 20 unrelated women (age from 15 to 27 years) with sporadic idiopathic CPP were assessed using a standardized biochemical protocol. All of these women were chronically treated with GnRH analogues. MKRN3 and DLK1 defects were excluded in this group of women with CPP. The research protocol was approved by the ethics committee of Sao Paulo University. Written informed consent was obtained from all participants.
Genetic analysis
Sanger and exome sequencing
Genomic DNA was collected from the index cases and family members. The entire coding region of DLK1 (GenBank accession number NM_003836) was amplified by PCR followed by automatic sequencing of the products using the Sanger method in 60 unrelated patients with CPP. Segregation analysis of the identified variants was also performed by Sanger sequencing.
Whole-exome sequencing was performed in the two affected sisters with PCOS and their unaffected mother (family 1) according to previously published protocols (21).
DLK1 serum measurements
Serum DLK1 levels were measured in affected patients and controls using a soluble DLK1 ELISA (IBL America, Minneapolis, MN). DLK1 levels in healthy human control serum range from 0.4 to >2.5 ng/mL. The lower limit of detection for the assay is <0.4 ng/mL with a mean intraassay variability of 5% and a mean interassay variability of 8.1%.
Statistics
Comparisons between women who have CPP due to DLK1 mutations and women with idiopathic CPP were performed by Fisher exact test or χ2 analysis for each categorical variable using InStat software (GraphPad, Inc., San Diego, CA). P < 0.05 was considered statistically significant.
Results
DNA sequencing
Three distinct loss-of-function mutations of the DLK1 gene (all located in exon 5) were identified in three families with CPP (families 1 to 3) from a cohort of 31 cases with familial CPP. In family 1, a heterozygous frameshift variant (c.594_594delC, p.Gly199Alafs*11) was identified by whole-exome sequencing in two sisters who had a history of precocious menarche (7 years), PCOS, metabolic alterations, and infertility. Their affected father harbored the same DLK1 variant, confirming the expected segregation for an imprinted gene [Fig. 1(a)]. This frameshift was absent in their unaffected mother, younger brother, and paternal aunt. In Family 2, a heterozygous frameshift variant (c.810_810delT, p.Val271Cysfs*14) was identified by Sanger sequencing in an English woman and her brother, who experienced CPP at infancy. Her asymptomatic son was a carrier of this defect as expected for the maternal imprinting pattern of DLK1 [Fig. 1(b)]. In family 3, a heterozygous variant (c.479_479delC, p.Pro160Leufs*50) was identified by Sanger sequencing in a woman and her paternal aunt; both had untreated CPP [Fig. 1(c)].
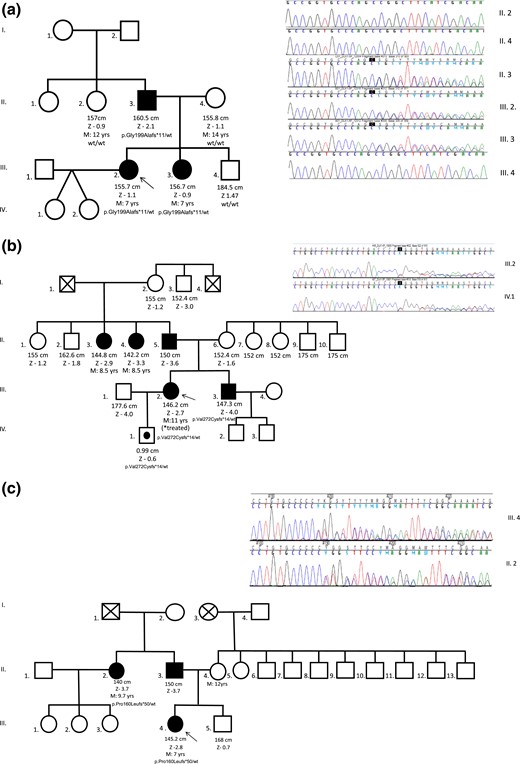
Pedigrees and sequencing data of three distinct families (panels a, b and c) with DLK1 mutations. Unaffected male family members (□) and female family members (○); clinically affected male family members (▪) and female family members (●); asymptomatic carriers (symbols with black circles); and deceased family members (symbols with X) are shown. Arrows indicate the proband in each family. DLK1 variants were confirmed and/or segregated in family members by Sanger sequencing. The DLK1 genotypes are shown for family members whose DNA was available for genetic analysis. M, age at menarche; Z, height z score.
These three frameshift mutations were located in the extracellular domain of DLK1, a region with EGF-like domains containing six conserved cysteines, which form three disulfide bonds (22). The p.Pro160Leufs*50 and p.Gly199Alafs*11 mutations were located in EGF like‒4 and EGF like‒5 domains, respectively, both in predicted disulfide bonds, whereas the p.Val271Cysfs*14 mutation was located in the extracellular domain but outside the EGF-like domains (Fig. 2) (22).
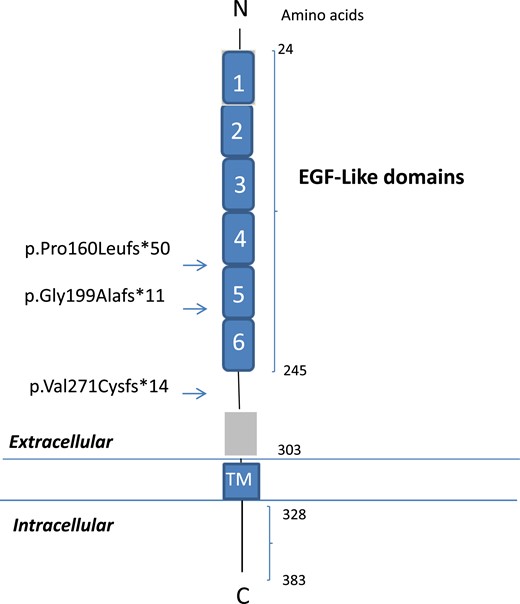
Schematic representation of the DLK1 protein containing EGF-like extracellular domains. The numbers correspond to the amino acid positions in the protein. DLK1 comprises six EGF-like extracellular domains (amino acids 24 to 245), an extracellular region (amino acids 246 to 303), a transmembrane region (amino acids 304 to 327), and a cytoplasmic region (amino acids 328 to 383). Arrows indicate the location of the DLK1 frameshift mutations. C, carboxyterminal; N, aminoterminal; TM, transmembrane.
