





Abstract
Although the physiology of sodium, water, and arginine vasopressin (AVP), also known as antidiuretic hormone, has long been known, accumulating data suggest that this system operates as a more complex network than previously thought.
English-language basic science and clinical studies of AVP and osmolarity on the development of kidney and cardiovascular disease and overall outcomes.
Apart from osmoreceptors and hypovolemia, AVP secretion is modified by novel factors such as tongue acid-sensing taste receptor cells and brain median preoptic nucleus neurons. Moreover, pharyngeal, esophageal, and/or gastric sensors and gut microbiota modulate AVP secretion. Evidence is accumulating that increased osmolarity, AVP, copeptin, and dehydration are all associated with worse outcomes in chronic disease states such as chronic kidney disease (CKD), diabetes, and heart failure. On the basis of these pathophysiological relationships, an AVP receptor 2 blocker is now licensed for CKD related to polycystic kidney disease.
From a therapeutic perspective, fluid intake may be associated with increased AVP secretion if it is driven by loss of urine concentration capacity or with suppressed AVP if it is driven by voluntary fluid intake. In the current review, we summarize the literature on the relationship between elevated osmolarity, AVP, copeptin, and dehydration with renal and cardiovascular outcomes and underlying classical and novel pathophysiologic pathways. We also review recent unexpected and contrasting findings regarding AVP physiology in an attempt to explain and understand some of these relationships.
Plasma osmolarity is proportional to the number of molecules per liter of solution. Plasma osmolarity is set within certain limits by homeostatic mechanisms given its vital importance for the organism. Increased osmolarity activates various physiological systems in an effort to lower plasma osmolarity toward normal, baseline levels (1). The key systems activated by hyperosmolarity are thirst and arginine vasopressin (AVP), which is also known as antidiuretic hormone. The reason to specify AVP is because pig vasopressin substitutes lysine for arginine at position 8 and is termed lysine vasopressin (2).
AVP is considered a beneficial hormone, because its primary action is to increase water reabsorption in the collecting duct under conditions of hyperosmolarity or extracellular volume depletion, thus decreasing urinary water losses and restoring homeostasis (3). However, recent evidence suggests that there is a “tradeoff” in which elevated AVP levels can be detrimental. Indeed, increased plasma osmolarity and AVP levels and dehydration were associated with worse renal and cardiovascular outcomes (1). In this review, we summarize the literature on the relationship of elevated osmolarity and AVP with renal and cardiovascular outcomes and the underlying pathophysiology.
Overview of AVP Physiology
AVP is not generated as a final product but rather first as a prepro-protein named prepro-vasopressin that is transcribed from the AVP gene. This preprotein then needs to be proteolysed, which yields AVP as well as copeptin and neurophysin II (as seen in Fig. 1). Biochemically, AVP is cyclical, a nonapeptide with a critical disulfide bridge that determines biological activity. AVP is mainly created in large (magnocellular) supraoptic and paraventricular neurons in the hypothalamus and are released arterially in the posterior pituitary. AVP is escorted by a carrier protein that is appropriately named neurophysin. The trip courses along the supraoptic hypophyseal tract to reach the posterior pituitary gland axonal terminals of the magnocellular neurons. Release of AVP can be stimulated physically (nausea, vomiting, pain, stress) or chemically (hypoxemia, acidosis). Separately, catecholamines can stimulate central adrenergic receptors to modulate AVP release with a variety of effects on vasopressin release. Most importantly, and teleologically most logically related to dietary intake, osmolarity is the major stimulus for AVP. Regardless of the stimulus, AVP is functionally a neurotransmitter acting upon AVP-sensitive neurons downstream of neurons sensing the above-mentioned stimuli (Fig. 2).
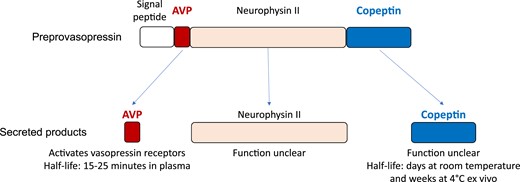
Preprovasopressin, AVP, and copeptin.
