





Abstract
Polycystic ovary syndrome (PCOS) is the most common endocrinopathy of reproductive-aged women. In pregnancy, women with PCOS experience increased risk of miscarriage, gestational diabetes, preeclampsia, and extremes of fetal birth weight, and their offspring are predisposed to reproductive and cardiometabolic dysfunction in adulthood. Pregnancy complications, adverse fetal outcomes, and developmental programming of long-term health risks are known to have placental origins. These findings highlight the plausibility of placental compromise in pregnancies of women with PCOS.
A comprehensive PubMed search was performed using terms “polycystic ovary syndrome,” “placenta,” “developmental programming,” “hyperandrogenism,” “androgen excess,” “insulin resistance,” “hyperinsulinemia,” “pregnancy,” and “pregnancy complications” in both human and animal experimental models.
There is limited human placental research specific to pregnancy of women with PCOS. Gestational androgen excess and insulin resistance are two clinical hallmarks of PCOS that may contribute to placental dysfunction and underlie the higher rates of maternal–fetal complications observed in pregnancies of women with PCOS. Additional research is needed to prevent adverse maternal and developmental outcomes in women with PCOS and their offspring.
Polycystic ovary syndrome (PCOS) is the most common endocrine disorder of reproductive-aged women (1, 2). With an estimated incidence of 5% to 15% (3), PCOS has reproductive, metabolic, and gynecologic implications for women throughout their lifespan. Women with PCOS can experience increased risks of abnormal uterine bleeding, infertility, depression, insulin resistance, type 2 diabetes, hyperlipidemia, hypertension, cardiovascular disease, and endometrial cancer (2). In the United States, health care expenditures related to PCOS in reproductive-aged women are estimated to exceed $4 billion annually (4).
The Rotterdam criteria are generally used for the clinical diagnosis of PCOS, requiring two of the following: (i) oligoovulation or anovulation, (ii) clinical or biochemical evidence of hyperandrogenism, and (iii) ultrasound findings of polycystic ovaries (5). Subsequently, four distinct PCOS phenotypes have been recognized, reflecting the clinical heterogeneity of the syndrome: type A (hyperandrogenism, oligo-anovulation, and polycystic ovarian morphology), type B (hyperandrogenism and oligo-anovulation), type C (hyperandrogenism and polycystic ovarian morphology), and type D (oligo-anovulation and polycystic ovarian morphology) (6). Importantly, the diagnosis of PCOS requires the exclusion of other conditions of androgen excess, including congenital adrenal hyperplasia, adrenal or ovarian neoplasm, and Cushing syndrome (5). The etiology of PCOS is largely unknown (2, 7), and there are currently no proven strategies for PCOS prevention or cure.
PCOS Is Associated With Adverse Pregnancy Outcomes
As a reproductive disorder, PCOS may be accompanied by a variety of fertility challenges. Women with PCOS may have decreased fecundity due to etiologies such as anovulation or oligoovulation, reduced oocyte competence, and impaired endometrial receptivity (8–10). Women with PCOS have a higher prevalence of overweight or obesity, both independent risk factors for subfertility (11–13). Women with oligoovulation or anovulation may require ovulation induction agents to achieve pregnancy (14, 15). When assisted reproductive technologies (ARTs) such as in vitro fertilization are used, women with PCOS may experience poorer embryo development, lower pregnancy rates, and higher risk of iatrogenic ovarian hyperstimulation syndrome, relative to women without PCOS (16). Importantly, once pregnancy is achieved, women with PCOS are also at greater risk of pregnancy complications, as summarized below.
Maternal consequences
Compared with controls without PCOS, women with PCOS can demonstrate a twofold to fourfold increased risk of pregnancy-induced hypertension, preeclampsia, gestational diabetes, spontaneous preterm labor, and increased need for cesarean delivery (17–19). Owing to the phenotypic heterogeneity of PCOS, some studies have investigated these maternal outcomes with regard to the specific phenotype expressed. However, it remains unclear whether these risks are different among women of varying PCOS phenotypes (especially hyperandrogenic vs normoandrogenic), and how the presence of additional risk factors such as obesity, metabolic disturbances, or need for assisted reproductive technology may exacerbate this risk (20–25).
Fetal and offspring consequences
Maternal PCOS is also associated with fetal and neonatal morbidity, including prematurity, intrauterine growth restriction (IUGR), extremes of birth weight (BW; small or large for gestational age), and a twofold higher rate of admission to the neonatal intensive care unit (17, 19). Epidemiologic studies have also demonstrated that prenatal exposure to excess androgens may directly predispose the offspring to develop a PCOS-like phenotype and associated reproductive, metabolic, cardiovascular, and behavioral dysfunctions in childhood and adulthood (26–28). These findings are encompassed within the developmental origins of health and disease concept arising from the Barker hypothesis, whereby in utero alterations in the nutritional, endocrine, metabolic, and inflammatory milieu may alter gene expression during fetal development and predispose to the development of noncommunicable disease (29, 30). Besides PCOS, conditions that may induce a hyperandrogenic state during pregnancy include maternal obesity, exposure to endocrine-disrupting chemicals, smoking, preeclampsia, black race, exogenous exposure to steroid hormones, and antiepileptic medications (27, 31–33).
The placenta as a mediator of adverse pregnancy outcomes
Across all species, the placenta serves a similar purpose: maintenance of pregnancy, exchange of nutrients, clearance of fetal waste products, and protection from immunologic, endocrine, and environmental insults (34). The placenta is a dynamic endocrine organ with the ability to adapt to evolving intrauterine conditions and is uniquely positioned to receive and adapt to both maternal and fetal input (35, 36). Early human placentation is characterized by the invasion of extravillous trophoblast cells into maternal decidua and myometrium, followed by remodeling of the maternal spiral uterine arteries into a low-resistance, uteroplacental vascular network (37, 38). Later in pregnancy, under optimal conditions, the placenta integrates maternal and fetal signals to optimize the well-being and growth potential of the fetus (35, 39). Nevertheless, the inner workings of the placenta remain elusive, which has reenergized research efforts into placental research (40). The US National Institute of Child Health and Human Development has recently called for expanded research into the placental pathways of pregnancy-related disorders, to ultimately develop targeted interventions and therapies to improve maternal and fetal outcomes (41).
A large body of literature implicates the placenta as a mediator of both pregnancy complications, birth outcomes, and developmental programming (37, 42–46). It is intuitive, therefore, that there are placental alterations that occur in women with PCOS, which may predispose to adverse maternal, offspring, and birth outcomes. At present, our understanding of PCOS-specific placental pathologies is limited. Owing to variability in PCOS phenotypic expression, in addition to possible confounding comorbidities, including obesity, diabetes, hypertension, and/or the need for ART to conceive a pregnancy, there are significant methodological challenges in investigating placental dysfunction in PCOS. Therefore, it has been difficult to identify singular, precise mechanisms by which placental compromise may occur in women with PCOS. However, there is increasing evidence that androgen excess and insulin resistance, both of which are clinical hallmarks of PCOS, may negatively impact placental function (Fig. 1). Thus, in this review, we detail our current understanding of placental pathophysiology in the settings of (i) androgen excess and (ii) insulin resistance. We summarize current knowledge of PCOS-related placental dysfunction and identify gaps in knowledge to direct future research investigation that leads to targeted therapies for the prevention of pregnancy complications in women with PCOS.