Clinical features of three DLK1-affected families
Family 1
Two Brazilian sisters, 1A and 1B, were first seen at 34 and 24 years old, respectively, for PCOS investigation. Patient 1A initially presented with thelarche at 5 years and menarche at 7 years. Menstrual irregularities began at 12 years followed by hirsutism and infertility (Table 1). At 27 years, she was diagnosed with type 2 diabetes mellitus. Her physical examination revealed a weight of 70.9 kg, a height of 155.7 cm, a body mass index (BMI) of 29.2 kg/m2, a waist circumference of 98 cm, and a Ferriman-Gallwey score of 16. Her initial biochemical profile showed uncontrolled type 2 diabetes mellitus (glycated hemoglobin, 9.5%) and hyperlipidemia (Table 2). Dual X-ray absorptiometry demonstrated increased (35.4%) fat mass. Her sister (1B) also experienced precocious pubertal development (thelarche at 5 years). She was irregularly treated, and spontaneous menarche occurred at 7 years of age. She subsequently developed irregular menses and infertility. Type 2 diabetes mellitus was diagnosed at 16 years of age. An initial physical examination revealed a weight of 66.8 kg, a height of 156.7 cm, a BMI of 27.2 kg/m2, and a waist circumference of 94 cm (Table 1). Her initial biochemical and hormonal profiles showed insulin resistance and high levels of testosterone (Table 2). Macromastia, acanthosis nigricans, and acne were detected in both sisters (Fig. 3). Pelvic ultrasonography showed micropolycystic ovarian morphology in these sisters (23), and abdominal ultrasonography showed hepatic steatosis. Their mother had normal pubertal development, with menarche at 14 years of age. Their father did not recall his age of pubertal development. His height was 160.5 cm with normal BMI.
Clinical Features of 10 Adult Women With Central Precocious Puberty Due to DLK1 Defects
| Family No. (origin) | Case No. | Age, y | Pubertal Development, y | CPP Treatment | Final Height, cm (SD) | BMI, kg/m2 | Additional Clinical Features | DLK1 Variant | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Thelarche | Menarche | cDNA | Protein | |||||||
| Family 1 (Brazil) | 1A | 34 | 5.0 | 7.0 | Oral medication | 155.7 (−1.1) | 29.2 | PCOS, infertility, type 2 DM (27 y), hepatic steatosis | c.594_594delC | p.Gly199Alafs*11 |
| 1B | 24 | 5.0 | 7.0 | Irregular | 156.7 (−0.9) | 27.2 | PCOS, infertility, type 2 DM (16 y), hepatic steatosis | |||
| Family 2 (United Kingdom) | 2A | 29 | 5.0 | 11 | Oral medication | 146.2 (−2.6) | 21.1 | — | c.810_810delT | p.Val271Cysfs*14 |
| Family 3 (Brazil) | 3A | 19 | 7.0 | 7.0 | No | 145.2 (−2.8) | 13.2 | — | c.479_479delC | p.Pro160Leufs*50 |
| 3B | 56 | 7.0 | 9.0 | No | 137.8 (−4.0) | 37.7 | Glucose intolerance | |||
| Hypercholesterolemia | ||||||||||
| Family 4a (Brazil) | 4A | 22 | 5.5 | 12 | GnRHa | 156.5 (−0.9) | 29.2 | Increased % fat mass | Complex genomic rearrangement/5′UTR and exon 1 deletion | Truncated protein |
| 4B | 21 | 5.0 | 12 | GnRHa | 159.7 (−0.4) | 22 | Increased % fat mass | |||
| 4C | 18 | 4.6 | 12 | GnRHa | 159.3 (−0.5) | 33 | Increased % fat mass | |||
| Glucose intolerance | ||||||||||
| 4D | 16 | 5.9 | 10.8 | GnRHa | 160.5 (−0.3) | 22 | Increased % fat mass | |||
| 4E | 63 | NA | ∼9.0 | No | 154.0 (−1.3) | 29.6 | Type 2 DM | |||
| Hypertension | ||||||||||
| Hypercholesterolemia | ||||||||||
| Family No. (origin) | Case No. | Age, y | Pubertal Development, y | CPP Treatment | Final Height, cm (SD) | BMI, kg/m2 | Additional Clinical Features | DLK1 Variant | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Thelarche | Menarche | cDNA | Protein | |||||||
| Family 1 (Brazil) | 1A | 34 | 5.0 | 7.0 | Oral medication | 155.7 (−1.1) | 29.2 | PCOS, infertility, type 2 DM (27 y), hepatic steatosis | c.594_594delC | p.Gly199Alafs*11 |
| 1B | 24 | 5.0 | 7.0 | Irregular | 156.7 (−0.9) | 27.2 | PCOS, infertility, type 2 DM (16 y), hepatic steatosis | |||
| Family 2 (United Kingdom) | 2A | 29 | 5.0 | 11 | Oral medication | 146.2 (−2.6) | 21.1 | — | c.810_810delT | p.Val271Cysfs*14 |
| Family 3 (Brazil) | 3A | 19 | 7.0 | 7.0 | No | 145.2 (−2.8) | 13.2 | — | c.479_479delC | p.Pro160Leufs*50 |
| 3B | 56 | 7.0 | 9.0 | No | 137.8 (−4.0) | 37.7 | Glucose intolerance | |||
| Hypercholesterolemia | ||||||||||
| Family 4a (Brazil) | 4A | 22 | 5.5 | 12 | GnRHa | 156.5 (−0.9) | 29.2 | Increased % fat mass | Complex genomic rearrangement/5′UTR and exon 1 deletion | Truncated protein |
| 4B | 21 | 5.0 | 12 | GnRHa | 159.7 (−0.4) | 22 | Increased % fat mass | |||
| 4C | 18 | 4.6 | 12 | GnRHa | 159.3 (−0.5) | 33 | Increased % fat mass | |||
| Glucose intolerance | ||||||||||
| 4D | 16 | 5.9 | 10.8 | GnRHa | 160.5 (−0.3) | 22 | Increased % fat mass | |||
| 4E | 63 | NA | ∼9.0 | No | 154.0 (−1.3) | 29.6 | Type 2 DM | |||
| Hypertension | ||||||||||
| Hypercholesterolemia | ||||||||||
Abbreviations: DM, diabetes mellitus; NA, not applicable.