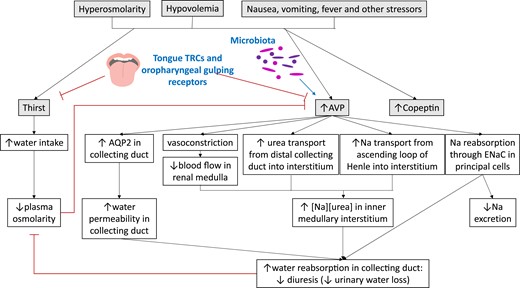
Regulation of thirst and AVP secretion and pathways engaged to restore plasma osmolarity. Novel regulators of thirst and AVP secretion are shown in bold blue. Copeptin is cosecreted with AVP, but its function remains unclear. TRC, taste receptor cell.
AVP release and osmolarity
Plasma osmolarity and AVP have a linear relationship, with a threshold for AVP release (4, 5). This threshold (6) determines when a reduced circulating volume and blood pressure modulate the osmoregulatory threshold. The threshold is more permissive in states of hypovolemia, and as hypovolemia progresses, nonosmotically regulated AVP release can persist despite significant hyponatremia (7).
Special vascular organ of lamina terminalis neurons are able to sense osmolarity of blood and cerebrospinal fluid by using aquaporin (AQP) 4 receptors. In hyperosmolar states, water flow through the aquaporin stretches the membrane and depolarizes the vascular organ of lamina terminalis neuron, sending a depolarization downstream that releases AVP. As water leaves the cell, TRPV (a family of transient receptor potential cation channels) cation channels sense the mechanical stretch and allow cation influx, which leads to stimulation of thirst and, again, AVP release (8).
In fact, AQP4 is abundant in the brain especially at the blood–brain barrier (which is logical, as it must sense osmolar changes in the bloodstream). AQP4 acts upon presynaptic glial cells to regulate water transport through AQP1 (which can be detected and is a surrogate marker of synaptic activation) (9) and helps control brain volume via its water content (10–12). AVP has a half-life of 5 to 25 minutes not regulated by osmolality. The molecule is degraded peripherally in the bloodstream (13).
AVP release and thirst
The physiology of AVP is closely linked to thirst. Serum osmolarity of 285 mOsm/L has been determined as the threshold for both thirst perception and AVP release (13). Drinking water reduces thirst and reduces osmolarity slightly, but it results in a disproportionately large drop in serum osmolarity that raises the possibility of a nonosmotic-dependent response (14).
Interestingly, recent evidence supports the existence of additional novel regulatory pathways of AVP secretion. For example, the “sour” receptors for taste in the tongue, the type 3 presynpatic acid-sensing taste receptor cells, express otopetrin 1, which mediates a downstream responses to water intake. Median preoptic nucleus neuron activity in the lamina terminalis is also proportional to the intensity of dehydration. It is possible that oropharyngeal gulp motions are sensed by the neurons and provide liquid-specific thirst modulation (15). Pharyngeal, esophageal, and/or gastric sensors could additionally convey organ-specific information of water ingestion by the sensory vagus nerve (16). Additionally, both dietary and endogenously produced fructose has recently been recognized as a nutrient that stimulates AVP synthesis and secretion as a consequence of its metabolism (17–20). Thus, AVP secretion is reduced before osmolality returns to normal limits: It decreases immediately after water comes into contact with the oral mucosa through afferent nerves signaling to the brain (21).
AVP release and gut microbiota
There are also data regarding the regulation of AVP secretion by gut microbiota. Indeed, antibiotic treatment of mice, which disrupts and depletes the gut microbiota, decreases hypothalamic AVP expression (22). The hypothalamus and gut microbiota may have a bidirectional modulation. When AVP is expressed, there are effects on stress response, inflammatory response, and behaviors that may alter gut microbiota. Heterozygous AVP knockout rats have different gut microbiota in between the homozygous mutant and wild-type rats, in a sex-specific fashion, possibly via antigenic mimicry of host neuropeptides to microbiotic receptors (23, 24). In vitro, AVP is stable in colonic environments without normal fecal microbiota (25), suggesting that microbiota may metabolize AVP (23). However, it is still unclear how or whether hypothalamus-secreted AVP reaches the gut lumen.
