





Abstract
Congenital hypopituitarism (CH) is rarely observed in combination with severe joint contractures (arthrogryposis). Schaaf-Yang syndrome (SHFYNG) phenotypically overlaps with Prader-Willi syndrome, with patients also manifesting arthrogryposis. L1 syndrome, a group of X-linked disorders that include hydrocephalus and lower limb spasticity, also rarely presents with arthrogryposis.
We investigated the molecular basis underlying the combination of CH and arthrogryposis in five patients.
The heterozygous p.Q666fs*47 mutation in the maternally imprinted MAGEL2 gene, previously described in multiple patients with SHFYNG, was identified in patients 1 to 4, all of whom manifested growth hormone deficiency and variable SHFYNG features, including dysmorphism, developmental delay, sleep apnea, and visual problems. Nonidentical twins (patients 2 and 3) had diabetes insipidus and macrocephaly, and patient 4 presented with ACTH insufficiency. The hemizygous L1CAM variant p.G452R, previously implicated in patients with L1 syndrome, was identified in patient 5, who presented with antenatal hydrocephalus.
Human embryonic expression analysis revealed MAGEL2 transcripts in the developing hypothalamus and ventral diencephalon at Carnegie stages (CSs) 19, 20, and 23 and in the Rathke pouch at CS20 and CS23. L1CAM was expressed in the developing hypothalamus, ventral diencephalon, and hindbrain (CS19, CS20, CS23), but not in the Rathke pouch.
We report MAGEL2 and L1CAM mutations in four pedigrees with variable CH and arthrogryposis. Patients presenting early in life with this combined phenotype should be examined for features of SHFYNG and/or L1 syndrome. This study highlights the association of hypothalamo-pituitary disease with MAGEL2 and L1CAM mutations.
Schaaf-Yang syndrome (SHFYNG) [Online Mendelian Inheritance in Man (OMIM): 615547] is a rare congenital disorder that is often misdiagnosed as Prader-Willi syndrome (PWS) (OMIM: 176270) but includes arthrogryposis within the phenotypic spectrum. Arthrogryposis multiplex congenita (OMIM: 208100), commonly known as arthrogryposis, occurs in 1 of 3000 live births and involves multiple congenital joint contractures in two or more areas of the body, resulting from reduced or absent fetal movement. Arthrogryposis multiplex congenita has been reported in a patient with pituitary ectopia who had seizures thought to be caused by hypoglycemia and was later found to have a small anterior pituitary and an ectopic posterior pituitary (PP); however, no genetic etiology was identified (1). The main overlapping characteristic features of SHFYNG and PWS are hypotonia, feeding difficulties during infancy, global developmental delay/intellectual disability, and sleep apnea (2–4). However, patients with SHFYNG lack certain stereotypical PWS features, such as hyperphagia and subsequent obesity. PWS is linked to the specific locus 15q11-q13 within the genome, where five maternally imprinted (paternally expressed) genes, namely MKRN3, MAGEL2, NDN, NPAP1, and SNURF-SNRPN, and six maternally imprinted small nucleolar RNA genes/clusters are located (3). Different deletions in this region give rise to variable PWS, with a combination of genes being responsible for different manifestations of the disease (5–7).
L1 syndrome describes a range of X-linked disorders including spastic paraplegia, MASA syndrome (mental retardation, aphasia, spasticity, and adducted thumbs; OMIM: 303350), X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (OMIM: 307000), and X-linked complicated corpus callosum agenesis (8). L1 syndrome occurs in 1 in 30,000 individuals and includes hydrocephalus, variably severe intellectual deficits, and spasticity of the lower limbs, with generalized contractures in rare cases. MASA syndrome, named after the characteristic phenotypes present in patients, also includes adducted thumbs in 50% of cases. A small number of patients (<20) have a combination of L1 syndrome and Hirschsprung disease, a rare disorder affecting the colon and leading to severe constipation and intestinal obstruction due to missing ganglion cells in the myenteric (Auerbach) plexus in the colon (9).
In this study, we sought to investigate genetic etiology in five patients from four unrelated families who presented with variable congenital hypopituitarism (CH) and arthrogryposis.
Materials and Methods
Exome sequencing of patients 1 to 5
The full coding regions of patients 1 to 5 were sequenced as follows: by GOSgene, London, UK (patients 1 and 5); by Great Ormond Street Hospital (GOSH) UK as part of the Deciphering Developmental Disorders (DDD) Study (patients 2 and 3); and by colleagues at the Pontificia Universidad Católica de Chile (patient 4). Raw sequencing data were mapped against the GRCh37/hg18 reference genome, and data were analyzed using Ingenuity® Variant Analysis™ software (https://www.qiagenbioinformatics.com/products/ingenuity-variant-analysis) from Qiagen, Inc (GOSgene Manchester, United Kingdom). All remaining filtered variants were considered potentially pathogenic disease-causing mutations. Exome sequencing and data analysis for patients 1 and 5 were performed by GOSgene as previously described (10), for patients 2 and 3 under the DDD study as previously described (11), and for patient 4 by Ambry Genetics (Aliso Viejo, CA) (www.ambrygen.com) using its standard protocol and filtering criteria. Mutations were confirmed in patients via Sanger sequencing using specifically designed exon-spanning primers that amplify the DNA region containing the variant (annealing temperatures and primer sequences are available upon request). A chromosome microarray was also performed on the twins, patients 2 and 3 (specific details of this protocol are available upon request). The appropriate ethical approval for the genetics and human embryonic tissue expression studies was obtained before this project took place. The patients/patient guardians gave full consent to all clinical and genetic studies carried out on their blood/DNA.
Human embryonic expression studies using in situ hybridization
Human embryonic tissue sections were obtained from the Human Developmental Biology Resource (http://hdbr.org) and were selected from Carnegie stages (CSs) 16, 19, 20, and 23 [equivalent to gestational ages (GAs) 5.5, 6, 7, and 8 weeks, respectively]. Digoxigenin RNA probes were made using purified vectors containing the full-length human cDNA of wild-type MAGEL2 (in the pCR4-TOPO vector; IMAGE ID: 8327725) and L1CAM (in the pCR-XL-TOPO vector; IMAGE ID: 8991945) (Source BioScience, Nottingham, United Kingdom) Gene expression studies were performed by in situ hybridization as previously described (12) to generate a human embryonic hypothalamo-pituitary expression profile for both MAGEL2 and L1CAM.
Results
Patient 1
A white European patient presented at the age of 3.2 years with short stature, hypoglycemia, and arthrogryposis with scoliosis and a flexion deformity of the knees. She had been hypotonic since birth and required nasal oxygen until 5 weeks of age. A respiratory collapse at 7 weeks of age necessitated a prolonged pediatric intensive care unit admission. She was also noted to have laryngeal polyps. She was diagnosed with growth hormone deficiency (GHD), with a peak GH of 6.4 μg/L and an undetectable IGF-I concentration at age 3.7 years (Table 1). GH treatment was commenced at 4 years of age [Fig. 1(A)]. Dysmorphic features were noted, including bulbar palsy, a long face, a prominent forehead, and micrognathia. She also had global developmental delay and a squint with mild optic nerve hypoplasia and cerebral visual impairment. She had central sleep apnea and gastroesophageal reflux. MRI of the brain was reported as normal [Fig. 2(A)].