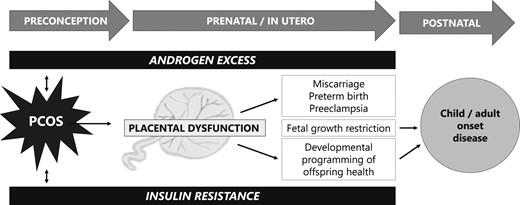
Placental dysfunction mediated by androgen excess and insulin resistance may lead to adverse pregnancy, birth, and long-term developmental outcomes in offspring.
Macroscopic and Microscopic Placental Changes Observed in Pregnancies of Women With PCOS
In routine practice, placental assessment by clinical pathologists involves an evaluation of both macroscopic and microscopic features. Here, we describe these methods and outline what is currently known in women with PCOS.
Placental assessments, as performed by clinical pathologists, routinely include placental weight (PW), diameter, thickness, shape, and location of umbilical cord insertion (47). A cross-sectional study of >11,000 healthy, singleton pregnancies delivered between 37 and 42 weeks provides reference ranges for average PW (48), although it is important to recognize that PW may be influenced by the method of placental collection, including mode of delivery, time to umbilical cord clamping, and subsequent handling of the sample (48, 49). Perhaps more clinically important than an isolated PW, is the ratio of fetal BW to PW (39). In simplified terms, the BW/PW ratio may infer how effectively the placenta has adapted to fetal growth needs. In the setting of fetal growth restriction, a diminished BW/PW ratio would indicate that the placenta was unable to functionally compensate for its small size, suggesting placental insufficiency as a mechanism for IUGR (39). Even in the absence of fetal growth restriction, impaired nutritional supply to the fetus may result in increased placental size relative to fetal size as a compensatory attempt to optimize nutritional delivery to the fetus (50). This suggests that a lower BW/PW ratio may still predict adverse pregnancy outcomes, even in the setting of BW appropriate for gestational age (51). Risk factors, including maternal diabetes and smoking, and outcomes, including lower neonatal Apgar scores and higher admission rates to the neonatal intensive care unit, have also been correlated with diminished BW/PW ratios (51–53).
Histological examinations are another important component of placental analysis (54). Broadly speaking, histologic lesions observed by placental pathologists may include either “vascular” or “villous” insults (54, 55), which may reflect a variety of immune, inflammatory, oxidative, or hypoxic conditions and provide insight into how pregnancy risk factors may translate into adverse fetal outcomes. However, limitations of placental examinations may include inherent observer bias in unblinded studies and interobserver variability among pathologists (55, 56).
In a case control study of pregnant women with PCOS (n = 30), matched with healthy pregnant controls (n = 60), the group with PCOS had a statistically significant reduction in PW, thickness, density, and volume (57). Furthermore, the derived BW/PW ratio was significantly lower in women with PCOS vs controls (5.12 ± 0.88 in women with PCOS, vs 5.90 ± 0.93 in controls). There was a trend toward decreased placental surface area in women with PCOS, although this did not reach statistical significance. Of note, the subjects with PCOS in this study only included nonobese women <35 years of age with spontaneous conceptions, uncomplicated pregnancies, and appropriate for gestational age neonates. Thus, although suggestive of an association between maternal PCOS and altered placental measures, these findings are not generalizable to a phenotypically diverse population with PCOS (57). In this same study, placentas of women with PCOS demonstrated higher rates of vascular lesions, chronic villitis, and villous immaturity than did placentas of women without PCOS, even in the absence of conditions such as maternal obesity, diabetes, prematurity, or small for gestational age neonates.
Meanwhile, Koster et al. (58) found no differences in PW or BW/PW ratio in PCOS (n = 73) vs non-PCOS controls (n = 209). In that study, subjects were not excluded for extremes of body mass index, mode of conception, or presence of pregnancy comorbidities, reflective of a more inclusive and heterogeneous sample of women with PCOS. In a subgroup analysis, the authors identified a trend toward a decreased BW/PW ratio in women who are hyperandrogenic vs normo-androgenic women (6.6 vs 7.3), although this did not reach statistical significance. Regarding histologic characteristics, women with PCOS compared with non-PCOS controls demonstrated higher rates of chorioamnionitis, funisitis, villitis, vascular thrombosis, infarction, and villous immaturity (58). These observations remained statistically significant after adjusting for confounding variables, including maternal age, ethnicity, parity, smoking status, preeclampsia, gestational diabetes, gestational age at delivery, and mode of delivery. A strength of this study is that all placental assessments were performed by a single experienced, blinded pathologist.
Mechanisms Underlying PCOS-Associated Placental Dysfunctions
There are several challenges in conducting mechanistic human placental research. Placental samples are generally collected at the time of delivery, in the third trimester or at term (59), and additional confounding factors may include timing of onset of labor and mode of delivery (60, 61). Owing to ethical considerations and the inherent risks of invasive placental sampling, placental tissue from early gestational time points is not easily obtained for research purposes.
Capitalizing on animal models to improve our understanding of placental pathology in PCOS
Fortunately, animal models of PCOS offer translational value for understanding the mechanisms of both normal and abnormal early placentation. One major benefit offered by animal models is the ability to investigate changes at the earliest stages of placentation and throughout gestation. The advantages and disadvantages of using experimental animal models for placental investigation have been extensively reviewed elsewhere (34, 62, 63). Although there are species-specific differences in placental histology and morphology (64), animal models are necessary for understanding the normal trajectory of placental development, in addition to identifying the timing and onset of placental dysfunction (34, 40). For PCOS-related placental work, most published research involves rodent, sheep, and nonhuman primate models (65), and the benefits and limitations of these models are briefly summarized below.
Human placentas are defined as “hemochorial.” In the early stages of placental development, the invading extravillous trophoblasts are in direct contact with the maternal vasculature. This is in contrast to “epitheliochorial” and “endotheliochorial” placentation, where the trophoblasts are in contact with the uterine epithelium or maternal vessel endothelium, respectively (38, 64, 66). Rodents, similar to humans, exhibit hemochorial placentation and are valuable for studying the early gestational processes of trophoblast invasion, differentiation, and spiral artery remodeling (38, 67). Advantages of rodent models include a short gestational length and affordability. However, they differ from humans in that their offspring are altricial and born in litters, and their small physical size may create challenges for invasive and noninvasive monitoring (34, 62).