Overall, recent data suggest that thirst and AVP release are regulated by factors beyond osmoreceptors, including the gastrointestinal system.
AVP actions
Renal actions
AVP regulates renal water resorption (Fig. 2). The nephrons are bathed in a gradient of osmolarity and express aquaporins that allow concentration of the urine as needed, based on osmolarity-dependent AVP action. Whereas basolateral membrane AQP3 and AQP4 are constitutively expressed, hypertonicity regulates AQP2 expression on the luminal collecting duct, which is responsive to AVP concentrations. Specifically, AVP-derived cAMP activates protein kinase A to phosphorylate vesicles that have preprepared AQP, sending them to the apical membrane. Water is recaptured, causing concentration of the urine. When vasopressin is not present, the urine is not concentrated via this pathway (8).
Additional kidney effects of AVP include the decrease of medullary blood flow and stimulation of urea recapture to the medullary interstitium (26–28). Sodium is also concentrated here, allowing a hypertonic gradient through which the loop of Henle passes (29), allowing water countercurrent exchange for water reabsorption modulation (13). Epithelial sodium channels also reabsorb water in response to AVP (30).
Glucose metabolism actions
AVP increases blood glucose concentration, leading to speculation about its metabolic value (31). When AVP binds V1a receptors in the liver, this initiates glycogenolysis by glycogen phosphporylase (32). In the liver, AVP acts through calcium-dependent pathways, distinguishing it from glucagon (33), although rat studies have also suggested that AVP actually stimulates glucagon secretion (34) to reach its metabolic endpoint.
AVP also causes release of ACTH from the anterior pituitary gland, stimulates corticotropin-releasing factor release from the hypothalamus via V1bR activation, and also stimulates cortisol release directly (35, 36). Both the α- and β-cells in the islets of Langerhans contain V1b receptors, so both insulin and glucagon are at play depending on the glucose concentration in the extracellular space. For example, normoglycemic individuals become hyperglycemic with AVP infusion (37). In sum, AVP may be a risk factor for hyperglycemia/metabolic syndrome/diabetes spectrum metabolic derangements.
Separately, AVP decreases circulating ketones (31) and suppresses lipolysis using the AVP V1A receptor in a calcium-dependent fashion (38). The exact mechanism of AVP-induced lipid metabolism remains understudied (39); however, V1A knockout mice hypermetabolize fat, possibly due to insulin-mediated effects on hormone-sensitive lipase activity (40).
Teleologically, blood loss is an important cause of hypovolemia, and it is logical that AVP stimulates release of procoagulant factors from endothelial cells (41). Vasopressin corrals platelets for aggregation using its V1a receptor (42). Divalent cations (calcium and magnesium) regulate the thrombotic effects of AVP to a lesser and greater extent, respectively. Desmopressin (DDAVP) is another osmolarity-related molecule that regulates platelet function via P-selectin expression at the endothelial level (41).
Hypertensive actions
AVP increases blood pressure via vasoconstriction and antidiuretic effects, and it is therefore complementary to the renin-angiotensin-aldosterone system (RAAS). Syndromes such as neurogenic hypertension, angiotensin II–related hypertension, and salt-sensitive hypertension as well as obesity-induced hypertension have all been seen to have elevated vasopressin activity (43). AVP stimulates aldosterone release directly and via RAAS stimulation, increases sympathetic activity and baroreflex sensitivity, and increases renal collecting ductal epithelial sodium channel activity (44, 45). Urine sodium excretion decreases after urine flow drops below 1 L, implying a threshold for sodium-specific AVP actions (46). AVP regulates glomerular filtration rate (GFR) via the V1a receptor using RAAS and vasopressin 2 receptor–AQP2 to determine fluid homeostasis (47).