Family 4 was previously reported Dauber et al. (18).
Clinical Features of 10 Adult Women With Central Precocious Puberty Due to DLK1 Defects
| Family No. (origin) | Case No. | Age, y | Pubertal Development, y | CPP Treatment | Final Height, cm (SD) | BMI, kg/m2 | Additional Clinical Features | DLK1 Variant | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Thelarche | Menarche | cDNA | Protein | |||||||
| Family 1 (Brazil) | 1A | 34 | 5.0 | 7.0 | Oral medication | 155.7 (−1.1) | 29.2 | PCOS, infertility, type 2 DM (27 y), hepatic steatosis | c.594_594delC | p.Gly199Alafs*11 |
| 1B | 24 | 5.0 | 7.0 | Irregular | 156.7 (−0.9) | 27.2 | PCOS, infertility, type 2 DM (16 y), hepatic steatosis | |||
| Family 2 (United Kingdom) | 2A | 29 | 5.0 | 11 | Oral medication | 146.2 (−2.6) | 21.1 | — | c.810_810delT | p.Val271Cysfs*14 |
| Family 3 (Brazil) | 3A | 19 | 7.0 | 7.0 | No | 145.2 (−2.8) | 13.2 | — | c.479_479delC | p.Pro160Leufs*50 |
| 3B | 56 | 7.0 | 9.0 | No | 137.8 (−4.0) | 37.7 | Glucose intolerance | |||
| Hypercholesterolemia | ||||||||||
| Family 4a (Brazil) | 4A | 22 | 5.5 | 12 | GnRHa | 156.5 (−0.9) | 29.2 | Increased % fat mass | Complex genomic rearrangement/5′UTR and exon 1 deletion | Truncated protein |
| 4B | 21 | 5.0 | 12 | GnRHa | 159.7 (−0.4) | 22 | Increased % fat mass | |||
| 4C | 18 | 4.6 | 12 | GnRHa | 159.3 (−0.5) | 33 | Increased % fat mass | |||
| Glucose intolerance | ||||||||||
| 4D | 16 | 5.9 | 10.8 | GnRHa | 160.5 (−0.3) | 22 | Increased % fat mass | |||
| 4E | 63 | NA | ∼9.0 | No | 154.0 (−1.3) | 29.6 | Type 2 DM | |||
| Hypertension | ||||||||||
| Hypercholesterolemia | ||||||||||
| Family No. (origin) | Case No. | Age, y | Pubertal Development, y | CPP Treatment | Final Height, cm (SD) | BMI, kg/m2 | Additional Clinical Features | DLK1 Variant | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Thelarche | Menarche | cDNA | Protein | |||||||
| Family 1 (Brazil) | 1A | 34 | 5.0 | 7.0 | Oral medication | 155.7 (−1.1) | 29.2 | PCOS, infertility, type 2 DM (27 y), hepatic steatosis | c.594_594delC | p.Gly199Alafs*11 |
| 1B | 24 | 5.0 | 7.0 | Irregular | 156.7 (−0.9) | 27.2 | PCOS, infertility, type 2 DM (16 y), hepatic steatosis | |||
| Family 2 (United Kingdom) | 2A | 29 | 5.0 | 11 | Oral medication | 146.2 (−2.6) | 21.1 | — | c.810_810delT | p.Val271Cysfs*14 |
| Family 3 (Brazil) | 3A | 19 | 7.0 | 7.0 | No | 145.2 (−2.8) | 13.2 | — | c.479_479delC | p.Pro160Leufs*50 |
| 3B | 56 | 7.0 | 9.0 | No | 137.8 (−4.0) | 37.7 | Glucose intolerance | |||
| Hypercholesterolemia | ||||||||||
| Family 4a (Brazil) | 4A | 22 | 5.5 | 12 | GnRHa | 156.5 (−0.9) | 29.2 | Increased % fat mass | Complex genomic rearrangement/5′UTR and exon 1 deletion | Truncated protein |
| 4B | 21 | 5.0 | 12 | GnRHa | 159.7 (−0.4) | 22 | Increased % fat mass | |||
| 4C | 18 | 4.6 | 12 | GnRHa | 159.3 (−0.5) | 33 | Increased % fat mass | |||
| Glucose intolerance | ||||||||||
| 4D | 16 | 5.9 | 10.8 | GnRHa | 160.5 (−0.3) | 22 | Increased % fat mass | |||
| 4E | 63 | NA | ∼9.0 | No | 154.0 (−1.3) | 29.6 | Type 2 DM | |||
| Hypertension | ||||||||||
| Hypercholesterolemia | ||||||||||
Abbreviations: DM, diabetes mellitus; NA, not applicable.