Sheep have been historically invaluable for the study of maternal and fetal physiology (34, 68). Benefits of sheep models include their large size relative to rodent species, allowing for repeated blood measures and ultrasound imaging techniques that have translational value to humans (68, 69). Sheep are a domesticated species and able to be kept in stress-free, natural flocks (70). Importantly, sheep are also a precocial species, similar to humans, and thus have translational relevance from a developmental standpoint (62, 70). In contrast to humans, sheep demonstrate epitheliochorial placentation, where chorionic villi interface with the epithelium of numerous (>50), discrete maternal placental units (placentomes). Nonetheless, there is considerable structural similarity between sheep and human placenta as reflected by the architecture of the fetal villous tree with stem, intermediate, and terminal vessels (71, 72). The terminal vessels, or capillary complex, which are the functional units of exchange, differ very little between sheep and humans. In both, the chorioallantoic villous tree builds up to fetal placental cotyledons. In sheep, each cotyledon penetrates the maternal septal system of the endometrial caruncle and, together, they form one of numerous structures called placentomes. In humans, cotyledons protrude into the extended maternal intervillous blood space of the single polycotyledonary placental disc. The presence of placentomes is an advantage, as it enables the study of small, clearly identified regions of interest.
Benefits of primate models, such as the rhesus macaque, include their large size and their close similarities to humans with regard to length of gestation, uterine anatomy, trophoblast invasion in maternal spiral artery remodeling, and parturition (34). Nonhuman primates also exhibit hemochorial placentation, with a villous structure similar to human placental villi (34, 64). Disadvantages of primate models include a long gestational period, high maintenance cost of primate colonies, and ethical concerns due to their close ancestral relationship to humans (62). Despite the limitations of these above-mentioned animal models, they provide important translational value for improving our understanding of human placental pathology.
Mediators of placental pathology in other disease states
Various mediators of placental pathophysiology (Fig. 2) have been observed in maternal conditions such as obesity, preeclampsia, diabetes, and using ART to conceive, as briefly summarized below. Although these placental mechanisms have yet to be proven in pregnancies of women with PCOS, we propose that they warrant further investigation in the population with PCOS and in animal models of PCOS as potential mediators of adverse pregnancy outcomes.
Hypoxia: Early placental development, even in normal conditions, occurs in a low-oxygen environment (73) until the end of the first trimester. In animal models, excess hypoxic stress in early pregnancy may negatively affect proliferation or differentiation of trophoblast cells (74). In corollary, markers of hypoxia, such as the transcription factor hypoxia-inducible factor-1α (HIF-1α), are elevated in the placenta of women who are preeclamptic (75) and are worth investigation in PCOS models.
Vasculopathy: As a vascular organ, the placenta boasts an estimated 15 m2 of capillary surface area in healthy human pregnancies (76). Vascular growth within the placenta is complex, balanced by complementary proangiogenic and antiangiogenic factors. Miscarriage, IUGR, and preeclampsia are thought to arise from disordered angiogenesis (76–78), and PCOS may likewise be characterized by abnormal vasculogenesis or angiogenesis.
Altered nutrient transport: Optimization of fetal growth depends on the availability and transport of maternal nutrients, such as lipids, amino acids, and glucose through the placenta into the developing fetus (79). Abnormalities in nutrient transport may occur in conditions of maternal undernutrition or overnutrition and may also have long-term effects on the programming of adult-onset disease (80–83). Prior work by Maliqueo et al. (84) has demonstrated that placental STAT3 signaling, a regulator for nutrient transport and fetal growth, is enhanced in placentas of women with PCOS vs non-PCOS controls, regardless of levels of circulating sex steroids. More research is warranted on the subject of placental nutrient transport in PCOS, particularly with regard to metabolic parameters such as body mass index and insulin resistance, as well as steroidal milieu such as androgen excess, particularly because infants born from women with PCOS are paradoxically at risk for both small for gestational age or large for gestational age.
Oxidative stress: The high metabolic activity of the placenta leads to continuous production of reactive oxidative species, and thus normal pregnancy is characterized by some degree of oxidative stress (85, 86). Disease states that may alter the balance between reactive oxygen species and antioxidants, to predispose to a persistent state of placental oxidative stress and cellular damage, include spontaneous abortion, IUGR, preeclampsia, and preterm labor (87, 88), all of which are observed at higher rates in women with PCOS.
Inflammation: Normal pregnancy is characterized by some degree of systemic maternal inflammation, particularly in the third trimester (89, 90). The placenta itself is characterized by a vast network of both inflammatory cytokines and adipokines that may be involved in physiologic regulation of maternal–fetal metabolism, fetal growth, and parturition (90, 91) and adverse outcomes, including gestational diabetes and spontaneous preterm birth (92–94), also seen at higher frequency in pregnancies of women with PCOS.
Lipotoxicity: Lipotoxicity is defined as lipid accumulation in nonadipose tissue (95). Placental lipotoxicity is associated with placental inflammation and oxidative stress, and it may be observed in conditions of maternal obesity, insulin resistance, and gestational diabetes (96–98), further highlighting the possibility of placental lipotoxicity in women with PCOS.
Fibrosis: Collagen deposition within tissue is a normal, restorative response to injury. In pathologic conditions, however, excess collagen deposition can occur, leading to fibrosis, loss of tissue function, and even tissue death (99). In the obstetric literature, placental fibrotic lesions have been associated with preeclampsia (100, 101) but not yet explored in PCOS.
Endocrinopathy: The placenta is a hormonally regulated organ, demonstrating receptors to steroid hormones (including sex steroids and glucocorticoids), peptides (such as GnRH), proteins (such as insulin, prolactin, and leptin), and glycoproteins (such as human chorionic gonadotropin) (102). Hormonal dysregulation in the setting of maternal thyroid disease, insulin resistance, obesity, or PCOS may impact placental development and function via the above-mentioned mechanisms.

Proposed mechanisms for placental dysfunction in women with PCOS.
Contribution of Androgen Excess and Insulin Resistance to Placental Dysfunction in PCOS
Androgen excess and insulin resistance are two clinical hallmarks of PCOS. Owing to the phenotypic heterogeneity of the condition, women with PCOS may demonstrate one, both, or neither of these features. Because placental studies are limited in women with PCOS, for purposes of this review, we additionally consider animal models of PCOS to outline the contributions of (i) androgen excess and (ii) insulin resistance on placental dysfunction.
Contribution of androgen excess
Similar to levels of estrogens and progesterone, androgen levels demonstrate a physiologic increase during normal pregnancy. They serve as substrate for placental aromatization to estrogens, and may even have a role in parturition (103, 104). Pregnant women with PCOS may demonstrate higher circulating levels of testosterone (T), dehydroepiandrosterone sulfate, and androstenedione compared with pregnant non-PCOS controls, regardless of fetal sex (105, 106). As such, maternal PCOS may confer an environment of androgen excess to the developing fetus, even if virilization of a female fetus is not observed (105). As maternal androgens are not thought to directly cross the placenta (107), and placental aromatase activity is thought to “protect” the fetus from androgen excess via conversion to estrogen, one possible mechanism is that maternal androgens may exert a programming effect on placental and/or fetal steroidogenesis to alter androgen levels within the fetal–placental unit (23, 108–110). In humans, this concept is supported by work from Maliqueo et al. (109), wherein placental tissue from women with PCOS demonstrated increased 3β-hydroxysteroid dehydrogenase 1 enzymatic activity and decreased aromatase activity compared with non-PCOS controls.