AVP and copeptin
Given the very short half-life of AVP, it is not practical to measure plasma AVP as a prognostic marker. Instead, copeptin is used for this purpose because it has a longer half-life (48). Copeptin is created at the same time as AVP, as described above (49, 50) (Fig. 1). Copeptin levels are also correlated with osmolarity. A recent study explored the impact of water intake on plasma copeptin in 82 healthy adults stratified by daily fluid intake volumes: 50% to 80%, 81% to 120%, or 121% to 200% of dietary reference values from the European Food Safety Authority. Following a baseline visit, those drinking less water were instructed to increase their water intake for 6 weeks to match those drinking more water. This was associated with a significant decrease of copeptin, urine osmolality, and urine specific gravity (51). Copeptin properties stimulated research into the significance of copeptin levels as a surrogate for AVP synthesis in various clinical conditions, including cardiovascular and kidney diseases (52). As discussed below, despite the physiological role of AVP, increased osmolarity and copeptin are associated with adverse outcomes (53).
Clinical Evidence
Preclinical and human observational studies have linked situations of higher AVP secretion to progression of chronic kidney disease (CKD) or adverse cardiovascular outcomes and, more recently, the vasopressin receptor 2 blocker tolvaptan was shown to slow the renal damage in the autosomal-dominant type of polycystic kidney disease (ADPKD). As summarized in Table 1, the implications of some of these different variables are context-dependent.
Plasma and Urine Osmolarity, Diuresis and Plasma AVP, and Copeptin in Diverse Pathophysiological Situations
| Plasma Osmolarity | Urine Osmolarity | Urine Volume | Plasma AVP | Plasma Copeptin | |
|---|---|---|---|---|---|
| Defective urine concentrating ability (e.g., CKD) | ↑ | ↔ | ↑ | ↑ | ↑ |
| ↓ Water intake | ↑ | ↑ | ↓ | ↑ | ↑ |
| ↑ Solute intake | ↑ | ↑ | ↓ | ↑ | ↑ |
| ↑ Fluid intake in response to thirst | ↑ | ↑ | ↓ | ↑ | ↑ |
| ↑ Spontaneous fluid intake | ↓ | ↓ | ↑ | ↓ | ↓ |
| Taste receptor cells (tongue type III presynaptic acid-sensing taste receptor cells) | ↓ (?) | ||||
| Gut microbiota | ↑ (?) |
| Plasma Osmolarity | Urine Osmolarity | Urine Volume | Plasma AVP | Plasma Copeptin | |
|---|---|---|---|---|---|
| Defective urine concentrating ability (e.g., CKD) | ↑ | ↔ | ↑ | ↑ | ↑ |
| ↓ Water intake | ↑ | ↑ | ↓ | ↑ | ↑ |
| ↑ Solute intake | ↑ | ↑ | ↓ | ↑ | ↑ |
| ↑ Fluid intake in response to thirst | ↑ | ↑ | ↓ | ↑ | ↑ |
| ↑ Spontaneous fluid intake | ↓ | ↓ | ↑ | ↓ | ↓ |
| Taste receptor cells (tongue type III presynaptic acid-sensing taste receptor cells) | ↓ (?) | ||||
| Gut microbiota | ↑ (?) |
Plasma and Urine Osmolarity, Diuresis and Plasma AVP, and Copeptin in Diverse Pathophysiological Situations
| Plasma Osmolarity | Urine Osmolarity | Urine Volume | Plasma AVP | Plasma Copeptin | |
|---|---|---|---|---|---|
| Defective urine concentrating ability (e.g., CKD) | ↑ | ↔ | ↑ | ↑ | ↑ |
| ↓ Water intake | ↑ | ↑ | ↓ | ↑ | ↑ |
| ↑ Solute intake | ↑ | ↑ | ↓ | ↑ | ↑ |
| ↑ Fluid intake in response to thirst | ↑ | ↑ | ↓ | ↑ | ↑ |
| ↑ Spontaneous fluid intake | ↓ | ↓ | ↑ | ↓ | ↓ |
| Taste receptor cells (tongue type III presynaptic acid-sensing taste receptor cells) | ↓ (?) | ||||
| Gut microbiota | ↑ (?) |
| Plasma Osmolarity | Urine Osmolarity | Urine Volume | Plasma AVP | Plasma Copeptin | |
|---|---|---|---|---|---|
| Defective urine concentrating ability (e.g., CKD) | ↑ | ↔ | ↑ | ↑ | ↑ |
| ↓ Water intake | ↑ | ↑ | ↓ | ↑ | ↑ |
| ↑ Solute intake | ↑ | ↑ | ↓ | ↑ | ↑ |
| ↑ Fluid intake in response to thirst | ↑ | ↑ | ↓ | ↑ | ↑ |
| ↑ Spontaneous fluid intake | ↓ | ↓ | ↑ | ↓ | ↓ |
| Taste receptor cells (tongue type III presynaptic acid-sensing taste receptor cells) | ↓ (?) | ||||
| Gut microbiota | ↑ (?) |
Increased osmolarity, dehydration, and cardiorenal outcome