Phenotypes of Patients With MAGEL2 and L1CAM Mutations
| Patient/Sex (M/F) | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 |
|---|---|---|---|---|---|
| F | F | F | M | M | |
| Ethnicity | White European | White European | White European | White Chilean | Afro-Caribbean |
| Mutation | c.1996dup, p.Q666Pfs*47 | c.1996dup, p.Q666Pfs*47 | c.1996dup, p.Q666Pfs*47 | c.1996dupC, p.Q666Pfs*47 | Hemizygous |
| MAGEL2, NM_019066 | MAGEL2, NM_019066 | MAGEL2, NM_019066 | MAGEL2, NM_019066 | c.1354G>A, p.G452R | |
| De novo | Not maternally inherited; paternal DNA was unavailable | Not maternally inherited; paternal DNA was unavailable | De novo | L1CAM, NM_000425 | |
| Chromosome microarray showed a 16q11 duplication, 45,186,600–45,416670); Nimblegen 135K Hg18 WG CGH v3.1. | Chromosome microarray: normal | Chromosome microarray: normal | Mother is a heterozygous carrier | ||
| Family history | Healthy younger siblings | Healthy older sister | Healthy older sister | Healthy older brother | |
| 2× miscarriages at 7/40 | 2× miscarriages at 7/40 | Mother had bilateral metatarsus varus and a history of recurrent shoulder dislocations | |||
| Prev. stillbirth at 37/40 | Prev. stillbirth at 37/40 | ||||
| Perinatal history | Normal antenatal scans | Polyhydramnios | IUGR and polyhydramnios | — | Antenatal ventriculomegaly |
| Cesarean; breech | DCDA twin pregnancy | DCDA twin pregnancy | Elective LSCS | ||
| Hypotonic and respiratory distress after birth needing oxygen | Cesarean; breech | Cesarean; breech | Born in good condition | ||
| Resuscitation at birth: APGARs – 2 at 1 min; 1 at 5 min | |||||
| Cooled for HIE | |||||
| Ventilated to day 4 of life | |||||
| Gestation, wk | 40 | 36 + 1/40 | 36 + 1/40 | 38 | 40 |
| Birth weight, kg (SDS) | NA | 2.7 (+0.14) | 2.0 (−1.67) | 3.5 (0.07) | 3.6 (+0.07) |
| Endocrine referral | Short stature and previous history of hypoglycemia | Short stature, diabetes insipidus | Short stature, diabetes insipidus | Growth evaluation, short stature | Hypoglycemic seizures |
| Age at referral, y | 3.2 | 1.10 | 1.10 | 2.8 | 0.7 |
| Height, SDS | −2.14 | −1.3 | −2.0 | −2.84 | −1.7 |
| BMI, SDS | 0.61 | +1.52 | −1.52 | −0.92 | −4.21 |
| Current age, y | 9.7 | 7.6 | 7.6 | 4.7 | 5.0 |
| Height, cm (SDS) | 114.8 (−3.21) | 120.1 (−0.58) | 120.8 (−0.45) | 99 (−1.8) | 99.3 (−1.95) |
| Weight, kg (SDS) | 23.5 (−1.76) | 20.55 (−1.3) | 21.15 (−1.1) | 12.2 (−3.53) | 12.8 (−3.55) |
| Dysmorphic facial features | Long face | Macrocephaly | Macrocephaly | Long face | Bilateral radial clubbed hands |
| Prominent forehead | Long face | Long face | Bitemporal narrowing | Plagiocephaly | |
| Micrognathia | Bitemporal narrowing | Bitemporal narrowing | Prominent forehead | ||
| Prominent forehead | Prominent forehead | Micrognathia | |||
| Hypotelorism | Hypotelorism | Glossoptosis | |||
| Scaphocephalic shape | Scaphocephalic shape | High arched palate | |||
| Micrognathia | Micrognathia | ||||
| Cleft palate | High arched palate | ||||
| Gastrointestinal | GERD | Constipation | Constipation | Gastrostomy for feeding difficulty, gastro-esophageal reflux; chronic constipation | PEG fed |
| Food allergy | Unsafe swallow | ||||
| Dysphagia | |||||
| Constipation | |||||
| Cardiac | No known cardiac pathology | No known cardiac pathology | No known cardiac pathology | Ostium secundum interauricular communication, spontaneous closure | Small VSD |
| Neurodevelopment | Global developmental delay | Severe global developmental delay | Global developmental delay | Global developmental delay | Global developmental delay |
| Bulbar palsy | Gross Motor Function Classification System Level 4 | Independently walking | Autism spectrum disorder | Generalized hypotonia | |
| Wheelchair dependent, nonverbal | Communicates using Makaton, eye gaze, and pointing | Hypotonia | |||
| Able to sit if put in sitting position; attempts rolling over | Some vocalization | Some vocalization | |||
| Some self-harm behavior: head banging | Walking with help | ||||
| Able to sit independently | |||||
| Skeletal system | Arthrogryposis | Distal arthrogryposis | Distal arthrogryposis | Contractures with limited extension of the elbows, knees, hips, and finger | Arthrogryposis |
| Scoliosis | Scoliosis | Hands/fingers tapering | Camptodactyly | Bilateral radial clubbed hands | |
| Flexion deformity of knees | Overlapping fifth finger, positions fingers 3/4/5 together | Small hands and feet | Right hip subluxation | ||
| Short long bones | Scoliosis | ||||
| 11 ribs | |||||
| Ophthalmology | Squint | Optic nerve hypoplasia and severe sight impairment | Mild optic nerve hypoplasia (fundoscopy) and sight impaired | Strabismus | Bilateral astigmatism, left divergent squint (visual impairment); wears glasses |
| Mild optic nerve hypoplasia with cerebral visual impairment | Nystagmus | ||||
| Myopic astigmatism | |||||
| Endocrine assessment | GHD | Central diabetes insipidus since birth – on DDAVP | Central diabetes insipidus since birth – on DDAVP | GHD stimulation test not performed as hypotensive | GHD |
| Rest of endocrinology normal | Severe GHD | Severe GHD | Short stature with deceleration of growth rate | ||
| Premature thelarche – resolved | Premature thelarche – resolved | Adrenal insufficiency: peak cortisol 10.2 μg/dL on stimulation; rest normal | |||
| Premature pubarche | Premature pubarche | Cryptorchidism with bilateral orchidopexies | |||
| Other features | N/A | Hypoxic ischemic encephalopathy at birth – cooled | Percutaneous endoscopic gastrostomy tube in situ for unsafe swallow | Central sleep apnea | Hydrocephalus (VP shunt soon after birth) |
| Overhanging epiglottis | Central sleep apnea and hypoxia – no need for supplemental oxygen | ||||
| Percutaneous endoscopic gastrostomy tube in situ for unsafe swallow | Submandibular gland excision for excessive drooling | ||||
| Previous ovarian cyst on ultrasonography | Previous ovarian cyst on ultrasonography | ||||
| Diaphragmatic eversion | |||||
| Pierre Robin sequence without cleft palate. | |||||
| GH axis, age, y | 3.7 | 0.8 | 0.8 | 0.3 | 0.8 |
| Stimulation test | Glucagon | Overnight GH profile | Overnight GH profile | Not performed because of hypotension | Glucagon |
| Basal, ug/L | 0.6 | — | — | — | 2.4 |
| Peak, ug/L (NR > 6.7) | 6.4 | 4.8 (only 1 peak max 4.8) | 3.2 (only 1 peak max 3.2) | — | 3.7 |
| IGF-1, ng/mL (NR) | <25 | Undetectable | Undetectable | 31.9 (49–289) | <25 |
| Thyroid axis, age, y | 3.7 | 0.8 | 0.8 | 0.3 | 0.6 |
| fT4 pmol/L (NR) | 14.4 (9.8–19) | 13.8 (9.8–19) | 11.3 (9.8–19) | 18.8 (10.3–27) | 12.8 (9.8–19) |
| TSH mU/L (NR) | 2.8 (<6.0) | 1.9 (<6.0) | 1.1 (<6.0) | 2.87 (<6.0) | 1.5 (<6.0) |
| Adrenal axis | |||||
| Stimulation test (age, y) | Glucagon (3.7) | (0.8) | (0.8) | Synacthen (2.8) | Glucagon (0.7) |
| Peak cortisol, nmol/L | 989 | — | — | 281 | 488 |
| Random cortisol nmol/L (time) | Basal 252 (9 am) and 442 (12 noon at the end of the fast) | Random cortisol 1192 (9 am) | Random cortisol 748 (9 am) | — | 173 (7:30 pm) |
| Pubertal axis | Prepubertal | Prepubertal | Prepubertal | Prepubertal | Prepubertal |
| Endocrine medication (age at start, y) | GH (4) | GH (1) | GH (1) | HC (2.9) | GH (1) |
| DDAVP (from birth) | DDAVP (from birth) | GH (3.5) | |||
| MRI brain imaging | Normal | Progressive global cerebral hemisphere atrophy with relative preservation of the posterior fossa structures; small posterior pituitary and small optic nerves; thin corpus callosum | Mature right parieto-occipital infarct; generalized underdevelopment of the brain; posterior pituitary normal; thin corpus callosum | Normal | Right ventricular parietal VP shunt remains in situ; very thin corpus callosum; no evidence of obstructive hydrocephalus; bulky tectum and thin corpus callosum |
| Most recent results, age, y | 7.7 | 7.6 | 7.6 | 4.7 | 4.6 |
| Cortisol, nmol/L (time) | 339 (9 am) | 173 (11:45 am) | 159 (11:45 am) | — | 276 (9 am) |
| LH, IU/L (NR) | 0.2 (0.7–2.0) | — | 0.1 (0.7–2.0) | — | <0.1 (0.7–6.5) |
| FSH, IU/L (NR) | 0.6 (0.2–5.8) | — | 2.1 (0.2–5.8) | — | 0.7 (0.1–5.8) |
| PRL, mU/L (NR) | 131 (45–466) | — | 261 (45–466) | 194 (57–717) | |
| fT4, pmol/L (NR) | 16.6 (10.8–19.0) | 10.9 (10.8–19.0) | 10.0 (10.8–19.0) | 18.8 (10.29–27.03) | 13.8 (10.8–19.0) |
| TSH, mU/L (NR) | 2.8 (<6.0) | 2.5 (<6.0) | 2.4 (<6.0) | 1.22 (0.7–5.7) | 1.4 (<6.0) |
| IGF-1, ng/mL (NR) | 118 (64–345) | 102 (80–233) | 172 (80–233) | 65 (22–208) | 74 (47–231) |
| IGFBP-3, mg/L (NR) | 2.66 (1.6–6.5) | 3.73 (1.6–6.5) | 4.38 (1.6–6.5) | — | 1.60 (1.1–5.2) |
| Estradiol, pmol/L, or testosterone, nmol/L | — | Estradiol <44 | Estradiol <44 | — | Testosterone <0.69 |
| Patient/Sex (M/F) | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 |
|---|---|---|---|---|---|
| F | F | F | M | M | |
| Ethnicity | White European | White European | White European | White Chilean | Afro-Caribbean |