Interestingly, in studies using umbilical cord blood to estimate fetal–placental androgen levels, there is conflicting evidence as to whether, and how, maternal androgen levels are correlated to those in the fetal–placental unit in pregnancies of women with PCOS. Some studies have suggested that neonatal umbilical cord levels of T and/or androstenedione may be elevated in pregnancies of women with PCOS, irrespective of infant sex (111–113). However, other studies suggest no difference in cord blood androgen levels of infants of women with PCOS vs controls, whereas other studies demonstrate decreased androstenedione levels in cord blood of female offspring of women with PCOS (109, 114–116). These discrepancies may be due to differences in study design, patient selection, and methodologic differences of cord blood collection.
Prenatal hyperandrogenic states in animal models of PCOS (117) are associated with fetal growth restriction and low BW (118, 119), suggesting an effect on placental function and efficiency. The biologic plausibility of this notion is supported by the fact that the androgen receptor is expressed in placenta of humans and animals (120–122). In animal studies, gestational hyperandrogenism can be achieved with maternal T treatment (70, 123, 124). Rodent models of PCOS generally involve T administration relatively late in pregnancy, so these findings do not account for any potential androgen effects at early fetal–placental developmental time points. Nevertheless, studies involving prenatally androgenized rats and sheep have provided evidence for the role of androgens on placental function and pathologies, as summarized in Table 1 (107, 108, 120, 121, 125–129). In one study, T treatment in late pregnancy was linked to increased levels of circulating androgens and estrogens, in addition to increased placental gene expression of estrogen receptor (ER)-α and ER-β, androgen receptor, and 17β-hydroxysteroid dehydrogenase 2 (121). In this model, T treatment was also accompanied by decreased placental and fetal weights, but no significant change in the BW/PW ratio. Notably, the offspring of T-treated dams developed metabolic dysfunction later in life, highlighting the role of androgen excess in the developmental programming of adult metabolic disease (121). Similarly, in another experimental model, T treatment in late gestation was linked to decreased fetal weights and PWs via decreased expression of placental amino acid transporter, but no difference in the BW/PW ratio (107). T treatment has also been associated with decreased uterine artery blood flow, increased expression of HIF-1α, and decreased fetal weights and PWs in rodent models (125). In sheep, T treatment during early gestational time points also leads to intrauterine growth restriction and low BW, particularly of female offspring. Placental insufficiency may underlie these observations, as T-treated sheep demonstrated advancement of placental differentiation as early as mid-gestation (108).
Animal Models of Gestational Androgen Excess and Its Effect on Placental Function
| Study (Ref.) | Animal | Androgen Intervention | Effect of Androgen Intervention on Placental Function |
|---|---|---|---|
| Sathishkumar et al., 2011 (107) | Rodent | 0.5 mg/kg T propionate daily, gestational days 15–19 | Decreased fetal weights and PWs, but no difference in BW/PW ratio |
| Decreased placental amino acid transport | |||
| Sun et al., 2012 (121) | Rodent | 5 mg T daily, gestational days 16–19 | Decreased fetal weights and PWs, but no difference in BW/PW ratio |
| Increased placental expression of ER-α and ER-β, androgen receptor, and 17β-hydroxysteroid dehydrogenase 2 | |||
| Chinnathambi et al., 2014 (126) | Rodent | 0.5 mg/kg T propionate daily, gestational days 15–19 | Decreased fetal weights and PWs |
| Increased gene and protein expression of hypoxia-responsive genes HIF-1α, Ankrd 37, and Egln | |||
| Hu et al., 2015 (127) | Rodent | 0.5 mg/kg T daily, gestational days 15–19, with or without tamoxifen 10 µg/kg daily | Decreased PW and increased STAT3 expression |
| Gopalakrishnan et al., 2016 (125) | Rodent | 0.5 mg/kg T propionate daily, gestational days 15–19 | Decreased fetal weights and PWs, but no difference in BW/PW ratio |
| Decreased uterine artery blood flow and elevated vascular resistance | |||
| Sex-specific alterations in utero placental vasculature | |||
| Fornes et al., 2017 (128) | Rodent | 100 µL of DHT daily, gestational days 15.5–17.5 | No difference in fetal weights or PWs |
| Control diet vs high-fat-high sucrose diet | Increased placental AR expression | ||
| Fornes et al., 2019 (129) | Rodent | 100 µL of DHT daily, gestational days 15.5–18.5 | Differential expression of phosphorylated catechol-O-methyltransferase depending on maternal diet |
| Control diet vs high-fat/high-sucrose diet | |||
| Beckett et al., 2014 (108) | Sheep | 100 mg IM of T propionate biweekly, gestational days 30–90 | Placental insufficiency at gestational day 140 (decreased BW/PW ratio) |
| Advanced placental differentiation | |||
| Cleys et al., 2015 (120) | Sheep | 100 mg IM of T propionate biweekly, gestational days 30–90 | Decreased fetal weight (female lambs only) at gestational day 90 |
| Advanced placental differentiation | |||
| Increased mRNA and protein expression of VEGF |
| Study (Ref.) | Animal | Androgen Intervention | Effect of Androgen Intervention on Placental Function |
|---|---|---|---|
| Sathishkumar et al., 2011 (107) | Rodent | 0.5 mg/kg T propionate daily, gestational days 15–19 | Decreased fetal weights and PWs, but no difference in BW/PW ratio |
| Decreased placental amino acid transport | |||
| Sun et al., 2012 (121) | Rodent | 5 mg T daily, gestational days 16–19 | Decreased fetal weights and PWs, but no difference in BW/PW ratio |
| Increased placental expression of ER-α and ER-β, androgen receptor, and 17β-hydroxysteroid dehydrogenase 2 | |||
| Chinnathambi et al., 2014 (126) | Rodent | 0.5 mg/kg T propionate daily, gestational days 15–19 | Decreased fetal weights and PWs |
| Increased gene and protein expression of hypoxia-responsive genes HIF-1α, Ankrd 37, and Egln | |||
| Hu et al., 2015 (127) | Rodent | 0.5 mg/kg T daily, gestational days 15–19, with or without tamoxifen 10 µg/kg daily | Decreased PW and increased STAT3 expression |
| Gopalakrishnan et al., 2016 (125) | Rodent | 0.5 mg/kg T propionate daily, gestational days 15–19 | Decreased fetal weights and PWs, but no difference in BW/PW ratio |
| Decreased uterine artery blood flow and elevated vascular resistance | |||
| Sex-specific alterations in utero placental vasculature | |||
| Fornes et al., 2017 (128) | Rodent | 100 µL of DHT daily, gestational days 15.5–17.5 | No difference in fetal weights or PWs |
| Control diet vs high-fat-high sucrose diet | Increased placental AR expression | ||
| Fornes et al., 2019 (129) | Rodent | 100 µL of DHT daily, gestational days 15.5–18.5 | Differential expression of phosphorylated catechol-O-methyltransferase depending on maternal diet |
| Control diet vs high-fat/high-sucrose diet | |||
| Beckett et al., 2014 (108) | Sheep | 100 mg IM of T propionate biweekly, gestational days 30–90 | Placental insufficiency at gestational day 140 (decreased BW/PW ratio) |
| Advanced placental differentiation | |||
| Cleys et al., 2015 (120) | Sheep | 100 mg IM of T propionate biweekly, gestational days 30–90 | Decreased fetal weight (female lambs only) at gestational day 90 |
| Advanced placental differentiation | |||
| Increased mRNA and protein expression of VEGF |
Abbreviation: VEGF, vascular endothelial growth factor.