In recent years, various studies have examined the relationship between variables such as serum and urine osmolarity, fluid or water intake and diuresis, and cardiorenal outcomes. Dehydration is traditionally accepted as a risk factor for conditions such as nephrolithiasis and elevated blood urea nitrogen. A recent comprehensive review investigated the relationship between hydration status and kidney diseases. The authors concluded that increasing water intake reduces AVP secretion, especially relevant in patients with ADPKD and in those at risk for CKD (54). Previous studies demonstrated that doubling water intake halved AVP levels in rats with CKD and slowed proteinuria and hypertension (55). However, low fluid intake (which may potentially result in increased plasma and urine osmolarity) could beget CKD and its progression (56–58). A relationship between serum osmolarity and development of CKD was observed in a retrospective 5-year cohort study of 12,041 Japanese subjects. Kuwabara et al. (53) demonstrated that serum osmolarity predicts CKD development with an increased risk of 24% for every 5 mOsm/L increase.
Urine osmolarity independently predicted dialysis initiation in a retrospective analysis of 273 patients with CKD, of whom 105 initiated dialysis after a median of 92 months, with a threshold time point of 72 months, after which maximal dialysis initiation was observed (59). Prevalence of dialysis initiation increased with osmolarity at 72 months.
Mesoamerican nephropathy or heat stress nephropathy has been proposed to depend on dehydration and increased osmolarity. This primarily occurs in male field laborers in humid conditions. There is high creatinine level, mild proteinuria, mild hypertension, and normal blood glucose and urinary sediment, arguing against glomerular injury. The predominant pathology is tubulointerstitial injury, and the major pathophysiologic mechanism is thought to be increased serum osmolarity (1, 58).
Regarding blood pressure, we recently investigated 10 healthy individuals not taking any medications with four separate soup challenges with increasing salt content. At each visit, plasma osmolarity, sodium and copeptin levels, and blood pressure were measured at baseline and hourly for 4 hours. Intake of 3 g of salt was associated with increased plasma osmolality (+6 mOsm/L) and sodium (2.5 mmol/L) and systolic blood pressure (10 mm Hg) at 2 hours. The concomitant intake of water prevented these changes. Thus, the acute increase in blood pressure following salt ingestion depends on the osmolarity change, not the absolute salt amount. This implies that concurrent intake of enough water to prevent the rise in serum osmolarity may protect from blood pressure elevation after salt intake. Clinically, increasing water intake may be an alternative to salt restriction (60). This issue is particularly important given the low adherence (10% to 20%) to low-salt diets (61). In health, the kidney excretes more sodium as the patient drinks more water, whereas conversely, AVP retains sodium (30). Despite these observations, studies are needed to explore whether concomitant water intake may have protective effects over salt-induced changes in blood pressure in the long term.
However, some contrary data exist. A population-based prospective study of ∼4000 patients ≥49 years of age was performed using a food questionnaire. After 13.1 years, estimated glomerular filtration rate (eGFR) was found to decrease 2.2 mL/min/1.73 m2 in the 1207 patients who had data available for this analysis. Importantly, this was not associated with fluid intake, raising the question of whether fluid intake is truly nephroprotective (62). However, the rate of eGFR loss with aging was slower than the expected rate of 1.0 mL/min per 1.73 m2/y, raising questions about the representativeness of the sample. Additionally, water was not included, which would be expected to have a higher vasopressin-lowering effect than electrolyte-containing food or beverages. Finally, the https://vwp-psapp01.dhe.duke.edu/RadPortal/ study assessed spontaneous fluid intake, which may be in response to thirst (and, thus, increased vasopressin levels) triggered by prior water losses and dehydration. Thus, a high fluid intake may not necessarily imply suppressed vasopressin secretion.
