





Abstract
Androgen insensitivity syndrome (AIS) is the most common cause of disorders of sex development in 46,XY individuals. It is an X-linked condition usually caused by pathogenic allelic variants in the androgen receptor (AR) gene. The phenotype depends on the AR variant, ranging from severe undervirilization (complete AIS) to several degrees of external genitalia undervirilization. Although 90% of those with complete AIS will have AR mutations, this will only be true for 40% of those with partial AIS (PAIS).
To identify the genetic etiology of AIS in a large multigenerational family with the PAIS phenotype.
Nine affected individuals with clinical and laboratory findings consistent with PAIS and a normal exonic AR sequencing
Endocrine clinic and genetic institute from two academic referral centers
Analysis of whole exons of the AR gene, including splicing regions, was performed, followed by sequencing of the 5′untranslated region (UTR) of the AR gene. Detailed phenotyping was performed at the initial diagnosis and long-term follow-up, and circulating levels of steroid gonadal hormones were measured in all affected individuals. AR expression was measured using RT-PCR and cultured fibroblasts.
All 46,XY family members with PAIS had inherited, in hemizygosity, a complex defect (∼1100 bp) in the 5′UTR region of the AR surrounded by a duplicated 18-bp sequence (target site duplication). This sequence is 99.7% similar to an active, long, interspersed element present on the X chromosome (AC002980; Xq22.2), which was inserted in the 5′UTR of the AR gene, severely reducing AR expression and leading to PAIS.
The molecular diagnosis of PAIS remains challenging. The genomic effect of retrotransposon mobilization should be considered a possible molecular cause of AIS and other AR diseases.
Androgen insensitivity syndrome (AIS) is an X-linked disorder and the most common etiology of disorders of sex development in 46,XY individuals (1). In this condition, normal androgen receptor (AR) action is lacking, resulting in several degrees of external genitalia undervirilization, which range from typically female external genitalia in the complete form of AIS to typically male external genitalia associated with infertility and/or gynecomastia in the mild form of AIS. Between these two extreme phenotypes exists a spectrum of male external genitalia undervirilization, characterized as a partial form of AIS (PAIS) (2). More than 500 mutations have been identified in the AR in AIS (3). Although AR sequencing will be able to identify allelic variants in >90% of patients with the complete form of AIS, this will be true for only 40% of patients with PAIS, suggesting that other factors outside the AR gene might lead to AIS (1, 3).
The human genome, similar to that of other eukaryotes, comprises largely interspersed repeats. Most of these repeats result from mobile DNA elements, which are DNA elements with the ability to move and insert themselves into other locations in the genome. Collectively, they are termed mobile DNA and constitute 50% of the human genome (4, 5). Their mobilization has unquestionably contributed to human evolution as a source of both heritable structural variation and somatic mutations in the genome. Transposable elements are divided into two major groups: DNA transposons and RNA transposons (retrotransposons); however, only the latter are active in human DNA. Long interspersed element 1 (LINE-1) retrotransposons are a substantial source of endogenous mutagenesis in humans. They use a “copy-and-paste” mechanism to retrotranspose, inserting themselves into new genomic loci. If, on the one hand, transposable elements are important players in genome evolution, on the other hand, they can cause human diseases by insertion, homologous recombination, genetic instability, and the introduction of sequences into cis-trans signals, affecting gene expression (4, 6, 7).
Subjects
We studied nine individuals with 46,XY karyotype from the same family with severe undervirilization of external genitalia at birth (Prader stage 3; enlarged phallus, single urogenital sinus, and almost complete fusion of the labia). Five of the nine individuals had been assigned and reared as female (III:8, III:13, III:14, III:15, and IV:1). The remaining four had been assigned as male at birth (II:1, II:2, II:16, and III:11). The older affected individuals had been assigned as male, and the younger individuals had been assigned as female. The decision to assign the younger individuals as female was because of the lack of virilization observed in the older individuals.
The family pedigree suggested an X-linked pattern of inheritance (Fig. 1). Patient III:11 (index case) had come to our hospital at 27 years old with a history of atypical genitalia at birth. He had been assigned as male at birth. His parents were consanguineous. At puberty, he had developed gynecomastia. At 18 years old, he had undergone bilateral orchiectomy and masculinizing genitoplasty, followed by mastectomy. He had been treated with high doses of intramuscular testosterone esters for 6 months between surgical procedures without any substantial virilization. On physical examination, he had a high-pitched voice, a complete absence of facial and corporal hair, and a penile size of 6.0 × 2.0 cm. No Müllerian derivatives were observed on the ultrasound examination.