Animal Models of Gestational Androgen Excess and Its Effect on Placental Function
| Study (Ref.) | Animal | Androgen Intervention | Effect of Androgen Intervention on Placental Function |
|---|---|---|---|
| Sathishkumar et al., 2011 (107) | Rodent | 0.5 mg/kg T propionate daily, gestational days 15–19 | Decreased fetal weights and PWs, but no difference in BW/PW ratio |
| Decreased placental amino acid transport | |||
| Sun et al., 2012 (121) | Rodent | 5 mg T daily, gestational days 16–19 | Decreased fetal weights and PWs, but no difference in BW/PW ratio |
| Increased placental expression of ER-α and ER-β, androgen receptor, and 17β-hydroxysteroid dehydrogenase 2 | |||
| Chinnathambi et al., 2014 (126) | Rodent | 0.5 mg/kg T propionate daily, gestational days 15–19 | Decreased fetal weights and PWs |
| Increased gene and protein expression of hypoxia-responsive genes HIF-1α, Ankrd 37, and Egln | |||
| Hu et al., 2015 (127) | Rodent | 0.5 mg/kg T daily, gestational days 15–19, with or without tamoxifen 10 µg/kg daily | Decreased PW and increased STAT3 expression |
| Gopalakrishnan et al., 2016 (125) | Rodent | 0.5 mg/kg T propionate daily, gestational days 15–19 | Decreased fetal weights and PWs, but no difference in BW/PW ratio |
| Decreased uterine artery blood flow and elevated vascular resistance | |||
| Sex-specific alterations in utero placental vasculature | |||
| Fornes et al., 2017 (128) | Rodent | 100 µL of DHT daily, gestational days 15.5–17.5 | No difference in fetal weights or PWs |
| Control diet vs high-fat-high sucrose diet | Increased placental AR expression | ||
| Fornes et al., 2019 (129) | Rodent | 100 µL of DHT daily, gestational days 15.5–18.5 | Differential expression of phosphorylated catechol-O-methyltransferase depending on maternal diet |
| Control diet vs high-fat/high-sucrose diet | |||
| Beckett et al., 2014 (108) | Sheep | 100 mg IM of T propionate biweekly, gestational days 30–90 | Placental insufficiency at gestational day 140 (decreased BW/PW ratio) |
| Advanced placental differentiation | |||
| Cleys et al., 2015 (120) | Sheep | 100 mg IM of T propionate biweekly, gestational days 30–90 | Decreased fetal weight (female lambs only) at gestational day 90 |
| Advanced placental differentiation | |||
| Increased mRNA and protein expression of VEGF |
| Study (Ref.) | Animal | Androgen Intervention | Effect of Androgen Intervention on Placental Function |
|---|---|---|---|
| Sathishkumar et al., 2011 (107) | Rodent | 0.5 mg/kg T propionate daily, gestational days 15–19 | Decreased fetal weights and PWs, but no difference in BW/PW ratio |
| Decreased placental amino acid transport | |||
| Sun et al., 2012 (121) | Rodent | 5 mg T daily, gestational days 16–19 | Decreased fetal weights and PWs, but no difference in BW/PW ratio |
| Increased placental expression of ER-α and ER-β, androgen receptor, and 17β-hydroxysteroid dehydrogenase 2 | |||
| Chinnathambi et al., 2014 (126) | Rodent | 0.5 mg/kg T propionate daily, gestational days 15–19 | Decreased fetal weights and PWs |
| Increased gene and protein expression of hypoxia-responsive genes HIF-1α, Ankrd 37, and Egln | |||
| Hu et al., 2015 (127) | Rodent | 0.5 mg/kg T daily, gestational days 15–19, with or without tamoxifen 10 µg/kg daily | Decreased PW and increased STAT3 expression |
| Gopalakrishnan et al., 2016 (125) | Rodent | 0.5 mg/kg T propionate daily, gestational days 15–19 | Decreased fetal weights and PWs, but no difference in BW/PW ratio |
| Decreased uterine artery blood flow and elevated vascular resistance | |||
| Sex-specific alterations in utero placental vasculature | |||
| Fornes et al., 2017 (128) | Rodent | 100 µL of DHT daily, gestational days 15.5–17.5 | No difference in fetal weights or PWs |
| Control diet vs high-fat-high sucrose diet | Increased placental AR expression | ||
| Fornes et al., 2019 (129) | Rodent | 100 µL of DHT daily, gestational days 15.5–18.5 | Differential expression of phosphorylated catechol-O-methyltransferase depending on maternal diet |
| Control diet vs high-fat/high-sucrose diet | |||
| Beckett et al., 2014 (108) | Sheep | 100 mg IM of T propionate biweekly, gestational days 30–90 | Placental insufficiency at gestational day 140 (decreased BW/PW ratio) |
| Advanced placental differentiation | |||
| Cleys et al., 2015 (120) | Sheep | 100 mg IM of T propionate biweekly, gestational days 30–90 | Decreased fetal weight (female lambs only) at gestational day 90 |
| Advanced placental differentiation | |||
| Increased mRNA and protein expression of VEGF |
Abbreviation: VEGF, vascular endothelial growth factor.
Prenatal androgens have also been implicated in placental vasculopathies (77, 130, 131). In pregnant rats, elevated androgens increase the vascular tone of the uterine arteries, compromising the primary blood supply to the pregnant uterus, and subsequently increase the expression of hypoxia-responsive genes (126). Additionally, in rat models, gestational hyperandrogenism has been shown to reduce gene expression of placental proangiogenic factors (107, 132, 133). In sheep, prenatal T treatment is associated with an increase in placental mRNA and protein expression of vascular endothelial growth factor (VEGF), an important regulator of placental angiogenesis (120). In this well-validated sheep model, T treatment is administered from an early gestational time point (between days 30 through 90, with lambing at gestational day 147), which provides the benefit of studying the effects of androgen excess during early, critical windows of fetal development (70, 108).