Family 4 was previously reported Dauber et al. (18).
Metabolic and Hormonal Data of Five Adult Women With DLK1 Defects
| Reference Values | Patient 1A | Patient 1B | Patient 3A | Patient 3B | Patient 4Ea | |
|---|---|---|---|---|---|---|
| Glucose, mg/dL (nmol/L) | 70–99 (3.9–5.5) | 173 (9.6) | 112 (6.2) | 85 (4.7) | 103 (5.7) | 124 (6.9) |
| HbA1c, % | 4.1–6.0 | 9.5 | 6.5 | — | 5.9 | 8.4 |
| Insulin, μU/mL | 2.6–24.9 | — | 51.6 | — | 13.6 | 21.1 |
| Total cholesterol, mg/dL (mmol/L) | <190 (<4.9) | 192 (5.0) | 177 (4.6) | 162 (4.2) | 206 (5.3) | 234 (6.1) |
| Triglycerides, mg/dL (mmol/L) | <150 (<1.7) | 638 (7.2) | 130 (1.5) | 62 (0.7) | 77 (0.9) | 93 (1.0) |
| LDL, mg/dL (mmol/L) | <130 (<3.4) | — | 111 (2.9) | 86 (2.2) | 106 (2.7) | 170 (4.4) |
| HDL, mg/dL (mmol/L) | >40 (>1.0) | 31 (0.8) | 41 (1.1) | 64 (1.7) | 83 (2.2) | 43 (1.1) |
| FSH, IU/L | 3.5–12.5 | |||||
| 25.8–134.8c | 5.9 | 7.1 | 7.7 | 65.5 | 51.5 | |
| LH, IU/L | 2.4–12.6 | 3.9 | 39.3 | 8.1 | 28.7 | 22.5 |
| Testosterone, ng/dL (nmol/L) | 20–49 y: ≤48 (1.7) >50 y: ≤41 (1.4) | 32 (1.1)b | 101 (3.5) | 24 (0.8) | — | 46 (1.6) |
| Androstenedione, ng/mL (nmol/L) | <2.2 (7.7) | — | 3.4 (11.9) | — | — | 0.75 (2.6) |
| Reference Values | Patient 1A | Patient 1B | Patient 3A | Patient 3B | Patient 4Ea | |
|---|---|---|---|---|---|---|
| Glucose, mg/dL (nmol/L) | 70–99 (3.9–5.5) | 173 (9.6) | 112 (6.2) | 85 (4.7) | 103 (5.7) | 124 (6.9) |
| HbA1c, % | 4.1–6.0 | 9.5 | 6.5 | — | 5.9 | 8.4 |
| Insulin, μU/mL | 2.6–24.9 | — | 51.6 | — | 13.6 | 21.1 |
| Total cholesterol, mg/dL (mmol/L) | <190 (<4.9) | 192 (5.0) | 177 (4.6) | 162 (4.2) | 206 (5.3) | 234 (6.1) |
| Triglycerides, mg/dL (mmol/L) | <150 (<1.7) | 638 (7.2) | 130 (1.5) | 62 (0.7) | 77 (0.9) | 93 (1.0) |
| LDL, mg/dL (mmol/L) | <130 (<3.4) | — | 111 (2.9) | 86 (2.2) | 106 (2.7) | 170 (4.4) |
| HDL, mg/dL (mmol/L) | >40 (>1.0) | 31 (0.8) | 41 (1.1) | 64 (1.7) | 83 (2.2) | 43 (1.1) |
| FSH, IU/L | 3.5–12.5 | |||||
| 25.8–134.8c | 5.9 | 7.1 | 7.7 | 65.5 | 51.5 | |
| LH, IU/L | 2.4–12.6 | 3.9 | 39.3 | 8.1 | 28.7 | 22.5 |
| Testosterone, ng/dL (nmol/L) | 20–49 y: ≤48 (1.7) >50 y: ≤41 (1.4) | 32 (1.1)b | 101 (3.5) | 24 (0.8) | — | 46 (1.6) |
| Androstenedione, ng/mL (nmol/L) | <2.2 (7.7) | — | 3.4 (11.9) | — | — | 0.75 (2.6) |
Abbreviations: HDL, high-density lipoprotein; LDL, low-density lipoprotein.
Clinical data of patients 4A, 4B, 4C, and 4D were previously reported Dauber et al. (18).
medroxyprogesterone.
Postmenopause levels.