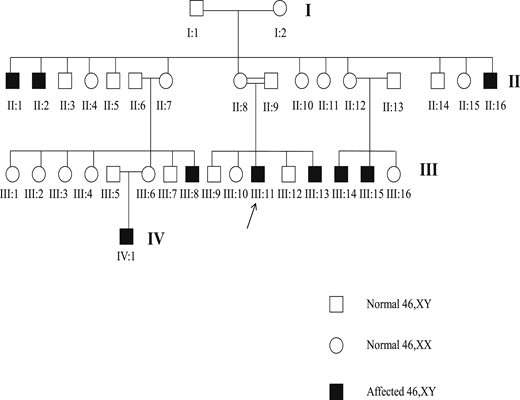
Familial pedigree. The individuals III:8, III:13, III:14, III:15, and IV:1 were assigned as female and the individuals II:1, II:2, II:16, and III:11 were assigned as male.
Patient III:8, a maternal cousin of the index case had come to our hospital at 9 years old. She had presented with atypical genitalia at birth. She had been assigned as female and had undergone bilateral gonadectomy at 6 months of age. At 9 years old, she had presented with atypical genitalia, with a 3.8 × 1.5 cm phallus and a single perineal opening. No breast tissue or pubic hair were found on physical examination. The genitogram showed a 2-cm blind vagina. Her karyotype was 46,XY.
Patient III:13, a younger sister of patient III:11, had come to our hospital at 4 years old, with a referral because of clitoral enlargement and the presence of a mass in the inguinal region. She had been assigned and raised as female. The physical examination showed clitoromegaly (3.2 × 1.0 cm) and a single perineal opening. Urethrocystography showed a blind vagina. At 5 years old, she underwent bilateral gonadectomy, and no malignancy was identified. At 11 years old, she started estrogen replacement, with satisfactory breast development.
Patient III:14, a maternal cousin of the index case, had come to our hospital at 17 years old because of clitoromegaly. At 3 years old, she had undergone bilateral gonadectomy. At 13 years old, she had started estrogen replacement, with the development of increased breast tissue. At our hospital, she underwent clitoroplasty and started estrogen replacement therapy.
Patient IV:1, a maternal cousin of patient III:11, had been assigned as female. The physical examination revealed a phallus measuring 2.2 × 0.9 cm and a single perineal opening. Urethrocystography showed a blind vagina, and no Müllerian derivatives were observed on ultrasound examination. Her karyotype was 46,XY. At 3 months of age, a human chorionic gonadotropin (hCG) test was performed using four intramuscular injections of 50 to 100 IU/kg each, with a 4-day interval between the injections. The serum levels of testosterone were measured before and 72 hours after the last hCG injection (Table 1). The basal level of LH was high. An increase in the testosterone levels was found after hCG testing, similar to that reported in patients with AIS.
Laboratory Data From Subject IV:1
| Variable | LH (U/L) | FSH (U/L) | Testosterone (ng/dL) | 17OHP (ng/mL) | Androstenedione (ng/mL) | DHT (ng/dL) | T/DHT Ratio |
|---|---|---|---|---|---|---|---|
| Basal | 16.4 | 4.8 | 191 | 0.73 | 0.29 | 24.8 | 7.7 |
| 48 Hours after hCG stimulation | 30.3 | <4 | 332 | 2.38 | 0.5 | 34.3 | 9.6 |
| Reference | |||||||
| Basal | <10 | 0–10 | <30 | 0.1–1.1 | 0.2–0.6 | 3–9 | — |
| After hCG | — | 150 | — | — | 7–22 | 14 ± 5 |
| Variable | LH (U/L) | FSH (U/L) | Testosterone (ng/dL) | 17OHP (ng/mL) | Androstenedione (ng/mL) | DHT (ng/dL) | T/DHT Ratio |
|---|---|---|---|---|---|---|---|
| Basal | 16.4 | 4.8 | 191 | 0.73 | 0.29 | 24.8 | 7.7 |
| 48 Hours after hCG stimulation | 30.3 | <4 | 332 | 2.38 | 0.5 | 34.3 | 9.6 |
| Reference | |||||||
| Basal | <10 | 0–10 | <30 | 0.1–1.1 | 0.2–0.6 | 3–9 | — |
| After hCG | — | 150 | — | — | 7–22 | 14 ± 5 |
Abbreviations: 17OHP, 17-hydroxyprogesterone; DHT, dihydrotestosterone; T/DHT, testosterone/dihydrotestosterone.