Preeclampsia, a syndrome of maternal hypertension, proteinuria, and vascular compromise, has also been linked to androgen excess (134–136). Preeclampsia is a leading cause of maternal and fetal morbidity and is thought to arise from abnormalities in early placental development. Maternal obesity, type 2 diabetes, and the use of ART, all of which are observed more frequently in women with PCOS, are established risk factors for the development of preeclampsia (137). Evidence suggests that aberrant trophoblast invasion and impaired remodeling of the maternal spiral arteries predispose to preeclampsia (42, 134). Epidemiologic studies have also demonstrated higher androstenedione and T levels in maternal serum of pregnancies of women with preeclamsia vs controls (138, 139). Interestingly, prior work by Palomba et al. (140) has demonstrated defective endovascular trophoblast invasion in late first-trimester placental samples from women with PCOS, compared with non-PCOS controls, providing support for an association between androgen excess and impairment of spiral artery remodeling.
There also exists a large body of literature related to third-trimester placental characteristics of preeclampsia. Placentas from mothers with preeclampsia at advanced gestational ages have more villous lesions (a finding that suggests maternal hypoperfusion), higher expression of inflammatory markers, and a greater proportion of antiangiogenic compared with proangiogenic factors (42). Additionally, placentas from women with preeclampsia have higher lipid content (134) and increased markers of oxidative stress (43). These investigations have highlighted several potential opportunities for the prevention of preeclampsia, including the use of antioxidants, anti-inflammatories, vasodilators, and even metformin (37, 141, 142). Although similar molecular targets have yet to be tested in PCOS-related placental pathologies, the possibility that androgen excess may underlie both PCOS and preeclampsia suggests that these two conditions may share similar preventive or therapeutic strategies.
Androgens vs estrogens
In normal pregnancy, placental production of estradiol is high (∼100 mg/d) (143). Unlike androgens, which are thought to increase uterine vascular tone and downregulate placental angiogenic genes, estrogens promote vascular relaxation and increase placental VEGF expression, particularly in early gestation (132). Estrogens also regulate genes involved in cholesterol supply to the placenta (143), important for fetal and placental steroid hormone production. The mechanisms underlying placental estrogen production are complex, but, in brief, they involve the conversion of fetal adrenal dehydroepiandrosterone into estrogens by the enzymatic activity of placental aromatase. In abnormal states, such as preeclampsia, estrogen levels are thought to be reduced, which may be linked to impaired angiogenesis through alterations in placental VEGF (144, 145).
Because androgens are precursors to estrogens, a methodical challenge is determining whether specific findings are mediated by androgenic vs estrogenic pathways (132). In animal studies, this particular challenge has been partially overcome by examining the direct effects of DHT, a nonaromatizable androgen (128, 129). In rodents, T treatment increased circulating levels of T and DHT, both of which were associated with decreased PW, suggesting that this effect may be androgen mediated (127). In this same study, cotreatment of T with tamoxifen, a selective ER modulator, also increased phosphorylated and total STAT3 protein expression, suggesting that ER is not involved in this same pathway that has been shown to be activated in women with PCOS (84, 127). In prenatally androgenized sheep models, wherein androgen excess results in IUGR through placental compromise, the distinct placental effects of T-treated vs DHT-treated pregnant sheep were investigated (108). In both experimental groups, accelerated placental aging was observed relative to controls, suggesting that the observed placental effects are more likely mediated by the androgenic, not estrogenic, actions of T. Importantly, note that DHT may be converted to 3-β-diol, which can bind ER-β and elicit downstream estrogenic effects. Thus, an alternative approach to elucidate whether placental changes are androgen or estrogen mediated is cotreatment of T with an androgen antagonist, such as flutamide. In sheep, cotreatment of T with flutamide partially prevented the observed placental advancement in this model (108), suggesting that the placental changes were mediated at least in part by androgenic action.
Contribution of insulin resistance
Normal pregnancy is characterized by decreased glucose uptake and increased insulin secretion, leading to a state of insulin resistance, which helps to provide adequate nutrition to the developing fetus (146, 147). Nevertheless, preconception insulin resistance is a clinical hallmark of PCOS (148, 149), and approximately one-third of women with obesity with PCOS demonstrate impaired glucose tolerance, independent from obesity (1). In pregnancy, women with PCOS compared with non-PCOS controls also demonstrate hyperinsulinemia and lipid abnormalities, including hypertriglyceridemia and increased low-density lipoprotein (150, 151). Maternal insulin resistance has been implicated in other complications, including miscarriage, pregnancy-induced hypertension, and growth restriction, and placental expression of insulin receptor has been widely demonstrated (152, 153), suggesting a link between insulin resistance and placental compromise (154, 155). Additionally, insulin, IGF-I, and IGF-II have been shown to inhibit the enzymatic activity of aromatase within human trophoblasts (156). These findings highlight the correlation between hyperinsulinemia and androgen excess in women with PCOS; the interaction of steroidogenic and metabolic disturbances in pregnancies of women with PCOS has been previously reviewed (157).
Insulin resistance may have negative effects on early placentation. Hyperglycemia in early pregnancy may increase rates of miscarriage, intrauterine growth restriction, and preeclampsia (158), likely due to impaired trophoblast invasion. In vitro studies of first-trimester human placental tissue have demonstrated that insulin resistance can impair IGF-1 signaling, which is important for trophoblast invasion of maternal spiral arteries. Conversely, addition of an insulin sensitizer can restore normal signaling and promote appropriate trophoblast migration and invasion (154). Relevant to PCOS, there is some evidence that women with PCOS display lower levels of IGF-1 in the first trimester of pregnancy, possibly due to insulin resistance (159, 160). Interestingly, in sheep, cotreatment of T with rosiglitazone, an insulin sensitizer, does not mitigate the observed advancement of placental aging in T-treated sheep, suggesting that insulin resistance may not be a critical mediator of placental insufficiency in this particular animal model of PCOS.
PCOS is also a known risk factor for the development of gestational diabetes mellitus (GDM) (161). GDM is generally not diagnosed until the second trimester; however, baseline insulin resistance may predispose to the development of GDM (142, 162, 163). Research on GDM-associated placental dysfunction is extensive and has highlighted several possible mechanisms, including reduced placental apoptosis, impaired vasculogenesis, increased ischemia, and villous immaturity relative to non-GDM controls (164–168). Table 2 (108, 169–174) highlights and briefly summarizes key placental findings in animal models of GDM. There is also emerging evidence that placental production and release of adipokines such as leptin, adiponectin, and resistin may be altered in pregnancies complicated by GDM, which in turn may be linked to placental oxidative stress and inflammation (91, 175–178). Because adipokines may be involved with regulating fetal growth, in addition to programming insulin sensitivity (179), their role in PCOS pregnancies warrants further investigation.