Metabolic and Hormonal Data of Five Adult Women With DLK1 Defects
| Reference Values | Patient 1A | Patient 1B | Patient 3A | Patient 3B | Patient 4Ea | |
|---|---|---|---|---|---|---|
| Glucose, mg/dL (nmol/L) | 70–99 (3.9–5.5) | 173 (9.6) | 112 (6.2) | 85 (4.7) | 103 (5.7) | 124 (6.9) |
| HbA1c, % | 4.1–6.0 | 9.5 | 6.5 | — | 5.9 | 8.4 |
| Insulin, μU/mL | 2.6–24.9 | — | 51.6 | — | 13.6 | 21.1 |
| Total cholesterol, mg/dL (mmol/L) | <190 (<4.9) | 192 (5.0) | 177 (4.6) | 162 (4.2) | 206 (5.3) | 234 (6.1) |
| Triglycerides, mg/dL (mmol/L) | <150 (<1.7) | 638 (7.2) | 130 (1.5) | 62 (0.7) | 77 (0.9) | 93 (1.0) |
| LDL, mg/dL (mmol/L) | <130 (<3.4) | — | 111 (2.9) | 86 (2.2) | 106 (2.7) | 170 (4.4) |
| HDL, mg/dL (mmol/L) | >40 (>1.0) | 31 (0.8) | 41 (1.1) | 64 (1.7) | 83 (2.2) | 43 (1.1) |
| FSH, IU/L | 3.5–12.5 | |||||
| 25.8–134.8c | 5.9 | 7.1 | 7.7 | 65.5 | 51.5 | |
| LH, IU/L | 2.4–12.6 | 3.9 | 39.3 | 8.1 | 28.7 | 22.5 |
| Testosterone, ng/dL (nmol/L) | 20–49 y: ≤48 (1.7) >50 y: ≤41 (1.4) | 32 (1.1)b | 101 (3.5) | 24 (0.8) | — | 46 (1.6) |
| Androstenedione, ng/mL (nmol/L) | <2.2 (7.7) | — | 3.4 (11.9) | — | — | 0.75 (2.6) |
| Reference Values | Patient 1A | Patient 1B | Patient 3A | Patient 3B | Patient 4Ea | |
|---|---|---|---|---|---|---|
| Glucose, mg/dL (nmol/L) | 70–99 (3.9–5.5) | 173 (9.6) | 112 (6.2) | 85 (4.7) | 103 (5.7) | 124 (6.9) |
| HbA1c, % | 4.1–6.0 | 9.5 | 6.5 | — | 5.9 | 8.4 |
| Insulin, μU/mL | 2.6–24.9 | — | 51.6 | — | 13.6 | 21.1 |
| Total cholesterol, mg/dL (mmol/L) | <190 (<4.9) | 192 (5.0) | 177 (4.6) | 162 (4.2) | 206 (5.3) | 234 (6.1) |
| Triglycerides, mg/dL (mmol/L) | <150 (<1.7) | 638 (7.2) | 130 (1.5) | 62 (0.7) | 77 (0.9) | 93 (1.0) |
| LDL, mg/dL (mmol/L) | <130 (<3.4) | — | 111 (2.9) | 86 (2.2) | 106 (2.7) | 170 (4.4) |
| HDL, mg/dL (mmol/L) | >40 (>1.0) | 31 (0.8) | 41 (1.1) | 64 (1.7) | 83 (2.2) | 43 (1.1) |
| FSH, IU/L | 3.5–12.5 | |||||
| 25.8–134.8c | 5.9 | 7.1 | 7.7 | 65.5 | 51.5 | |
| LH, IU/L | 2.4–12.6 | 3.9 | 39.3 | 8.1 | 28.7 | 22.5 |
| Testosterone, ng/dL (nmol/L) | 20–49 y: ≤48 (1.7) >50 y: ≤41 (1.4) | 32 (1.1)b | 101 (3.5) | 24 (0.8) | — | 46 (1.6) |
| Androstenedione, ng/mL (nmol/L) | <2.2 (7.7) | — | 3.4 (11.9) | — | — | 0.75 (2.6) |
Abbreviations: HDL, high-density lipoprotein; LDL, low-density lipoprotein.
Clinical data of patients 4A, 4B, 4C, and 4D were previously reported Dauber et al. (18).
medroxyprogesterone.
Postmenopause levels.
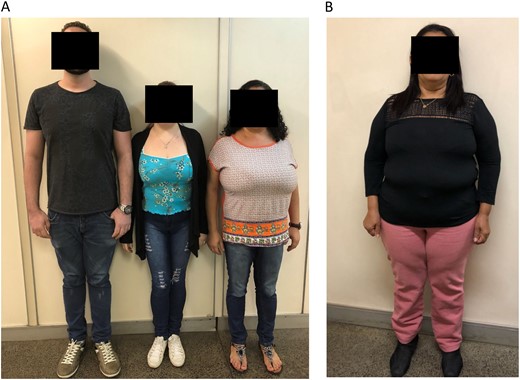
Pictures of patients who presented with CPP due to DLK1 defects. (A) Two affected sisters (patients 1A and 1B) who presented with truncal obesity and macromastia from family 1 are shown in the left panel. (B) An affected female patient (patient 3B) with short stature and obesity from family 3 is shown in the right panel.
Family 2
Genomic DNA from two members, a mother (2A) and her son (2B), of a British family (London, UK) were referred by a geneticist (coauthor M.L.) to Sao Paulo University because of a strong history of familial CPP associated with short stature. Patient 2A was a 29-year-old mother who was concerned about the possibility that her 4-year-old son, a normal child without growth or pubertal alterations, would develop precocious puberty. She developed thelarche at 5 years old and was treated with oral medications until 11 years (Table 1). Menarche occurred after treatment was discontinued. In addition, her father, older brother, and two paternal aunts had a history of precocious puberty, starting at around 5 years. Her brother also experienced CPP and was treated with a GnRH analogue (intranasal spray).
Family 3
A 19-year-old woman (3A) was referred to the Endocrinology Unit because of a pituitary microadenoma that was discovered by MRI during previous investigation of precocious puberty. She presented with thelarche and menarche around 7 years. At that time, hormonal investigation demonstrated activation of the reproductive axis (basal LH: 1.8 IU/L and peak GnRH-stimulated LH: 11.5 IU/L) with advanced bone age (Table 1). MRI excluded anatomical lesions in the hypothalamic region, but a pituitary microadenoma was observed. She did not receive adequate treatment. At adult age, her height was 145.5 cm (SDS −2.7 cm), and her BMI was 13 kg/m2 (Table 1). Biochemical and hormonal profiles were completely normal (Table 2). The patient’s father (52 years) also had short stature (150 cm; standard deviation score, −3.7 cm), with a BMI of 24.4 kg/m2 and high cholesterol levels; however, he did not remember the age at which his pubertal development started. Her paternal aunt (3B) also has a history of precocious menarche (around 9 years). No endocrine investigation or treatment was performed at that time. She apparently had a normal reproductive life (regular menstrual cycles and four spontaneous pregnancies). At age 56 years, she presented with short stature (137.8 cm; SDS, −4.0 cm), obesity (BMI: 36.8 kg/m2), a waist circumference of 114 cm associated with glucose intolerance, hypercholesterolemia, and a cardiac arrhythmia (Fig. 3B;Tables 1 and 2).