| Mutation | c.1996dup, p.Q666Pfs*47 | c.1996dup, p.Q666Pfs*47 | c.1996dup, p.Q666Pfs*47 | c.1996dupC, p.Q666Pfs*47 | Hemizygous |
| MAGEL2, NM_019066 | MAGEL2, NM_019066 | MAGEL2, NM_019066 | MAGEL2, NM_019066 | c.1354G>A, p.G452R | |
| De novo | Not maternally inherited; paternal DNA was unavailable | Not maternally inherited; paternal DNA was unavailable | De novo | L1CAM, NM_000425 | |
| Chromosome microarray showed a 16q11 duplication, 45,186,600–45,416670); Nimblegen 135K Hg18 WG CGH v3.1. | Chromosome microarray: normal | Chromosome microarray: normal | Mother is a heterozygous carrier | ||
| Family history | Healthy younger siblings | Healthy older sister | Healthy older sister | Healthy older brother | |
| 2× miscarriages at 7/40 | 2× miscarriages at 7/40 | Mother had bilateral metatarsus varus and a history of recurrent shoulder dislocations | |||
| Prev. stillbirth at 37/40 | Prev. stillbirth at 37/40 | ||||
| Perinatal history | Normal antenatal scans | Polyhydramnios | IUGR and polyhydramnios | — | Antenatal ventriculomegaly |
| Cesarean; breech | DCDA twin pregnancy | DCDA twin pregnancy | Elective LSCS | ||
| Hypotonic and respiratory distress after birth needing oxygen | Cesarean; breech | Cesarean; breech | Born in good condition | ||
| Resuscitation at birth: APGARs – 2 at 1 min; 1 at 5 min | |||||
| Cooled for HIE | |||||
| Ventilated to day 4 of life | |||||
| Gestation, wk | 40 | 36 + 1/40 | 36 + 1/40 | 38 | 40 |
| Birth weight, kg (SDS) | NA | 2.7 (+0.14) | 2.0 (−1.67) | 3.5 (0.07) | 3.6 (+0.07) |
| Endocrine referral | Short stature and previous history of hypoglycemia | Short stature, diabetes insipidus | Short stature, diabetes insipidus | Growth evaluation, short stature | Hypoglycemic seizures |
| Age at referral, y | 3.2 | 1.10 | 1.10 | 2.8 | 0.7 |
| Height, SDS | −2.14 | −1.3 | −2.0 | −2.84 | −1.7 |
| BMI, SDS | 0.61 | +1.52 | −1.52 | −0.92 | −4.21 |
| Current age, y | 9.7 | 7.6 | 7.6 | 4.7 | 5.0 |
| Height, cm (SDS) | 114.8 (−3.21) | 120.1 (−0.58) | 120.8 (−0.45) | 99 (−1.8) | 99.3 (−1.95) |
| Weight, kg (SDS) | 23.5 (−1.76) | 20.55 (−1.3) | 21.15 (−1.1) | 12.2 (−3.53) | 12.8 (−3.55) |
| Dysmorphic facial features | Long face | Macrocephaly | Macrocephaly | Long face | Bilateral radial clubbed hands |
| Prominent forehead | Long face | Long face | Bitemporal narrowing | Plagiocephaly | |
| Micrognathia | Bitemporal narrowing | Bitemporal narrowing | Prominent forehead | ||
| Prominent forehead | Prominent forehead | Micrognathia | |||
| Hypotelorism | Hypotelorism | Glossoptosis | |||
| Scaphocephalic shape | Scaphocephalic shape | High arched palate | |||
| Micrognathia | Micrognathia | ||||
| Cleft palate | High arched palate | ||||
| Gastrointestinal | GERD | Constipation | Constipation | Gastrostomy for feeding difficulty, gastro-esophageal reflux; chronic constipation | PEG fed |
| Food allergy | Unsafe swallow | ||||
| Dysphagia | |||||
| Constipation | |||||
| Cardiac | No known cardiac pathology | No known cardiac pathology | No known cardiac pathology | Ostium secundum interauricular communication, spontaneous closure | Small VSD |
| Neurodevelopment | Global developmental delay | Severe global developmental delay | Global developmental delay | Global developmental delay | Global developmental delay |
| Bulbar palsy | Gross Motor Function Classification System Level 4 | Independently walking | Autism spectrum disorder | Generalized hypotonia | |
| Wheelchair dependent, nonverbal | Communicates using Makaton, eye gaze, and pointing | Hypotonia | |||
| Able to sit if put in sitting position; attempts rolling over | Some vocalization | Some vocalization | |||
| Some self-harm behavior: head banging | Walking with help | ||||
| Able to sit independently | |||||
| Skeletal system | Arthrogryposis | Distal arthrogryposis | Distal arthrogryposis | Contractures with limited extension of the elbows, knees, hips, and finger | Arthrogryposis |
| Scoliosis | Scoliosis | Hands/fingers tapering | Camptodactyly | Bilateral radial clubbed hands | |
| Flexion deformity of knees | Overlapping fifth finger, positions fingers 3/4/5 together | Small hands and feet | Right hip subluxation | ||
| Short long bones | Scoliosis | ||||
| 11 ribs | |||||
| Ophthalmology | Squint | Optic nerve hypoplasia and severe sight impairment | Mild optic nerve hypoplasia (fundoscopy) and sight impaired | Strabismus | Bilateral astigmatism, left divergent squint (visual impairment); wears glasses |
| Mild optic nerve hypoplasia with cerebral visual impairment | Nystagmus | ||||
| Myopic astigmatism | |||||
| Endocrine assessment | GHD | Central diabetes insipidus since birth – on DDAVP | Central diabetes insipidus since birth – on DDAVP | GHD stimulation test not performed as hypotensive | GHD |
| Rest of endocrinology normal | Severe GHD | Severe GHD | Short stature with deceleration of growth rate | ||
| Premature thelarche – resolved | Premature thelarche – resolved | Adrenal insufficiency: peak cortisol 10.2 μg/dL on stimulation; rest normal | |||
| Premature pubarche | Premature pubarche | Cryptorchidism with bilateral orchidopexies | |||
| Other features | N/A | Hypoxic ischemic encephalopathy at birth – cooled | Percutaneous endoscopic gastrostomy tube in situ for unsafe swallow | Central sleep apnea | Hydrocephalus (VP shunt soon after birth) |
| Overhanging epiglottis | Central sleep apnea and hypoxia – no need for supplemental oxygen | ||||
| Percutaneous endoscopic gastrostomy tube in situ for unsafe swallow | Submandibular gland excision for excessive drooling | ||||
| Previous ovarian cyst on ultrasonography | Previous ovarian cyst on ultrasonography | ||||
| Diaphragmatic eversion | |||||
| Pierre Robin sequence without cleft palate. | |||||
| GH axis, age, y | 3.7 | 0.8 | 0.8 | 0.3 | 0.8 |
| Stimulation test | Glucagon | Overnight GH profile | Overnight GH profile | Not performed because of hypotension | Glucagon |
| Basal, ug/L | 0.6 | — | — | — | 2.4 |
| Peak, ug/L (NR > 6.7) | 6.4 | 4.8 (only 1 peak max 4.8) | 3.2 (only 1 peak max 3.2) | — | 3.7 |
| IGF-1, ng/mL (NR) | <25 | Undetectable | Undetectable | 31.9 (49–289) | <25 |
| Thyroid axis, age, y | 3.7 | 0.8 | 0.8 | 0.3 | 0.6 |
| fT4 pmol/L (NR) | 14.4 (9.8–19) | 13.8 (9.8–19) | 11.3 (9.8–19) | 18.8 (10.3–27) | 12.8 (9.8–19) |
| TSH mU/L (NR) | 2.8 (<6.0) | 1.9 (<6.0) | 1.1 (<6.0) | 2.87 (<6.0) | 1.5 (<6.0) |
| Adrenal axis | |||||
| Stimulation test (age, y) | Glucagon (3.7) | (0.8) | (0.8) | Synacthen (2.8) | Glucagon (0.7) |
| Peak cortisol, nmol/L | 989 | — | — | 281 | 488 |
| Random cortisol nmol/L (time) | Basal 252 (9 am) and 442 (12 noon at the end of the fast) | Random cortisol 1192 (9 am) | Random cortisol 748 (9 am) | — | 173 (7:30 pm) |
| Pubertal axis | Prepubertal | Prepubertal | Prepubertal | Prepubertal | Prepubertal |
| Endocrine medication (age at start, y) | GH (4) | GH (1) | GH (1) | HC (2.9) | GH (1) |
| DDAVP (from birth) | DDAVP (from birth) | GH (3.5) | |||
| MRI brain imaging | Normal | Progressive global cerebral hemisphere atrophy with relative preservation of the posterior fossa structures; small posterior pituitary and small optic nerves; thin corpus callosum | Mature right parieto-occipital infarct; generalized underdevelopment of the brain; posterior pituitary normal; thin corpus callosum | Normal | Right ventricular parietal VP shunt remains in situ; very thin corpus callosum; no evidence of obstructive hydrocephalus; bulky tectum and thin corpus callosum |
| Most recent results, age, y | 7.7 | 7.6 | 7.6 | 4.7 | 4.6 |
| Cortisol, nmol/L (time) | 339 (9 am) | 173 (11:45 am) | 159 (11:45 am) | — | 276 (9 am) |
| LH, IU/L (NR) | 0.2 (0.7–2.0) | — | 0.1 (0.7–2.0) | — | <0.1 (0.7–6.5) |
| FSH, IU/L (NR) | 0.6 (0.2–5.8) | — | 2.1 (0.2–5.8) | — | 0.7 (0.1–5.8) |
| PRL, mU/L (NR) | 131 (45–466) | — | 261 (45–466) | 194 (57–717) | |
| fT4, pmol/L (NR) | 16.6 (10.8–19.0) | 10.9 (10.8–19.0) | 10.0 (10.8–19.0) | 18.8 (10.29–27.03) | 13.8 (10.8–19.0) |
| TSH, mU/L (NR) | 2.8 (<6.0) | 2.5 (<6.0) | 2.4 (<6.0) | 1.22 (0.7–5.7) | 1.4 (<6.0) |
| IGF-1, ng/mL (NR) | 118 (64–345) | 102 (80–233) | 172 (80–233) | 65 (22–208) | 74 (47–231) |
| IGFBP-3, mg/L (NR) | 2.66 (1.6–6.5) | 3.73 (1.6–6.5) | 4.38 (1.6–6.5) | — | 1.60 (1.1–5.2) |
| Estradiol, pmol/L, or testosterone, nmol/L | — | Estradiol <44 | Estradiol <44 | — | Testosterone <0.69 |
Abbreviations: BMI, body mass index; DCDA, dichorionic diamniotic; DDAVP, desmopressin; fT4, free thyroxine; GERD, gastroesophageal reflux disease; HC, hydrocortisone; HIE, Hypoxic ischemic encephalopathy; IGFBP-3, insulin-like growth factor‒binding protein 3; IUGR, intrauterine growth restriction; LSCS, lower segment cesarean section; N/A, not applicable; PEG, percutaneous endoscopic gastrostomy; Prev, previous; PRL, prolactin; SDS, standard deviation score; VP, ventricular parietal; VSD, ventricular septal defect.