Laboratory Data From Subject IV:1
| Variable | LH (U/L) | FSH (U/L) | Testosterone (ng/dL) | 17OHP (ng/mL) | Androstenedione (ng/mL) | DHT (ng/dL) | T/DHT Ratio |
|---|---|---|---|---|---|---|---|
| Basal | 16.4 | 4.8 | 191 | 0.73 | 0.29 | 24.8 | 7.7 |
| 48 Hours after hCG stimulation | 30.3 | <4 | 332 | 2.38 | 0.5 | 34.3 | 9.6 |
| Reference | |||||||
| Basal | <10 | 0–10 | <30 | 0.1–1.1 | 0.2–0.6 | 3–9 | — |
| After hCG | — | 150 | — | — | 7–22 | 14 ± 5 |
| Variable | LH (U/L) | FSH (U/L) | Testosterone (ng/dL) | 17OHP (ng/mL) | Androstenedione (ng/mL) | DHT (ng/dL) | T/DHT Ratio |
|---|---|---|---|---|---|---|---|
| Basal | 16.4 | 4.8 | 191 | 0.73 | 0.29 | 24.8 | 7.7 |
| 48 Hours after hCG stimulation | 30.3 | <4 | 332 | 2.38 | 0.5 | 34.3 | 9.6 |
| Reference | |||||||
| Basal | <10 | 0–10 | <30 | 0.1–1.1 | 0.2–0.6 | 3–9 | — |
| After hCG | — | 150 | — | — | 7–22 | 14 ± 5 |
Abbreviations: 17OHP, 17-hydroxyprogesterone; DHT, dihydrotestosterone; T/DHT, testosterone/dihydrotestosterone.
The X-linked pattern of inheritance, absence of Müllerian derivatives, atypical genitalia, and the presence of gynecomastia at puberty were suggestive of AIS as the cause of the 46,XY disorders of sex development in this family. Patients II:1, II:2, II:16, and III:15 were not evaluated at our hospital.
Methods
Hormonal analysis
Serum LH and FSH were determined using a commercial 125I double antibody RIA (Diagnostic Products Corp., Los Angeles, CA) or by immunofluorometric assays (AutoDelfia; Wallac, Inc., Turku, Finland). The serum 17-hydroxyprogesterone, dehydroepiandrosterone, androstenedione, testosterone, and estradiol levels were determined after extraction with ethyl acetate and/or hexane, without previous chromatography. Serum dihydrotestosterone was measured by RIA after extraction with ethyl acetate and hexane and purification on Celite columns.
Molecular studies
Genomic DNA was obtained from peripheral blood leukocytes using the proteinase K-SDS salting-out method. The entire AR (NC_000023.11/ENSG00000169083/ENST00000374690) coding region, the exon–intron boundary areas of exons 1 to 8, and the AR promoter region were PCR amplified using specific primers. The primers used for the AR were designed using the Primer3 software (http://bioinfo.ut.ee/primer3-0.4.0/). The PCR products were sequenced using the protocol of the ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction Kit (Life Technologies Corp., Carlsbad, CA) and submitted to the ABI Prism Genetic Analyzer 3130XL (Life Technologies Corp.).
Quantitative RT-PCR
Fibroblasts were obtained from a genital skin biopsy (3-mm diameter punch) of the III:11 patient’s forearm and from a normal 46,XY male individual. These tissues were cultured in a 75-cm2 flask containing DMEM, 10% fetal bovine serum, and 1% antibiotic. The RNA was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol, with the use of DNAse I. RT-PCR was performed using the High Capacity cDNA Archive Kit (Applied Biosystems, Thermo Fisher Scientific, Inc., Waltham, MA). The AR gene quantitative expression level was determined using quantitative RT-PCR using the TaqMan system (Hs00171172.m1; Applied Biosystems, Thermo Fisher Scientific Inc.). The genes ACTB (4326315E), CYC, and TBP (4326322E; Applied Biosystems, Thermo Fisher Scientific Inc.) were used as endogenous controls. The AriaMx real-time PCR system (Agilent Technologies Inc., Santa Clara, CA) was used to perform the reaction. The experiments were performed in triplicate and were repeated in three independent assays. The relative expression levels were analyzed using the 2−∆∆Ct method. The mean expression of the target genes in the control’s skin fibroblast culture was assigned an expression value of 1.0, and a onefold decrease in the expression levels was determined for the patient’s skin fibroblast culture.