Circulating DLK1 concentrations
To investigate the effect of the variants on DLK1 production, serum DLK1 levels were measured in three affected women (patients 1A, 1B, and 3B) and five female controls with a history of normal pubertal timing. The father of patients 1A and 1B was also studied. DLK1 serum levels ranged from 3.2 to 11.3 ng/mL in the control group. In contrast, all affected individuals (patients 1A, 1B, and 3B) had undetectable DLK1 levels (<0.4 ng/mL), supporting the notion that the identified mutations lead to a complete lack of circulating DLK1 form in these individuals. The father of patients 1A and 1B, who harbored the same defect of DLK1, had an undetectable level, indicating that he was also affected, which was presumed given his short stature. Undetectable DLK1 concentrations were previously reported in affected members from family 4 (18).
Anthropometric and metabolic profile of DLK1-deficient and idiopathic women with CPP
DLK1-deficient women had a greater prevalence of short stature (30%), overweight/obesity (60%), hyperlipidemia (50%), insulin resistance (70%), type 2 diabetes mellitus (30%), and PCOS (20%) phenotype (Table 3) than 20 unrelated adult women with idiopathic CPP who were chronically treated with GnRH analogues.
Human and Mice Phenotypes Due to DLK1 Deficiency
| Human Phenotype | Mouse Phenotype | Comparison (DLK1 Mutated vs Idiopathic CPPa) | |||
|---|---|---|---|---|---|
| Temple Syndrome | Monogenic CPP Due to DLK1 Defects | Idiopathic CPP | Dlk1 KO | Pa | |
| Origin of study | Japan/United Kingdom | Brazil/United Kingdom | Brazil | United States | |
| Number of women | 32/51 | 10 | 20 | — | |
| Median age (range), y | Children and adults | 23 (16–63) | 16.7 (15–27) | ||
| Genetic basis | UPD(14)mat, epimutation, microdeletion | DLK1 deletion and frameshifts | Unknown | Generation of DLK1-null mice | |
| Short stature, % | 79–94 | 30 | 0 | Pre-postnatal growth retardation | Not applicable |
| Eye and bone alterations, % | — | 10 (syndactyly) | — | Blepharophimosis and rib alterations | — |
| Overweight/obesity, % | 11–49 | 60 | 35 | + | <0.001 |
| Hyperlipidemia, % | 10–23 | 50 | 0 | + | Not applicable |
| Insulin resistance % (HOMA-IR >2.7) | + | 70 | 15 | + | <0.001 |
| Type 2 diabetes mellitus % | 11–20 | 30 | 0 | + | Not applicable |
| CPP, % | 76–86 | 100 | 100 | — | |
| Menarche, y | Untreated: 7·8b | 12 | |||
| Treated: 11·6b | |||||
| PCOS/infertility, % | Not applicable | 20 | 10 | — | 0.047 |
| References | Kagami et al. (16)Ioannides et al. (15) | Dauber et al. (18) Current study | Current study | Moon et al. (19)Sul (25) | Current study |
| Human Phenotype | Mouse Phenotype | Comparison (DLK1 Mutated vs Idiopathic CPPa) | |||
|---|---|---|---|---|---|
| Temple Syndrome | Monogenic CPP Due to DLK1 Defects | Idiopathic CPP | Dlk1 KO | Pa | |
| Origin of study | Japan/United Kingdom | Brazil/United Kingdom | Brazil | United States | |
| Number of women | 32/51 | 10 | 20 | — | |
| Median age (range), y | Children and adults | 23 (16–63) | 16.7 (15–27) | ||
| Genetic basis | UPD(14)mat, epimutation, microdeletion | DLK1 deletion and frameshifts | Unknown | Generation of DLK1-null mice | |
| Short stature, % | 79–94 | 30 | 0 | Pre-postnatal growth retardation | Not applicable |
| Eye and bone alterations, % | — | 10 (syndactyly) | — | Blepharophimosis and rib alterations | — |
| Overweight/obesity, % | 11–49 | 60 | 35 | + | <0.001 |
| Hyperlipidemia, % | 10–23 | 50 | 0 | + | Not applicable |
| Insulin resistance % (HOMA-IR >2.7) | + | 70 | 15 | + | <0.001 |
| Type 2 diabetes mellitus % | 11–20 | 30 | 0 | + | Not applicable |
| CPP, % | 76–86 | 100 | 100 | — | |
| Menarche, y | Untreated: 7·8b | 12 | |||
| Treated: 11·6b | |||||
| PCOS/infertility, % | Not applicable | 20 | 10 | — | 0.047 |
| References | Kagami et al. (16)Ioannides et al. (15) | Dauber et al. (18) Current study | Current study | Moon et al. (19)Sul (25) | Current study |
Metabolic features were compared between women with DLK1 mutations and idiopathic CPP.
Abbreviations: +, present; −, absent; HOMA-IR, homeostatic model assessment of insulin resistance; KO, knockout; UPD, maternal uniparental disomy of chromosome 14.
DLK1-mutated vs idiopathic CPP (χ2 test).
Age (y) of the menarche in treated and untreated affected women.