Phenotypes of Patients With MAGEL2 and L1CAM Mutations
| Patient/Sex (M/F) | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 |
|---|---|---|---|---|---|
| F | F | F | M | M | |
| Ethnicity | White European | White European | White European | White Chilean | Afro-Caribbean |
| Mutation | c.1996dup, p.Q666Pfs*47 | c.1996dup, p.Q666Pfs*47 | c.1996dup, p.Q666Pfs*47 | c.1996dupC, p.Q666Pfs*47 | Hemizygous |
| MAGEL2, NM_019066 | MAGEL2, NM_019066 | MAGEL2, NM_019066 | MAGEL2, NM_019066 | c.1354G>A, p.G452R | |
| De novo | Not maternally inherited; paternal DNA was unavailable | Not maternally inherited; paternal DNA was unavailable | De novo | L1CAM, NM_000425 | |
| Chromosome microarray showed a 16q11 duplication, 45,186,600–45,416670); Nimblegen 135K Hg18 WG CGH v3.1. | Chromosome microarray: normal | Chromosome microarray: normal | Mother is a heterozygous carrier | ||
| Family history | Healthy younger siblings | Healthy older sister | Healthy older sister | Healthy older brother | |
| 2× miscarriages at 7/40 | 2× miscarriages at 7/40 | Mother had bilateral metatarsus varus and a history of recurrent shoulder dislocations | |||
| Prev. stillbirth at 37/40 | Prev. stillbirth at 37/40 | ||||
| Perinatal history | Normal antenatal scans | Polyhydramnios | IUGR and polyhydramnios | — | Antenatal ventriculomegaly |
| Cesarean; breech | DCDA twin pregnancy | DCDA twin pregnancy | Elective LSCS | ||
| Hypotonic and respiratory distress after birth needing oxygen | Cesarean; breech | Cesarean; breech | Born in good condition | ||
| Resuscitation at birth: APGARs – 2 at 1 min; 1 at 5 min | |||||
| Cooled for HIE | |||||
| Ventilated to day 4 of life | |||||
| Gestation, wk | 40 | 36 + 1/40 | 36 + 1/40 | 38 | 40 |
| Birth weight, kg (SDS) | NA | 2.7 (+0.14) | 2.0 (−1.67) | 3.5 (0.07) | 3.6 (+0.07) |
| Endocrine referral | Short stature and previous history of hypoglycemia | Short stature, diabetes insipidus | Short stature, diabetes insipidus | Growth evaluation, short stature | Hypoglycemic seizures |
| Age at referral, y | 3.2 | 1.10 | 1.10 | 2.8 | 0.7 |
| Height, SDS | −2.14 | −1.3 | −2.0 | −2.84 | −1.7 |
| BMI, SDS | 0.61 | +1.52 | −1.52 | −0.92 | −4.21 |
| Current age, y | 9.7 | 7.6 | 7.6 | 4.7 | 5.0 |
| Height, cm (SDS) | 114.8 (−3.21) | 120.1 (−0.58) | 120.8 (−0.45) | 99 (−1.8) | 99.3 (−1.95) |
| Weight, kg (SDS) | 23.5 (−1.76) | 20.55 (−1.3) | 21.15 (−1.1) | 12.2 (−3.53) | 12.8 (−3.55) |
| Dysmorphic facial features | Long face | Macrocephaly | Macrocephaly | Long face | Bilateral radial clubbed hands |
| Prominent forehead | Long face | Long face | Bitemporal narrowing | Plagiocephaly | |
| Micrognathia | Bitemporal narrowing | Bitemporal narrowing | Prominent forehead | ||
| Prominent forehead | Prominent forehead | Micrognathia | |||
| Hypotelorism | Hypotelorism | Glossoptosis | |||
| Scaphocephalic shape | Scaphocephalic shape | High arched palate | |||
| Micrognathia | Micrognathia | ||||
| Cleft palate | High arched palate | ||||
| Gastrointestinal | GERD | Constipation | Constipation | Gastrostomy for feeding difficulty, gastro-esophageal reflux; chronic constipation | PEG fed |
| Food allergy | Unsafe swallow | ||||
| Dysphagia | |||||
| Constipation | |||||
| Cardiac | No known cardiac pathology | No known cardiac pathology | No known cardiac pathology | Ostium secundum interauricular communication, spontaneous closure | Small VSD |
| Neurodevelopment | Global developmental delay | Severe global developmental delay | Global developmental delay | Global developmental delay | Global developmental delay |
| Bulbar palsy | Gross Motor Function Classification System Level 4 | Independently walking | Autism spectrum disorder | Generalized hypotonia | |
| Wheelchair dependent, nonverbal | Communicates using Makaton, eye gaze, and pointing | Hypotonia | |||
| Able to sit if put in sitting position; attempts rolling over | Some vocalization | Some vocalization | |||
| Some self-harm behavior: head banging | Walking with help | ||||
| Able to sit independently | |||||
| Skeletal system | Arthrogryposis | Distal arthrogryposis | Distal arthrogryposis | Contractures with limited extension of the elbows, knees, hips, and finger | Arthrogryposis |
| Scoliosis | Scoliosis | Hands/fingers tapering | Camptodactyly | Bilateral radial clubbed hands | |
| Flexion deformity of knees | Overlapping fifth finger, positions fingers 3/4/5 together | Small hands and feet | Right hip subluxation | ||
| Short long bones | Scoliosis | ||||
| 11 ribs | |||||
| Ophthalmology | Squint | Optic nerve hypoplasia and severe sight impairment | Mild optic nerve hypoplasia (fundoscopy) and sight impaired | Strabismus | Bilateral astigmatism, left divergent squint (visual impairment); wears glasses |
| Mild optic nerve hypoplasia with cerebral visual impairment | Nystagmus | ||||
| Myopic astigmatism | |||||
| Endocrine assessment | GHD | Central diabetes insipidus since birth – on DDAVP | Central diabetes insipidus since birth – on DDAVP | GHD stimulation test not performed as hypotensive | GHD |
| Rest of endocrinology normal | Severe GHD | Severe GHD | Short stature with deceleration of growth rate | ||
| Premature thelarche – resolved | Premature thelarche – resolved | Adrenal insufficiency: peak cortisol 10.2 μg/dL on stimulation; rest normal | |||
| Premature pubarche | Premature pubarche | Cryptorchidism with bilateral orchidopexies | |||
| Other features | N/A | Hypoxic ischemic encephalopathy at birth – cooled | Percutaneous endoscopic gastrostomy tube in situ for unsafe swallow | Central sleep apnea | Hydrocephalus (VP shunt soon after birth) |
| Overhanging epiglottis | Central sleep apnea and hypoxia – no need for supplemental oxygen | ||||
| Percutaneous endoscopic gastrostomy tube in situ for unsafe swallow | Submandibular gland excision for excessive drooling | ||||
| Previous ovarian cyst on ultrasonography | Previous ovarian cyst on ultrasonography | ||||
| Diaphragmatic eversion | |||||
| Pierre Robin sequence without cleft palate. | |||||
| GH axis, age, y | 3.7 | 0.8 | 0.8 | 0.3 | 0.8 |
| Stimulation test | Glucagon | Overnight GH profile | Overnight GH profile | Not performed because of hypotension | Glucagon |
| Basal, ug/L | 0.6 | — | — | — | 2.4 |
| Peak, ug/L (NR > 6.7) | 6.4 | 4.8 (only 1 peak max 4.8) | 3.2 (only 1 peak max 3.2) | — | 3.7 |
| IGF-1, ng/mL (NR) | <25 | Undetectable | Undetectable | 31.9 (49–289) | <25 |
| Thyroid axis, age, y | 3.7 | 0.8 | 0.8 | 0.3 | 0.6 |
| fT4 pmol/L (NR) | 14.4 (9.8–19) | 13.8 (9.8–19) | 11.3 (9.8–19) | 18.8 (10.3–27) | 12.8 (9.8–19) |
| TSH mU/L (NR) | 2.8 (<6.0) | 1.9 (<6.0) | 1.1 (<6.0) | 2.87 (<6.0) | 1.5 (<6.0) |
| Adrenal axis | |||||
| Stimulation test (age, y) | Glucagon (3.7) | (0.8) | (0.8) | Synacthen (2.8) | Glucagon (0.7) |
| Peak cortisol, nmol/L | 989 | — | — | 281 | 488 |