Animal Models of Gestational Diabetes and/or Hyperglycemia and Their Effects on Placental Function
| Study (Ref.) | Animal | Model for Gestational Diabetes | Effect of Hyperglycemia on Placental Function |
|---|---|---|---|
| Ericsson et al., 2007 (169) | Rodent | Single or multiple IP glucose injections on gestational day 10 | Increased PW and fetal weight on gestational day 21 |
| Downregulation of placental SNAT2 (system A transport) | |||
| Bobadilla et al., 2010 (170) | Rodent | Heterozygotes for defective leptin receptors (db/+) | Impaired trophoblast invasion in db/+ vs wild-type mice |
| Exogenous TNF-α treatment increases placental apoptosis in db/+ mice | |||
| Salbaum et al., 2011 (171) | Rodent | Streptozotocin injections prior to mating | Reduced junctional zone and labyrinth size |
| Altered gene expression profile (158 genes) | |||
| Li et al., 2013 (172) | Rodent | High-fat diet for 14 wk prior to mating and throughout gestation | Increased markers of hypoxia and oxidative stress |
| Increased mRNA expression of VEGF, TNF-α, and IL-6 | |||
| Cisse et al., 2013 (173) | Rodent | Single injection of nicotinamide plus streptozotocin prior to mating | Increased lipoprotein lipase-1 expression in macrosomic pups |
| Dela Justina et al., 2014 (174) | Rodent | Streptozotocin injection prior to mating | Increased PW |
| Increased nuclear factor κB activation | |||
| Increased expression of TNF-α and IL-6 | |||
| Beckett et al., 2014 (108) | Sheep | 100 mg IM of T propionate biweekly plus rosiglitazone 8 mg/kg orally daily, gestational days 30–90 | Addition of insulin sensitizer did not alter T-mediated placental advancement |
| Study (Ref.) | Animal | Model for Gestational Diabetes | Effect of Hyperglycemia on Placental Function |
|---|---|---|---|
| Ericsson et al., 2007 (169) | Rodent | Single or multiple IP glucose injections on gestational day 10 | Increased PW and fetal weight on gestational day 21 |
| Downregulation of placental SNAT2 (system A transport) | |||
| Bobadilla et al., 2010 (170) | Rodent | Heterozygotes for defective leptin receptors (db/+) | Impaired trophoblast invasion in db/+ vs wild-type mice |
| Exogenous TNF-α treatment increases placental apoptosis in db/+ mice | |||
| Salbaum et al., 2011 (171) | Rodent | Streptozotocin injections prior to mating | Reduced junctional zone and labyrinth size |
| Altered gene expression profile (158 genes) | |||
| Li et al., 2013 (172) | Rodent | High-fat diet for 14 wk prior to mating and throughout gestation | Increased markers of hypoxia and oxidative stress |
| Increased mRNA expression of VEGF, TNF-α, and IL-6 | |||
| Cisse et al., 2013 (173) | Rodent | Single injection of nicotinamide plus streptozotocin prior to mating | Increased lipoprotein lipase-1 expression in macrosomic pups |
| Dela Justina et al., 2014 (174) | Rodent | Streptozotocin injection prior to mating | Increased PW |
| Increased nuclear factor κB activation | |||
| Increased expression of TNF-α and IL-6 | |||
| Beckett et al., 2014 (108) | Sheep | 100 mg IM of T propionate biweekly plus rosiglitazone 8 mg/kg orally daily, gestational days 30–90 | Addition of insulin sensitizer did not alter T-mediated placental advancement |
Animal Models of Gestational Diabetes and/or Hyperglycemia and Their Effects on Placental Function
| Study (Ref.) | Animal | Model for Gestational Diabetes | Effect of Hyperglycemia on Placental Function |
|---|---|---|---|
| Ericsson et al., 2007 (169) | Rodent | Single or multiple IP glucose injections on gestational day 10 | Increased PW and fetal weight on gestational day 21 |
| Downregulation of placental SNAT2 (system A transport) | |||
| Bobadilla et al., 2010 (170) | Rodent | Heterozygotes for defective leptin receptors (db/+) | Impaired trophoblast invasion in db/+ vs wild-type mice |
| Exogenous TNF-α treatment increases placental apoptosis in db/+ mice | |||
| Salbaum et al., 2011 (171) | Rodent | Streptozotocin injections prior to mating | Reduced junctional zone and labyrinth size |
| Altered gene expression profile (158 genes) | |||
| Li et al., 2013 (172) | Rodent | High-fat diet for 14 wk prior to mating and throughout gestation | Increased markers of hypoxia and oxidative stress |
| Increased mRNA expression of VEGF, TNF-α, and IL-6 | |||
| Cisse et al., 2013 (173) | Rodent | Single injection of nicotinamide plus streptozotocin prior to mating | Increased lipoprotein lipase-1 expression in macrosomic pups |
| Dela Justina et al., 2014 (174) | Rodent | Streptozotocin injection prior to mating | Increased PW |
| Increased nuclear factor κB activation | |||
| Increased expression of TNF-α and IL-6 | |||
| Beckett et al., 2014 (108) | Sheep | 100 mg IM of T propionate biweekly plus rosiglitazone 8 mg/kg orally daily, gestational days 30–90 | Addition of insulin sensitizer did not alter T-mediated placental advancement |
| Study (Ref.) | Animal | Model for Gestational Diabetes | Effect of Hyperglycemia on Placental Function |
|---|---|---|---|
| Ericsson et al., 2007 (169) | Rodent | Single or multiple IP glucose injections on gestational day 10 | Increased PW and fetal weight on gestational day 21 |
| Downregulation of placental SNAT2 (system A transport) | |||
| Bobadilla et al., 2010 (170) | Rodent | Heterozygotes for defective leptin receptors (db/+) | Impaired trophoblast invasion in db/+ vs wild-type mice |
| Exogenous TNF-α treatment increases placental apoptosis in db/+ mice | |||
| Salbaum et al., 2011 (171) | Rodent | Streptozotocin injections prior to mating | Reduced junctional zone and labyrinth size |
| Altered gene expression profile (158 genes) | |||
| Li et al., 2013 (172) | Rodent | High-fat diet for 14 wk prior to mating and throughout gestation | Increased markers of hypoxia and oxidative stress |
| Increased mRNA expression of VEGF, TNF-α, and IL-6 | |||
| Cisse et al., 2013 (173) | Rodent | Single injection of nicotinamide plus streptozotocin prior to mating | Increased lipoprotein lipase-1 expression in macrosomic pups |
| Dela Justina et al., 2014 (174) | Rodent | Streptozotocin injection prior to mating | Increased PW |
| Increased nuclear factor κB activation | |||
| Increased expression of TNF-α and IL-6 | |||
| Beckett et al., 2014 (108) | Sheep | 100 mg IM of T propionate biweekly plus rosiglitazone 8 mg/kg orally daily, gestational days 30–90 | Addition of insulin sensitizer did not alter T-mediated placental advancement |
Future Directions in PCOS Placental Research
Gene environment interactions (epigenetic contributions)
Beyond efforts to understand how the human genome contributes to placental growth and development (180), there is emerging interest into the epigenetic mechanisms, including DNA methylation, histone modification, and miRNA and circular RNA expression, which may compromise placental function (Fig. 3) (181–187). For instance, a brief review of the preeclampsia literature highlights the potential role of epigenetic dysregulation, at the level of the placenta, in pregnancies with preeclampsia vs control pregnancies (188–193). Similarly, alterations in epigenetic programming have been identified in placental samples from women with GDM (194, 195).