Human and Mice Phenotypes Due to DLK1 Deficiency
| Human Phenotype | Mouse Phenotype | Comparison (DLK1 Mutated vs Idiopathic CPPa) | |||
|---|---|---|---|---|---|
| Temple Syndrome | Monogenic CPP Due to DLK1 Defects | Idiopathic CPP | Dlk1 KO | Pa | |
| Origin of study | Japan/United Kingdom | Brazil/United Kingdom | Brazil | United States | |
| Number of women | 32/51 | 10 | 20 | — | |
| Median age (range), y | Children and adults | 23 (16–63) | 16.7 (15–27) | ||
| Genetic basis | UPD(14)mat, epimutation, microdeletion | DLK1 deletion and frameshifts | Unknown | Generation of DLK1-null mice | |
| Short stature, % | 79–94 | 30 | 0 | Pre-postnatal growth retardation | Not applicable |
| Eye and bone alterations, % | — | 10 (syndactyly) | — | Blepharophimosis and rib alterations | — |
| Overweight/obesity, % | 11–49 | 60 | 35 | + | <0.001 |
| Hyperlipidemia, % | 10–23 | 50 | 0 | + | Not applicable |
| Insulin resistance % (HOMA-IR >2.7) | + | 70 | 15 | + | <0.001 |
| Type 2 diabetes mellitus % | 11–20 | 30 | 0 | + | Not applicable |
| CPP, % | 76–86 | 100 | 100 | — | |
| Menarche, y | Untreated: 7·8b | 12 | |||
| Treated: 11·6b | |||||
| PCOS/infertility, % | Not applicable | 20 | 10 | — | 0.047 |
| References | Kagami et al. (16)Ioannides et al. (15) | Dauber et al. (18) Current study | Current study | Moon et al. (19)Sul (25) | Current study |
| Human Phenotype | Mouse Phenotype | Comparison (DLK1 Mutated vs Idiopathic CPPa) | |||
|---|---|---|---|---|---|
| Temple Syndrome | Monogenic CPP Due to DLK1 Defects | Idiopathic CPP | Dlk1 KO | Pa | |
| Origin of study | Japan/United Kingdom | Brazil/United Kingdom | Brazil | United States | |
| Number of women | 32/51 | 10 | 20 | — | |
| Median age (range), y | Children and adults | 23 (16–63) | 16.7 (15–27) | ||
| Genetic basis | UPD(14)mat, epimutation, microdeletion | DLK1 deletion and frameshifts | Unknown | Generation of DLK1-null mice | |
| Short stature, % | 79–94 | 30 | 0 | Pre-postnatal growth retardation | Not applicable |
| Eye and bone alterations, % | — | 10 (syndactyly) | — | Blepharophimosis and rib alterations | — |
| Overweight/obesity, % | 11–49 | 60 | 35 | + | <0.001 |
| Hyperlipidemia, % | 10–23 | 50 | 0 | + | Not applicable |
| Insulin resistance % (HOMA-IR >2.7) | + | 70 | 15 | + | <0.001 |
| Type 2 diabetes mellitus % | 11–20 | 30 | 0 | + | Not applicable |
| CPP, % | 76–86 | 100 | 100 | — | |
| Menarche, y | Untreated: 7·8b | 12 | |||
| Treated: 11·6b | |||||
| PCOS/infertility, % | Not applicable | 20 | 10 | — | 0.047 |
| References | Kagami et al. (16)Ioannides et al. (15) | Dauber et al. (18) Current study | Current study | Moon et al. (19)Sul (25) | Current study |
Metabolic features were compared between women with DLK1 mutations and idiopathic CPP.
Abbreviations: +, present; −, absent; HOMA-IR, homeostatic model assessment of insulin resistance; KO, knockout; UPD, maternal uniparental disomy of chromosome 14.
DLK1-mutated vs idiopathic CPP (χ2 test).
Age (y) of the menarche in treated and untreated affected women.
Discussion
In the current study, we identified three loss-of-function mutations in DLK1 (p.Gly199Alafs*11, p.Val271Cysfs*14, and p.Pro160Leufs*50) in three distinct families whose affected adult members had a clear history of CPP. Initial breast development (thelarche) started from 4.6 to 7 years (median: 5 years), whereas menarche occurred from 7 to 9 years of age in the untreated women. Serum DLK1 concentrations were undetectable in three affected women from families 1 and 3, suggesting that identified mutations impaired DLK1 expression because of increased RNA decay or increased abnormal protein degradation; alternatively, antibody used in the ELISA assay could be targeted to a portion of the protein that is downstream of the frameshifts, and therefore this portion was absent. Similar results were also observed in five affected members of family 4 in our previous study (18). Therefore, serum DLK1 measurement appears to be an accurate screening tool to detect DLK1 deletions or frameshift mutations. The role of serum DLK1 measurement to detect missense mutations, resulting in protein synthesis but impaired function, has yet to be established. These current genetic findings indicate that DLK1 defects represent a definitive genetic cause of familial CPP.
Interestingly, the majority of women who had CPP due to DLK1 deficiency presented with metabolic alterations. The prevalence of overweight/obesity (60%), insulin resistance (70%), type 2 diabetes mellitus (30%), and hyperlipidemia (50%) at adulthood was significantly elevated in the 10 women with DLK1 deficiency who belong to families 1, 2, and 3 from the current description and family 4 from the previous report (18). Notably, the two affected sisters from family 1 who carry the p.Gly199Alafs*11 mutation exhibited truncal obesity associated with type 2 diabetes mellitus, which started before 30 years, and it was disproportionate to their overweight degree. Two other affected women with DLK1 abnormalities, patients 3B and 4E, had obesity and glucose intolerance or type 2 diabetes mellitus, respectively. In addition, four previously reported women (4A, 4B, 4C, and 4D) who carried a complex DLK1 deletion had increased body fat percentage demonstrated by electrical impedance analysis with predominance of visceral abdominal fat (18). The abnormal metabolic phenotype was more frequent in women carrying DLK1 defects than in a group of 20 adult women who had idiopathic CPP (no MKRN3 or DLK1 defects) that was adequately treated with GnRH analogues. The unfavorable metabolic profile associated with DLK1 defects is also more prevalent than in a large previously reported cohort of women with treated and untreated CPP (27 to 50 years) (24). The metabolic findings in adult women with paternally inherited DLK1 deficiency are very similar to those of Dlk1-null mice, which gained body weight more rapidly, had major fat depots and enlarged fatty liver, and showed increased circulating levels of triglycerides, cholesterol, and free fatty acids (Table 3) (19). In addition, when fed with a high-fat diet, these Dlk1-null mice developed impaired insulin resistance and glucose intolerance compared with wild-type mice on the same diet (25).