| Random cortisol nmol/L (time) | Basal 252 (9 am) and 442 (12 noon at the end of the fast) | Random cortisol 1192 (9 am) | Random cortisol 748 (9 am) | — | 173 (7:30 pm) |
| Pubertal axis | Prepubertal | Prepubertal | Prepubertal | Prepubertal | Prepubertal |
| Endocrine medication (age at start, y) | GH (4) | GH (1) | GH (1) | HC (2.9) | GH (1) |
| DDAVP (from birth) | DDAVP (from birth) | GH (3.5) | |||
| MRI brain imaging | Normal | Progressive global cerebral hemisphere atrophy with relative preservation of the posterior fossa structures; small posterior pituitary and small optic nerves; thin corpus callosum | Mature right parieto-occipital infarct; generalized underdevelopment of the brain; posterior pituitary normal; thin corpus callosum | Normal | Right ventricular parietal VP shunt remains in situ; very thin corpus callosum; no evidence of obstructive hydrocephalus; bulky tectum and thin corpus callosum |
| Most recent results, age, y | 7.7 | 7.6 | 7.6 | 4.7 | 4.6 |
| Cortisol, nmol/L (time) | 339 (9 am) | 173 (11:45 am) | 159 (11:45 am) | — | 276 (9 am) |
| LH, IU/L (NR) | 0.2 (0.7–2.0) | — | 0.1 (0.7–2.0) | — | <0.1 (0.7–6.5) |
| FSH, IU/L (NR) | 0.6 (0.2–5.8) | — | 2.1 (0.2–5.8) | — | 0.7 (0.1–5.8) |
| PRL, mU/L (NR) | 131 (45–466) | — | 261 (45–466) | 194 (57–717) | |
| fT4, pmol/L (NR) | 16.6 (10.8–19.0) | 10.9 (10.8–19.0) | 10.0 (10.8–19.0) | 18.8 (10.29–27.03) | 13.8 (10.8–19.0) |
| TSH, mU/L (NR) | 2.8 (<6.0) | 2.5 (<6.0) | 2.4 (<6.0) | 1.22 (0.7–5.7) | 1.4 (<6.0) |
| IGF-1, ng/mL (NR) | 118 (64–345) | 102 (80–233) | 172 (80–233) | 65 (22–208) | 74 (47–231) |
| IGFBP-3, mg/L (NR) | 2.66 (1.6–6.5) | 3.73 (1.6–6.5) | 4.38 (1.6–6.5) | — | 1.60 (1.1–5.2) |
| Estradiol, pmol/L, or testosterone, nmol/L | — | Estradiol <44 | Estradiol <44 | — | Testosterone <0.69 |
| Patient/Sex (M/F) | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 |
|---|---|---|---|---|---|
| F | F | F | M | M | |
| Ethnicity | White European | White European | White European | White Chilean | Afro-Caribbean |
| Mutation | c.1996dup, p.Q666Pfs*47 | c.1996dup, p.Q666Pfs*47 | c.1996dup, p.Q666Pfs*47 | c.1996dupC, p.Q666Pfs*47 | Hemizygous |
| MAGEL2, NM_019066 | MAGEL2, NM_019066 | MAGEL2, NM_019066 | MAGEL2, NM_019066 | c.1354G>A, p.G452R | |
| De novo | Not maternally inherited; paternal DNA was unavailable | Not maternally inherited; paternal DNA was unavailable | De novo | L1CAM, NM_000425 | |
| Chromosome microarray showed a 16q11 duplication, 45,186,600–45,416670); Nimblegen 135K Hg18 WG CGH v3.1. | Chromosome microarray: normal | Chromosome microarray: normal | Mother is a heterozygous carrier | ||
| Family history | Healthy younger siblings | Healthy older sister | Healthy older sister | Healthy older brother | |
| 2× miscarriages at 7/40 | 2× miscarriages at 7/40 | Mother had bilateral metatarsus varus and a history of recurrent shoulder dislocations | |||
| Prev. stillbirth at 37/40 | Prev. stillbirth at 37/40 | ||||
| Perinatal history | Normal antenatal scans | Polyhydramnios | IUGR and polyhydramnios | — | Antenatal ventriculomegaly |
| Cesarean; breech | DCDA twin pregnancy | DCDA twin pregnancy | Elective LSCS | ||
| Hypotonic and respiratory distress after birth needing oxygen | Cesarean; breech | Cesarean; breech | Born in good condition | ||
| Resuscitation at birth: APGARs – 2 at 1 min; 1 at 5 min | |||||
| Cooled for HIE | |||||
| Ventilated to day 4 of life | |||||
| Gestation, wk | 40 | 36 + 1/40 | 36 + 1/40 | 38 | 40 |
| Birth weight, kg (SDS) | NA | 2.7 (+0.14) | 2.0 (−1.67) | 3.5 (0.07) | 3.6 (+0.07) |
| Endocrine referral | Short stature and previous history of hypoglycemia | Short stature, diabetes insipidus | Short stature, diabetes insipidus | Growth evaluation, short stature | Hypoglycemic seizures |
| Age at referral, y | 3.2 | 1.10 | 1.10 | 2.8 | 0.7 |
| Height, SDS | −2.14 | −1.3 | −2.0 | −2.84 | −1.7 |
| BMI, SDS | 0.61 | +1.52 | −1.52 | −0.92 | −4.21 |
| Current age, y | 9.7 | 7.6 | 7.6 | 4.7 | 5.0 |
| Height, cm (SDS) | 114.8 (−3.21) | 120.1 (−0.58) | 120.8 (−0.45) | 99 (−1.8) | 99.3 (−1.95) |
| Weight, kg (SDS) | 23.5 (−1.76) | 20.55 (−1.3) | 21.15 (−1.1) | 12.2 (−3.53) | 12.8 (−3.55) |
| Dysmorphic facial features | Long face | Macrocephaly | Macrocephaly | Long face | Bilateral radial clubbed hands |
| Prominent forehead | Long face | Long face | Bitemporal narrowing | Plagiocephaly | |
| Micrognathia | Bitemporal narrowing | Bitemporal narrowing | Prominent forehead | ||
| Prominent forehead | Prominent forehead | Micrognathia | |||
| Hypotelorism | Hypotelorism | Glossoptosis | |||
| Scaphocephalic shape | Scaphocephalic shape | High arched palate | |||
| Micrognathia | Micrognathia | ||||
| Cleft palate | High arched palate | ||||
| Gastrointestinal | GERD | Constipation | Constipation | Gastrostomy for feeding difficulty, gastro-esophageal reflux; chronic constipation | PEG fed |
| Food allergy | Unsafe swallow | ||||
| Dysphagia | |||||
| Constipation | |||||
| Cardiac | No known cardiac pathology | No known cardiac pathology | No known cardiac pathology | Ostium secundum interauricular communication, spontaneous closure | Small VSD |
| Neurodevelopment | Global developmental delay | Severe global developmental delay | Global developmental delay | Global developmental delay | Global developmental delay |
| Bulbar palsy | Gross Motor Function Classification System Level 4 | Independently walking | Autism spectrum disorder | Generalized hypotonia | |
| Wheelchair dependent, nonverbal | Communicates using Makaton, eye gaze, and pointing | Hypotonia | |||
| Able to sit if put in sitting position; attempts rolling over | Some vocalization | Some vocalization | |||
| Some self-harm behavior: head banging | Walking with help | ||||
| Able to sit independently | |||||
| Skeletal system | Arthrogryposis | Distal arthrogryposis | Distal arthrogryposis | Contractures with limited extension of the elbows, knees, hips, and finger | Arthrogryposis |
| Scoliosis | Scoliosis | Hands/fingers tapering | Camptodactyly | Bilateral radial clubbed hands | |
| Flexion deformity of knees | Overlapping fifth finger, positions fingers 3/4/5 together | Small hands and feet | Right hip subluxation | ||
| Short long bones | Scoliosis | ||||
| 11 ribs | |||||
| Ophthalmology | Squint | Optic nerve hypoplasia and severe sight impairment | Mild optic nerve hypoplasia (fundoscopy) and sight impaired | Strabismus | Bilateral astigmatism, left divergent squint (visual impairment); wears glasses |
| Mild optic nerve hypoplasia with cerebral visual impairment | Nystagmus | ||||
| Myopic astigmatism | |||||
| Endocrine assessment | GHD | Central diabetes insipidus since birth – on DDAVP | Central diabetes insipidus since birth – on DDAVP | GHD stimulation test not performed as hypotensive | GHD |