Consent
All the subjects included in the present study had provided informed consent.
Results
Whole exon sequencing of the AR gene, including all eight exons and splice site regions, was normal in all affected patients. However, sequencing of the AR 5′untranslated region (UTR) found a large insertion. The length of the band indicated by agarose gel electrophoresis was 1100 bp larger than that of the control. All the affected subjects had presented with the same insertion in the 5′UTR of the AR gene, and all nonaffected 46,XY individuals (II:5, III:7, and III:12) studied from the same family had a normal 5′UTR AR sequence.
The insertion was flanked by a duplication of the AR 5′UTR sequence of 18 bp (5′-AAGATCTGGGCATCTTT-3′), which was compatible with target site duplication (TSD) (Fig. 2). Long-range PCR was able to determine an insertion of 804 bp of a LINE-1 sequence, followed by a long poly(A) tail (∼200 bp). This long poly(A) tail was additional evidence that this was a retrotransposon (Fig. 2). The insertion and the long poly(A) tail had just one nucleotide change from the 3′ end of a full-length, young LINE-1 sequence present on Xp22.2 (AC002980), suggesting the presence of a 5′UTR truncated LINE-1 sequence (Fig. 2). A canonical LINE-1 endonuclease cleavage site was identified immediately before the TSD (5′-TTTT/AA-3′). The 5′ end of the insertion contained 11 thymines (Ts), suggesting that a rare inversion had occurred with the poly(A) tail (8). The AR gene expression in the patient’s fibroblast culture was 10-fold less than that of the control (0.0976-fold; SD = 0.026; P = 0.03; Fig. 2).
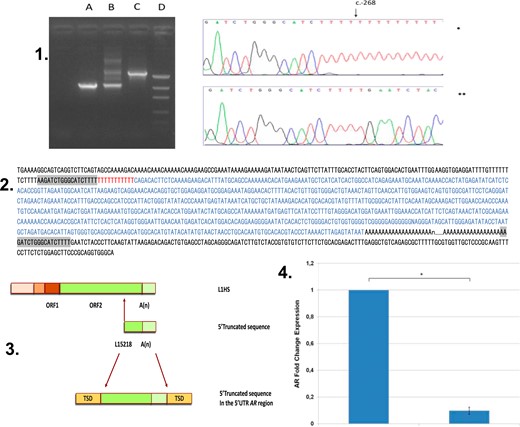
Molecular evidences of LINE-1 insertion. (Upper; 1) Analysis of the LINE-1 insertion site. (Left) Lane A, Agarose gel, normal 46,XY control; lane B, mother of affected individual; lane C, affected individual; and lane D, DNA molecular weight marker. (Right) Sanger sequencing of the 5′UTR AR gene (5′-3′). The insertion starts as a poly(T) at c.-268 position (*index case; **normal male as control); (Middle; 2) Sequencing of the 5′UTR region of AR in the propositus. The 18-bp TSD is marked in gray. The entire inserted LINE-1 sequence is marked in blue. The 11-bp poly(T) is shown in red. This insertion is composed of 804 nt plus poly(A) sequence. This sequence is 99.7% identical to the 3′ end of the putative parental LINE-1HS sequence (AC002980). (3. Schematic representation of the full-length human LINE-1HS. (Upper; 3) The 5′-truncated form of the LINE-1 (nucleotides 5218 to 6021) with 3′-poly(A) tracts. (Middle; 3) Insertion in the 5′UTR region in the AR gene flanked by 18-bp TSD (5′-AAGATCTGGGCATCTTTT-3′) (lower; 3). (4) AR gene expression comparison between affected individual and control.
Discussion
The familial pedigree of these patients clearly suggested X-linked inheritance. The diagnosis of AIS was corroborated by clinical evidence of external genitalia undervirilization, hormonal levels, the absence of Müllerian derivatives, and spontaneous gynecomastia. However, the sequencing of the AR gene was normal in all the affected individuals of this family.