In vitro studies showed that DLK1 was involved in the differentiation of pancreatic ductal cells into β-like cells; consequently, it favors insulin synthesis and secretion through activation of akt signaling (26). The impairment of pancreatic β-cell differentiation and function associated with increased adipogenesis and insulin resistance caused by DLK1 deficiency could support the finding of early onset of type 2 diabetes mellitus in women who are overweight.
Short stature is a well-known consequence of untreated or late diagnosis of different forms of precocious puberty in humans. A historical series of a limited number of untreated patients with precocious puberty showed mean heights of 152 cm in girls and 156 cm in boys, a loss of approximately 10 cm in girls and 20 cm in boys (27). Here, we demonstrated substantial short stature in the three untreated or undertreated women with familial CPP due to DLK1 pathogenic variants (mean SD height, −3.1 cm). The short stature associated with untreated CPP caused by DLK1 defects in these families seems to be more severe than that reported in historical series. A null mouse model of Dlk1 deficiency resulted in decreased prenatal and postnatal growth in the surviving mice, suggesting a potential direct effect of DLK1 on growth, independent of early puberty (19).
Another intriguing clinical aspect evidenced in family 1 was the diagnosis of PCOS in the two affected sisters with early menarche. PCOS is a complex and heterogeneous disease with a strong genetic influence, characterized by a wide range of components including hyperandrogenism, ovulatory dysfunction, altered gonadotropins secretion, and insulin resistance (28). In a subset of patients, the neuroendocrine abnormality in PCOS may include increased GnRH pulse frequency, which increases the frequency and pulse amplitude of LH over FSH production (29). The abnormality in GnRH pulsatility is well characterized in girls with CPP and could connect both entities. Indeed, it is suggested that patients with CPP are prone to developing PCOS (24, 30). Franceschi et al. (30) demonstrated that 15% of young women with previous idiopathic CPP had oligomenorrhea, and 28% to 48% had hyperandrogenism (30). Lazar et al. (24) reported a higher risk of hyperandrogenism with oligomenorrhea among women with untreated CPP. However, data are still conflicting on the long-term risk of PCOS in women with treated and untreated CPP (31). In the two sisters with a DLK1 pathogenic variant, it is possible that the major ovarian stimulus was determined by the premature hyperactivation of the reproductive axis, and it was worsened by the presence of high insulin levels (insulin resistance) and metabolic alterations. In the face of premature sexual development and PCOS association, we consider that a history of CPP or precocious menarche should be investigated in women with PCOS. In addition, loss-of-function variants of DLK1 should be screened in women with PCOS and a history of CPP or precocious menarche, especially when associated with a poor metabolic profile. However, menstrual cycles and fertility were preserved in the other women from families 2, 3, and 4, indicating that reproductive alterations could occur in a low prevalence in women with DLK1 deficiency or may depend on other genetic and environmental factors. Phenotypic variability could explain the distinct frequencies of some clinical components of the human DLK1 deficiency. Although CPP was diagnosed in 100% of patients with a mutation, PCOS, a condition that can cause infertility, was found in only 20%. Notably, two women (patients 3B and 4E) with a DLK1 deficiency had a history of menopause after 50 years.
Prior to this report, only a single family was reported with isolated DLK1 deficiency causing CPP (18). Here, we present an additional five women from three families, thereby establishing that loss-of-function mutations in DLK1 are a definitive cause of familial CPP. We also report metabolic and reproductive abnormalities in adult family members harboring DLK1 mutations, in particular central obesity, early type 2 diabetes mellitus, and hyperlipidemia aligned with the phenotype of Dlk1 deficiency already described in a null-mouse model (19, 25). The high prevalence of metabolic alterations in adult women who have experienced CPP due to DLK1 defects suggests that this antiadipogenic factor represents a link between reproduction and metabolism.
Abbreviations:
- BMI
body mass index
- CPP
central precocious puberty
- DLK1
delta-like homolog 1
- EGF
epidermal growth factor
- MKRN3
makorin ring finger 3
- PCOS
polycystic ovary syndrome
Acknowledgments
Financial Support: This work was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (302849/2015-7CNPq) and Fundação de Amparo à Pesquisa do Estado de São Paulo (2016/14803-6 FAPESP) (to A.C.L); (2015/17350-0 FAPESP) Coordenação de Aperfeiçoamento de Pessoas de Nível Superior (2014/1459789 CAPES; to L.G.G.); and Conselho Nacional de Desenvolvimento Científico e Tecnológico (153237/2016-3 CNPq; to R.P.C.), (301871/2016-7 CNPq; to A.A.L.J.), (303002/2016-6; to B.B.M), and [2014/50137-5; to Laboratório de sequenciamento em larga escala (SELA)].
Disclosure Summary: The authors have nothing to disclose.
References
Author notes
L.G.G., M.C.-S., V.N.B., and A.C.L. contributed equally to this study.

{kind=link}
{kind=link}
{kind=link}