| Rest of endocrinology normal | Severe GHD | Severe GHD | Short stature with deceleration of growth rate | ||
| Premature thelarche – resolved | Premature thelarche – resolved | Adrenal insufficiency: peak cortisol 10.2 μg/dL on stimulation; rest normal | |||
| Premature pubarche | Premature pubarche | Cryptorchidism with bilateral orchidopexies | |||
| Other features | N/A | Hypoxic ischemic encephalopathy at birth – cooled | Percutaneous endoscopic gastrostomy tube in situ for unsafe swallow | Central sleep apnea | Hydrocephalus (VP shunt soon after birth) |
| Overhanging epiglottis | Central sleep apnea and hypoxia – no need for supplemental oxygen | ||||
| Percutaneous endoscopic gastrostomy tube in situ for unsafe swallow | Submandibular gland excision for excessive drooling | ||||
| Previous ovarian cyst on ultrasonography | Previous ovarian cyst on ultrasonography | ||||
| Diaphragmatic eversion | |||||
| Pierre Robin sequence without cleft palate. | |||||
| GH axis, age, y | 3.7 | 0.8 | 0.8 | 0.3 | 0.8 |
| Stimulation test | Glucagon | Overnight GH profile | Overnight GH profile | Not performed because of hypotension | Glucagon |
| Basal, ug/L | 0.6 | — | — | — | 2.4 |
| Peak, ug/L (NR > 6.7) | 6.4 | 4.8 (only 1 peak max 4.8) | 3.2 (only 1 peak max 3.2) | — | 3.7 |
| IGF-1, ng/mL (NR) | <25 | Undetectable | Undetectable | 31.9 (49–289) | <25 |
| Thyroid axis, age, y | 3.7 | 0.8 | 0.8 | 0.3 | 0.6 |
| fT4 pmol/L (NR) | 14.4 (9.8–19) | 13.8 (9.8–19) | 11.3 (9.8–19) | 18.8 (10.3–27) | 12.8 (9.8–19) |
| TSH mU/L (NR) | 2.8 (<6.0) | 1.9 (<6.0) | 1.1 (<6.0) | 2.87 (<6.0) | 1.5 (<6.0) |
| Adrenal axis | |||||
| Stimulation test (age, y) | Glucagon (3.7) | (0.8) | (0.8) | Synacthen (2.8) | Glucagon (0.7) |
| Peak cortisol, nmol/L | 989 | — | — | 281 | 488 |
| Random cortisol nmol/L (time) | Basal 252 (9 am) and 442 (12 noon at the end of the fast) | Random cortisol 1192 (9 am) | Random cortisol 748 (9 am) | — | 173 (7:30 pm) |
| Pubertal axis | Prepubertal | Prepubertal | Prepubertal | Prepubertal | Prepubertal |
| Endocrine medication (age at start, y) | GH (4) | GH (1) | GH (1) | HC (2.9) | GH (1) |
| DDAVP (from birth) | DDAVP (from birth) | GH (3.5) | |||
| MRI brain imaging | Normal | Progressive global cerebral hemisphere atrophy with relative preservation of the posterior fossa structures; small posterior pituitary and small optic nerves; thin corpus callosum | Mature right parieto-occipital infarct; generalized underdevelopment of the brain; posterior pituitary normal; thin corpus callosum | Normal | Right ventricular parietal VP shunt remains in situ; very thin corpus callosum; no evidence of obstructive hydrocephalus; bulky tectum and thin corpus callosum |
| Most recent results, age, y | 7.7 | 7.6 | 7.6 | 4.7 | 4.6 |
| Cortisol, nmol/L (time) | 339 (9 am) | 173 (11:45 am) | 159 (11:45 am) | — | 276 (9 am) |
| LH, IU/L (NR) | 0.2 (0.7–2.0) | — | 0.1 (0.7–2.0) | — | <0.1 (0.7–6.5) |
| FSH, IU/L (NR) | 0.6 (0.2–5.8) | — | 2.1 (0.2–5.8) | — | 0.7 (0.1–5.8) |
| PRL, mU/L (NR) | 131 (45–466) | — | 261 (45–466) | 194 (57–717) | |
| fT4, pmol/L (NR) | 16.6 (10.8–19.0) | 10.9 (10.8–19.0) | 10.0 (10.8–19.0) | 18.8 (10.29–27.03) | 13.8 (10.8–19.0) |
| TSH, mU/L (NR) | 2.8 (<6.0) | 2.5 (<6.0) | 2.4 (<6.0) | 1.22 (0.7–5.7) | 1.4 (<6.0) |
| IGF-1, ng/mL (NR) | 118 (64–345) | 102 (80–233) | 172 (80–233) | 65 (22–208) | 74 (47–231) |
| IGFBP-3, mg/L (NR) | 2.66 (1.6–6.5) | 3.73 (1.6–6.5) | 4.38 (1.6–6.5) | — | 1.60 (1.1–5.2) |
| Estradiol, pmol/L, or testosterone, nmol/L | — | Estradiol <44 | Estradiol <44 | — | Testosterone <0.69 |
Abbreviations: BMI, body mass index; DCDA, dichorionic diamniotic; DDAVP, desmopressin; fT4, free thyroxine; GERD, gastroesophageal reflux disease; HC, hydrocortisone; HIE, Hypoxic ischemic encephalopathy; IGFBP-3, insulin-like growth factor‒binding protein 3; IUGR, intrauterine growth restriction; LSCS, lower segment cesarean section; N/A, not applicable; PEG, percutaneous endoscopic gastrostomy; Prev, previous; PRL, prolactin; SDS, standard deviation score; VP, ventricular parietal; VSD, ventricular septal defect.
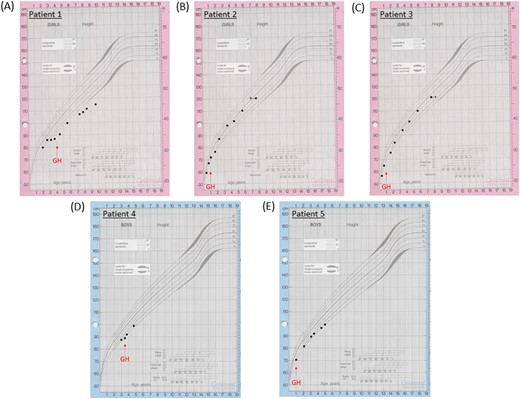
Growth charts of (A) patient 1, (B) patient 2, (C) patient 3, (D) patient 4, and (E) patient 5. The red arrows indicate when GH treatment commenced in the patients.
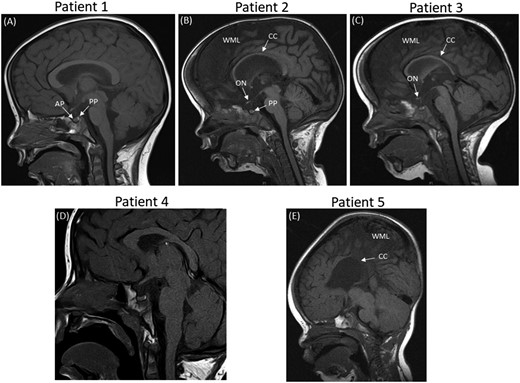
Sagittal MRIs for patients 1–5 showing abnormalities in patients 2, 3, and 5. (A) MRI of patient 1 shows a normal anterior and posterior pituitary, with no other anomalies. (B) MRI of patient 2 (a twin) reveals global cerebral hemisphere atrophy with a small posterior pituitary, a thin corpus callosum, and small optic nerves. (C) MRI of patient 3 (a twin) reveals generalized underdevelopment of the brain. The posterior pituitary is normal with small optic nerves and a thin corpus callosum. (D) MRI of patient 4 shows a normal anterior and posterior pituitary, with no other anomalies. (E) MRI of patient 5 shows generalized underdevelopment of the brain and a very thin corpus callosum. AP, anterior pituitary; CC, corpus callosum; ON, optic nerve; PP, posterior pituitary; WML, white matter loss.
Patients 2 and 3
These white European female nonidentical twins with distal arthrogryposis were initially referred with hypernatremia and were then diagnosed with diabetes insipidus (DI) shortly after birth. Subsequent short stature led to a diagnosis of GHD [peak GH to stimulation of 4.8 μg/L and 3.2 μg/L in patient 2 and patient 3, respectively, with an undetectable IGF-I concentration at 0.8 year (Table 1)]. Their DI had been treated with desmopressin since birth, and GH treatment commenced after 1 year of age [Fig. 1(B) and 1(C)]. The patients had distinctive features including macrocephaly, a long face with bitemporal narrowing, frontal bossing, scaphocephaly, micrognathia, and a cleft/high arched palate. Patient 2 had nystagmus with optic nerve atrophy and was severely sight impaired, whereas her sister had optic nerve hypoplasia with visual impairment. They both had global developmental delay. Patient 2 is wheelchair bound and unable to speak, whereas patient 3 is able to stand and has basic vocalization. The twins also had central sleep apnea and scoliosis. Patient 2 had chronic lung disease with supplemental oxygen requirement at night and had a tracheostomy until the age of 6 years. Patient 3 had a tracheostomy until 20 months of age. On MRI, patient 2 showed evidence of progressive global cerebral hemisphere atrophy with relative preservation of the posterior fossa structures, with a thin corpus callosum, a small PP, and optic nerve hypoplasia [Fig. 2(A)]. Patient 3 had generalized underdevelopment of the brain with a mature right parieto-occipital infarct and a thin corpus callosum, optic nerve hypoplasia, and a normal PP [Fig. 2(B)].