Molecular diagnosis remains a challenge for individuals with mutated-negative AR and clinical evidence of AIS. However, the study of these individuals presents an opportunity to expand our understanding of how the AR gene works. Some individuals with mutated-negative AR showed reduced transcriptional activity of the AR gene. Collectively, this condition was named AIS type II, which has been more the rule than the exception among individuals with PAIS, suggesting that factors outside the AR gene might be able to have an effect on AR function (9). However, the results from high-throughput DNA tests failed to increase the frequency of the molecular diagnosis of AIS (10).
Because the family we have studied showed strong evidence of AIS, we sequenced the 5′UTR region of the AR gene and found a large insertion with an X-linked pattern of segregation (Fig. 1). Analysis of AR expression showed highly reduced expression levels in the affected individuals compared with that of the controls, reinforcing the pathogenicity of this insertion.
The AR 5′UTR insertion had hallmarks of retrotransposition, including a TSD composed of 18 nucleotides, the presence of a poly(A) tail, and an endonuclease cleavage motif (5′-TTTT/AA-3′) at the 5′ end of the insertion. The whole insertion had only one nucleotide change from the putative parental LINE-1 sequence on Xp22.2 (AC002980), a canonical LINE-1 sequence that belongs to the Ta-1d group of human LINE-1s, which had the highest activity among the LINE-1 sequences in a cultured cell assay (11).
L1 sequences in the human genome have usually been polymorphic (33% to –49%), and most LINE-1-related diseases have been caused by 5′-truncated insertions (12). In humans, LINE-1 sequences are widespread along all chromosomes (7). Moreover, the density of transposable elements in the X chromosome is twice (26%) that in the autosomes (∼13%) (13). Mobile elements are likely more frequent in the X chromosome owing to the high A+T content of this chromosome. As a consequence, approximately one third of LINE-1-inherited conditions are X-linked (14). Among 124 inherited human diseases associated with retrotransposition events, there is only one LINE-1 sequence inserted in the 5′UTR region of a gene, the DMD gene, causing X-linked dilated cardiomyopathy (15).
In the AIS family studied, a T (polyT) stretch with 11 bp of nontemplate thymine was present between the 5′ TSD and the LINE-1 sequence. Genome-wide analysis demonstrated the presence of homopolymeric poly(T) stretches upstream of several truncated LINE-1 insertions (8). A computational analysis of several poly(T) stretches of 5′ sense-oriented LINE-1s suggested that these poly(T) stretches are an antisense poly(A) tail of a complex retrotransposon insertion involving an inverted L1 sequence (8). This interesting phenomenon likely results from a dual priming event, which involves priming of both the bottom and the top-strand nicks by two different open reading frame 2p reverse transcription, resulting in homopolymeric events, such as poly(T) stretches. A previous study found 5′ poly(T) stretches ranging from 14 to 39 bp, with an average of 23 bp (8). The poly(T) stretch found in the present study was slightly shorter than those previously reported.
In conclusion, the molecular diagnosis of PAIS remains a challenge. We have presented a case of PAIS due to retrotransposon insertion, characterizing this syndrome as an L1-inherited condition. Examples of LINE-1-related human diseases have been increasing owing to improvements in the detection of such events. This has helped to expand the genetic causes of inherited diseases, especially those of an unknown etiology. These findings have expanded the possibilities of a molecular diagnosis for AIS. Consequently, the genomic effect of retrotransposon mobilization should be considered as a possible molecular cause of AIS and other AR diseases.
Acknowledgments
We thank Alisha Soares for her technical support.
Financial Support: The present study was partially supported by Grant 303002/2016-6 to B.B.M. H.H.K. was supported by Grant 5R01GM099875-08 from the National Institute of General Medical Studies, National Institutes of Health, and J.L.G. by Grant 1R21AG056840-01A1 from the National Institute on Aging, National Institutes of Health.
Data Availability: All data generated or analyzed during this study are included in this published article or in the data repositories listed in References.
Abbreviations:
- AIS
androgen insensitivity syndrome
- AR
androgen receptor
- hCG
human chorionic gonadotropin
- LINE-1
long interspersed element 1
- PAIS
partial androgen insensitivity syndrome
- T
thymine
- TSD
target site duplication
- UTR
untranslated region
References and Notes
Author notes
E.M.F.C., H.H.K., and B.B.M. share last authorship for the present report.

{kind=link}
{kind=link}