Future directions in the field of PCOS placental research. Images used were sourced from open-source resources: www.pixabay.com, www.kisspng.com, www.wikipedia.org, and www.openclipart.org.
With regard to women with PCOS, recent studies have demonstrated that DNA methylation and miRNA expression may be altered in certain endocrine and metabolic tissues of women with PCOS, including adipose and ovaries (196–198). Meanwhile, altered epigenetic regulation of genes related to glucose homeostasis, lipid metabolism, and inflammation has been proposed based on a pilot study of umbilical cord blood in offspring of women with PCOS vs non-PCOS controls (199). However, many questions remain unanswered with regard to epigenetic patterns at the placental level, specific to women with PCOS, particularly because the placenta is a transitory organ. Because epigenetic alterations in both germ cells and placenta are thought to have an important role in developmental programming of offspring and intergenerational outcomes (200, 201), more research is needed to understand how epigenetic modifications in women with PCOS may be translated into adverse health implications for future generations.
Impact of fetal sex
A growing interest in the developmental programming of disease has expanded our awareness of the role of fetal sex on placental development. For instance, prior work has demonstrated that fetal sex impacts placental adaptations to external insults, including undernutrition or overnutrition and prenatal EDC exposure (202–206). In sheep models of PCOS, using exogenous T treatment during early developmental time points, fetal growth restriction is observed more frequently in female offspring, compared with males (108), highlighting the possibility of sex-specific placental alterations in pregnancies of women with PCOS. A recent review also highlights the potential role of the androgen receptor in mediating sex-specific fetal development (104). Additional research is needed to fully elucidate how the sex of the fetus may regulate or dysregulate placental function in pregnant women with PCOS (Fig. 3).
Capitalizing on animal models
As previously mentioned, animal models are of the utmost translational importance, as they offer the opportunity to identify both mechanisms and timing of onset of placental pathologies. Prior work in rodent and sheep models has already identified several key mechanisms that may underlie placental pathophysiology in PCOS (Tables 1 and 2). Animal models will continue to be invaluable for enhancing our understanding of, and developing intervention strategies for, PCOS-related placental compromise (Fig. 3).
Placental microbiome
The human microbiome and its role in health, disease, and developmental programming is another rapidly expanding area of research investigation (207). In pregnancy, distinct maternal microbiomes have been identified, each with a unique ability to influence pregnancy and neonatal outcomes: (i) placental, (ii) vaginal, and (iii) gastrointestinal (oral/gut) (208, 209). Characteristics of placental and vaginal microbial communities have been linked to pregnancy and birth outcomes, such as gestational diabetes and preterm birth (209–211). Additionally, bacteria are detected within newborn meconium, which may reflect exposure to the maternal gastrointestinal, placental, and/or vaginal microflora, further challenging the concept of the “sterile womb” (212). In turn, the intestinal flora of the newborn has been linked to a variety of long-term health outcomes, including immune or metabolic dysfunctions, inflammatory bowel disease, and childhood obesity (213), suggesting that in utero microbiota exposures might influence pathways for fetal developmental programming.
With regard to PCOS, research has suggested that androgen excess may contribute to altered gastrointestinal bacterial diversity in women with PCOS (214–216). Furthermore, prenatally androgenized rats have been shown to develop gut dysbiosis later in life (217). Research on the placental microbiome in women with PCOS vs non-PCOS controls is lacking. However, knowing that the diversity of the placental microbiome may influence birth outcomes such as BW, this could become an exciting avenue for future investigation in women with PCOS (Fig. 3).
“Omics” approaches: genomics, proteomics, and epigenomics
Using state-of-the-art “-omic” technologies, researchers are currently working to identify novel biomarkers of PCOS to aid in the early diagnosis and appropriate treatment of the disorder (218, 219). Identifying new, objective markers to predict the onset of maternal or fetal morbidity in pregnancy is also an exciting area of research at this time. In the context of this review, we advocate for specific research efforts to identify biomarkers, specific to each PCOS phenotype, that may predict adverse pregnancy outcomes (Fig. 3). Owing to the invasive nature of chorionic villous sampling, the identification of placental biomarkers is not practical. However, the application of -omic technologies to currently available screening tests (such as fetal cell-free DNA) to predict the onset of maternal or fetal morbidity may have relevance to women with PCOS.
Distinction of PCOS phenotypes
As previously mentioned, not all PCOS is created equal, and a woman’s particular phenotype may predispose her to greater or lesser pregnancy risk (220, 221). One must recognize the variability among PCOS phenotypes, in both the research and clinical settings, to provide evidence-based and appropriate counseling and surveillance to pregnant women with PCOS. Additionally, it is important to recognize how other individual factors, such as environment and ethnicity, may uniquely modulate a woman’s pregnancy risk (222).
Principal Points
Pregnancies of women with PCOS can be characterized by high maternal and fetal morbidity. The placenta is a key mediator of pregnancy and birth outcomes, highlighting the plausibility of placental dysfunction in pregnancies of women with PCOS. Because of the high prevalence of PCOS in reproductive-aged women, more human placental research is urgently needed to develop intervention strategies to improve outcomes for women with PCOS and their offspring.
Animal models of PCOS provide excellent translational value, especially in identifying the timing of onset of placental pathology, and they should be capitalized on for understanding placental alterations that may occur in states of gestational androgen excess or insulin resistance.
Most human placentas are discarded after delivery. Given the critical role of the placenta in supporting fetal growth and development, in addition to programming of child- and adult-onset disease, we encourage the development and expansion of human tissue biorepositories to facilitate placental research in women with PCOS.
Acknowledgments
We thank Dr. Muraly Puttabyatappa for assistance in generating Fig. 3.
Financial Support: This work was supported by National Institutes of Health Grant P01 HD44232 (to V.P.).
Additional Information
Disclosure Summary: The authors have nothing to disclose.
Data Availability: Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations:
- ART
assisted reproductive technology
- BW
birth weight
- ER
estrogen receptor
- GDM
gestational diabetes mellitus
- HIF-1α
hypoxia-inducible factor-1α
- IUGR
intrauterine growth restriction
- PCOS
polycystic ovary syndrome
- PW
placental weight
- T
testosterone
- VEGF
vascular endothelial growth factor

{kind=link}
{kind=link}
{kind=link}