Patient 4
A white male patient from Chile presented with short stature and a deceleration in growth rate at the age of 2.8 years. He was diagnosed with GHD (a stimulation test was not performed because of hypotension), adrenal insufficiency with a peak stimulated cortisol level of 281 nmol/L (Table 1), transient hyperprolactinemia, and arthrogryposis. The latter consisted of contractures, shortening of the extremities, and limited extension of the elbows, knees, hips, and fingers, namely camptodactyly. He was started on hydrocortisone at 2.9 years of age and began GH treatment at 3.5 years [Fig. 1(D)]. He had strabismus, global developmental delay with autism spectrum disorder (ASD), generalized hypotonia, and dysmorphic features including a long face with bitemporal narrowing, a prominent forehead, micrognathia, glossoptosis, and a high arched palate. He had gastroesophageal reflux and central sleep apnea, with respiratory complications leading to a tracheostomy. Cardiac complications included an ostium secundum interauricular communication with spontaneous closure. Cryptorchidism resolved with a bilateral orchidopexy. His MRI was normal [Fig. 2(D)].
Patient 5
An Afro-Caribbean male patient presented with antenatal ventriculomegaly and dysmorphic features, including bilateral radial clubbed hands and plagiocephaly. Flexion deformities that affected both the wrists and hands were noted antenatally, and he was diagnosed with distal arthrogryposis with adducted thumbs and flexion deformities of his digits postnatally. A ventriculo-peritoneal shunt was inserted at 4 days of age, and hypoglycemic seizures ensued at the age of 0.7 year. He was later diagnosed with GHD (peak GH, 3.7 μg/L; undetectable IGF-I concentration), and GH treatment was commenced from 1 year of age [Fig. 1(E); Table 1]. Gastrointestinal problems included dysphagia, and the patient was fed via a percutaneous endoscopic gastrostomy. Other phenotypic features in this patient included a ventricular septal defect, severe obstructive sleep apnea, global developmental delay, generalized hypotonia, right hip subluxation, and scoliosis. Bilateral astigmatism with a left divergent squint and subsequent visual impairment was apparent upon eye examination. His MRI revealed a bulky tectum, generalized white matter loss, and a thin corpus callosum, with no evidence of obstructive hydrocephalus [Fig. 2(C)].
Genetic analysis of patients 1 to 5
Whole exome sequencing was performed on the five patients with CH and arthrogryposis at three different institutions. The results identified the MAGEL2 c.1996dupC, p.Q666Pfs*47 truncation mutation in patient 1 (GOSgene), patients 2 and 3 (GOSH UK as part of the DDD Study), and patient 4 (Pontificia Universidad Católica de Chile). A chromosome microarray was also performed on the twins (patients 2 and 3), which revealed the 16q11 duplication 45,186,600-45,416,670 in patient 2 only. A hemizygous L1CAM c.1354G>A, p.G452R variant was identified in patient 5 (GOSgene), who also had hydrocephalus and other features consistent with L1 syndrome. The p.G452R variant is located at a highly conserved residue across multiple species and is located within the Ig5 extracellular domain of the L1 protein. Both MAGEL2 p.Q666Pfs*47 and L1CAM p.G452R are absent from control databases, including the gnomAD browser (http://gnomad.broadinstitute.org/).
Human embryonic expression profile of MAGEL2 and L1CAM using in situ hybridization
MAGEL2
At the early embryonic stage of CS16, there was no MAGEL2 expression in the developing hypothalamus or Rathke pouch (RP; the primordium of the anterior pituitary). However, there was strong transcript staining specifically in the inferior ganglion of the vagus nerve and the spinal ganglia. At CS19, MAGEL2 mRNA transcripts appeared in the hypothalamus and the spinal cord but were undetectable in the RP. At CS20, strong expression was present throughout the ventral diencephalon and in both the RP and the PP. This expression was maintained within the hypothalamus and the RP at CS23 and was noted in the trigeminal ganglia (Fig. 3). No staining was visualized using the sense control probe on equivalent sections at any stage.
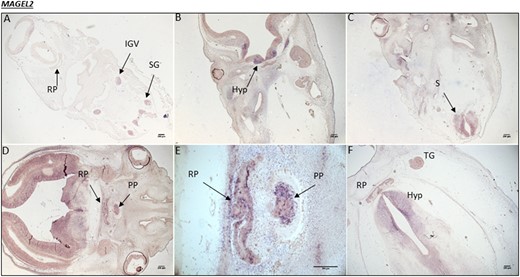
Human MAGEL2 expression during embryonic development. (A) CS16, the equivalent of 5.5 weeks into embryonic development. MAGEL2 expression is noted in the inferior ganglion of the vagus (IGV) nerve and the spinal ganglia (SG). (B) At CS19, 6 weeks into development, there are high levels of mRNA transcripts in the developing hypothalamus (Hyp), ventral diencephalon (VD), and (C) spinal cord (S). (D) At CS20, 7 weeks into development, strong transcript staining is present throughout the VD and in both the RP and PP. (E) A magnified image of the RP and PP from image (D). (F) At CS23, 8 weeks into development, MAGEL2 expression is maintained in the Hyp and RP, with some expression in the trigeminal ganglia (TG).
L1CAM
There was no L1CAM mRNA transcript staining at CS16 in the human embryonic brain sections incorporating the hypothalamus and RP. At CS19, there was strong expression in the hypothalamus and trigeminal ganglia, but not in the RP. Staining was also noted in the metencephalon and throughout the ventral diencephalon at this stage. L1CAM expression was maintained in the hypothalamus and forebrain as well as the hindbrain during CS20 and CS23 (Fig. 4). No staining was observed in the RP or in the PP at any stage analyzed in this study. The sense control probe produced no staining at any stage.
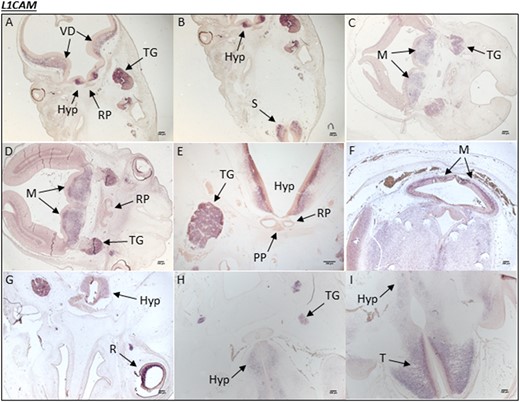
Human L1CAM expression during embryonic development. (A and B) A human embryonic section from CS19 showing L1CAM mRNA transcripts in the developing hypothalamus (Hyp), ventral diencephalon (VD), and trigeminal ganglia (TG). (B) mRNA transcripts can be seen in the spinal cord (S). In a different embryo section at (C) CS19 and at (D) CS20, L1CAM expression is noted throughout the metencephalon (M) and again in the TG. There is no mRNA transcript staining in the RP at either stage. (E) In a different embryo section at CS20, specific expression is seen throughout the hypothalamus and in the TG. (F) At CS23, L1CAM expression is observed ubiquitously throughout the brain, particularly in the M; (G) it is also present in the retina (R) of the eye. (H) A different embryo section at CS23 shows that L1CAM expression is partially maintained in the Hyp and TG. (I) At CS23, there is strong expression in the telencephalon (T) (forebrain).
Discussion
MAGEL2 is a member of the type II MAGE gene family involved in neurogenesis and brain function (13, 14). It is thought to enhance ubiquitin ligase activity (15), act as a regulator of retrograde transport, and promote endosomal F-actin assembly, and it is involved in the regulation of the circadian clock (16). In humans, loss of function point mutations causing truncations in the MAGEL2 gene were initially implicated in the etiology of variable PWS-like features and contractures of the small finger joints, a phenotype now commonly referred to as SHFYNG syndrome (3).
Magel2-null mice present with features similar to those of PWS in humans, including neonatal growth retardation, excessive weight gain after weaning, impaired hypothalamic regulation, reduced fertility, and excess fat with decreased muscle mass (17–20). In addition, Magel2-knockout mice elicit altered social phenotypes and impaired ability to distinguish between known and novel partners (21). Recent studies concluded that Pro-opiomelanocortin neuron activity and its communication with downstream targets are significantly compromised (22) and that oxytocin neuronal activity is suppressed (23) in Magel2-deficient mice.
Specific association of the MAGEL2 gene with PWS was first suggested following expression studies using northern blotting, where MAGEL2 was expressed in the adult human brain, notably the hypothalamus, and in the fetal brain (however, details were not specific), lung, and kidney (24). The authors concluded that loss of MAGEL2 may explain abnormalities in brain development in individuals with PWS. Expression analysis performed in the current study further characterized the location of MAGEL2 transcripts within the developing human fetal brain. We have shown that MAGEL2 is highly expressed in the developing hypothalamus from 6 to at least 8 weeks GA and in the developing pituitary gland (RP) at 7 to 8 weeks GA (Fig. 3), supporting the hypothesis that this gene plays a critical role during embryonic brain development.
The MAGEL2 mutation c.1996dupC, p.Q666Pfs*47 identified in patients 1 to 4 was previously identified in two siblings diagnosed with a neurodevelopmental disorder including hypotonia, ASD, hyperinsulinemic hypoglycemia, and features of arthrogryposis (25). Subsequently, the c.1996delC deletion, causing a frameshift in the same location, p.Q666Sfs*36, was described in three patients with a lethal form of arthrogryposis (26). Both the c.1996delC deletion and c.1996dupC duplication have since been identified in multiple patients with SHFYNG. These data widened the phenotypic spectrum of SHFYNG, expanding the range to include fetal akinesia and arthrogryposis (27, 28). In previous reports of patients harboring MAGEL2 truncating mutations, intellectual disability varied from mild to severe, and ASD was not always present. The majority of affected patients had arthrogryposis (varying in severity), short stature, and hypogonadism, which are all common features in SHFYNG (3, 27, 28), with one female patient manifesting hypogonadotropic hypogonadism (27). Interestingly, a recent report described the first patient with SHFYNG and early-onset obesity to harbor a MAGEL2 truncation (de novo c.1850G>A, p.Trp617*) (29).
GHD has frequently been identified in SHFYNG; however, other pituitary deficits have not been described until recently. Two siblings and an unrelated female patient with SHFYNG, arthrogryposis, and severe respiratory difficulties were found to carry the truncating MAGEL2 variants p.Q638* and p.S1044*, respectively, and manifested variable hypopituitarism (30). One of the siblings was diagnosed with central DI and gonadotrophin deficiency, whereas the unrelated patient was diagnosed with panhypopituitarism including GHD, central hypothyroidism, adrenal insufficiency, and gonadotrophin deficiency, with a hypoplastic anterior pituitary gland on MRI (30). Patients 2 and 3 from the current study manifested DI, and patient 4 has multiple pituitary hormone deficiencies, including GHD and ACTH insufficiency, indicating an association of the p.Q666Pfs*47 frameshift with endocrinopathies in patients with SHFYNG. Together with the previous report (30), these findings further highlight how different MAGEL2 truncations seem to play a role in the etiology of both DI and combined pituitary hormone deficiency as part of SHFYNG syndrome, which until recently were not major phenotypic features reported in such patients. Another recent case report identified the novel MAGEL2 p.Q1007* truncation in a patient with SHFYNG and GHD, hypothyroidism, and hyperprolactinemia (31), again suggesting that variable CH is increasingly identified in these patients. Of note, a previous report described a patient with Moebius syndrome, GHD, and arthrogryposis (32). Although no genetic mutations were identified in this patient, it demonstrates the link between these diverse phenotypes.
A recent publication reported the association of MAGEL2 truncation mutations with Chitayat-Hall syndrome (OMIM: 208080), which has a strong phenotypic overlap with SHFYNG (33). Chitayat-Hall syndrome is characterized by distal arthrogryposis, intellectual disability, dysmorphic features, and hypopituitarism, with GHD present in all reported cases to date (34). The same p.Q666Pfs*47 MAGEL2 truncation was present in one of the reported patients with Chitayat-Hall syndrome, demonstrating how variable overlapping phenotypes between SHFYNG and Chitayat-Hall syndromes arise from the same genotype and suggesting that full-length MAGEL2 is crucial for normal development of the human brain and for normal hypothalamo-pituitary function. Chitayat-Hall syndrome and SHFYNG may in fact be the same syndrome, albeit with variable penetrance, with some patients having sleep apnea, currently noted as a characteristic feature of SHFYNG but not Chitayat-Hall syndrome. An increasing number of patients with SHFYNG with MAGEL2 mutations (28) have not had their hypothalamo-pituitary function tested, suggesting that pituitary dysfunction may be a more frequent feature of SHFYNG, as is observed with Chitayat-Hall syndrome. Early endocrine diagnosis is crucial if endocrine morbidity is to be prevented and is therefore essential for improved quality of life for patients with these complex conditions.
Mutations in L1CAM, located on the X chromosome (Xp28) and encoding the L1 protein, have been implicated in the etiology of L1 syndrome (8). Female carriers may also manifest minor features of this syndrome, such as adducted thumbs or mild intellectual deficits (35). L1 is an axonal glycoprotein cell adhesion molecule that plays a role in neuronal migration and differentiation, including axon fasciculation (36), neurite outgrowth (37), synapse formation (38), and myelination (39). L1CAM-null mice have hydrocephalus, a smaller hippocampus and cerebellum, corpus callosal hypoplasia, hyperfasciculation of the corticothalamic tracts, and pyramidal tract abnormalities (40–45). Mutations within the cytoplasmic domain of the L1 protein have been described in MASA syndrome, which led to murine studies with L1 protein cytoplasmic domain disruption. Surprisingly, these mice had normal brain morphology, although they had defects in motor function (46). The hemizygous L1CAM mutation p.G452R identified in patient 5 was described previously in a patient with severe hydrocephalus (47). This mutation lies within, and is predicted to affect, the structure of the L1 extracellular domain required for correct folding of the protein and is subsequently thought to affect binding through the distortion of domain conformation (48). Further investigations have supported this, with a decreased ligand-binding ability in the presence of L1CAM p.G452R (49).
In rodents, L1cam is expressed in migrating neuron cell bodies from embryonic stage 9.5 and is later expressed in growing and regenerating axons. Myelinating Schwann cells express L1CAM during embryonic and postnatal development, whereas nonmyelinating Schwann cells express L1CAM through adulthood (50–53). The human L1CAM expression profile generated in this study revealed high transcript expression in the hypothalamus from 6 to 8 weeks of development (Fig. 4). However, no expression was visible in the RP or the PP, suggesting that this gene is hypothalamic and plays a critical role in this region during brain development. Patient 5 is the first patient to our knowledge with an L1CAM mutation and manifested GHD with pituitary dysfunction associated with features of L1 syndrome.
The trigeminal ganglia are sensory ganglia of the trigeminal nerve, responsible for sensation in the face and for motor functions. Both MAGEL2 and L1CAM expression within these specific tissues and during midline craniofacial development may suggest that sensation in the face is impaired in patients with mutations in these genes. However, the presence of global developmental delays did not allow assessment of this function. Limited availability of human embryonic sections did not allow analysis of expression beyond 8 weeks of gestation.
To summarize, our data suggest that patients with SHFYNG and L1 syndromes should all be screened and monitored for hypothalamo-pituitary abnormalities. Furthermore, patients with CH and accompanying joint contractures should be screened for MAGEL2 and L1CAM mutations and evaluated/monitored for additional phenotypes commonly present in SHFYNG or L1 syndrome, respectively. Our data and previously published data on SHFYNG and L1 syndromes suggest that MAGEL2 and L1CAM, respectively, should be screened for mutations using Sanger sequencing before next-generation techniques are conducted, as there is a high chance that a mutation lies within these genes in such patients. This would be the most cost-effective approach in screening for the most likely genetic diagnosis. However, in those cases in which a mutation is not identified in either of these genes, either whole exome or genome sequencing may be performed.
Acknowledgments
Financial Support: GOSH charity and the Medical Research Foundation (MRF) (Grant 535963) funded this study (to L.C.G.). The human embryonic and fetal material was provided by the Joint MRC/Wellcome Trust (Grant MR/R006237/1) Human Developmental Biology Resource (http://hdbr.org) (to L.C.G.). The research in this study is in part supported by the National Institute for Health Research, GOSH, and the Biomedical Research Center. The views expressed are those of the author(s) and not necessarily those of the National Health Service, the National Institute for Health Research, or the Department of Health. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (Grant HICF-1009-003), a parallel funding partnership between Wellcome and the Department of Health, and the Wellcome Sanger Institute (Grant WT098051; to L.C.G.). The views expressed in this publication are those of the author(s) and not necessarily those of Wellcome Trust or the Department of Health. The study has the UK Research Ethics Committee (REC) approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12, granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for Health Research through the Comprehensive Clinical Research Network.
Disclosure Summary: The authors have nothing to disclose.
Data Availability: All data generated or analyzed during this study are included in this published article or in the data repositories listed in References.
Abbreviations:
- ASD
autism spectrum disorder
- CH
congenital hypopituitarism
- CS
Carnegie stage
- DDD
Deciphering Developmental Disorders
- DI
diabetes insipidus
- GA
gestational age
- GHD
growth hormone deficiency
- GOSH
Great Ormond Street Hospital
- OMIM
Online Mendelian Inheritance in Man
- PP
posterior pituitary
- PWS
Prader-Willi syndrome
- RP
Rathke pouch
- SHFYNG
Schaaf-Yang syndrome

{kind=link}
{kind=link}
{kind=link}
{kind=link}