ADPKD has been a topic of research, given the preclinical and clinical evidence of beneficial effects of vasopressin signaling blockade that results in decreased cyst growth and other nephroprotective actions (63). In a retrospective analysis of 139 CKD patients with PKD and 442 without PKD during a follow-up period of 2.3 years, a higher mean 24-hour urine volume and lower urine osmolarity were associated with more rapid GFR decline. PKD increased this rate of decline mildly, but fluid intake did not slow renal decompensation (64). Given this, we should again differentiate between a high fluid intake secondary to prior fluid losses (e.g., secondary to partial nephrogenic diabetes insipidus associated with the urine concentration defect that may be found in CKD and, specially, in PKD), which is associated with a high vasopressin secretion, and preemptive fluid ingestion that also increases urinary losses and decreases urine osmolarity, but is associated with suppressed vasopressin secretion.
A very recent prospective study assessed the relationship between fasting urinary osmolality, CKD progression, and mortality in 2084 adults with CKD stages 1 to 4. At a median of 6 years, 225 patients died before end-stage renal disease (ESRD) occurred, and 380 ESRD events occurred. Having a lower baseline urine osmolality protected against ESRD but not death. Again, lower fasting urine osmolality may reflect a suboptimal urine concentration ability, a condition associated with higher vasopressin secretion.
Any study of nephroprotective water intake has many confounders (1). A randomized clinical trial of water supplementation vs control therapy in 631 patients with CKD (mean eGFR, 43 mL/min/1.73 m2) from different causes tested whether adding 1.0 to 1.5 L water daily to usual beverage intake for 12 months may significantly reduce the decline of GFR (65). Hydrated patients had a worse mean change in eGFR (−2.2 vs −1.9 mL/min/1.73 m2) despite having mean urine volumes change of +0.6 L/d (66). Thus, intervention patients did not comply with the 1.0 to 1.5 L additional water intake, although plasma copeptin decreased by 1.4 pmol/L (8%) from baseline. Unfortunately, drinking water without thirst is not easy, and noncompliance is expected to be a major obstacle for studies of similar design. Etiology-specific water intake trials are ongoing or have been recently completed, but results not yet reported, such a DRINK feasibility trial (NCT02933268) and the PREVENT-ADPKD (ANZCTR12614001216606) trials on ADPKD (67, 68).
AVP, copeptin, and cardiorenal outcomes
As observational studies of fluid intake or urine volume or osmolarity may be marred by the factors driving fluid intake or urine volume (e.g., voluntary fluid intake vs thirst-driven fluid intake or CKD-associated spontaneous polyuria), assessment of copeptin as a surrogate for AVP may provide better insight into pathophysiological mechanisms. In this regard, copeptin levels have been associated with adverse clinical outcomes in kidney and cardiovascular disease (Fig. 3).
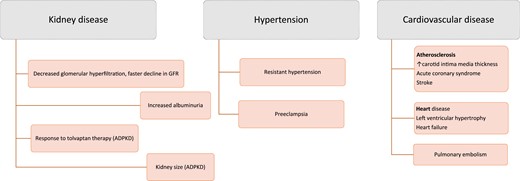
Clinical associations of high copeptin levels with negative outcomes in kidney and cardiovascular disease.
Experimentally, AVP accelerated CKD progression by causing glomerular hyperfiltration and albuminuria (55, 69). There are clinical data pointing in the same direction. High copeptin was associated with left ventricular hypertrophy and carotid intima thickening in a multivariate analysis among 86 patients with CKD not on renal replacement (70). A French study during 9 years of ∼1200 patients confirmed that copeptin elevations were associated with CKD development (71). Approximately 3200 patients in the Malmö Diet and Cancer–Cardiovascular Cohort demonstrated that copeptin was independently associated with eGFR decline (72). In three European general population cohorts (France, Sweden, and the Netherlands with median follow-up of 8.5, 16.5, and 11.3 years, respectively), the upper tertile of plasma copeptin levels was significantly and independently associated with higher risk for stage 3 CKD, kidney function decline in accordance with KDIGO criteria, rapid kidney function decline, and microalbuminuria (49%, P < 0.0001; 64%, P < 0.0001; 79%, P < 0.0001; and 24%, P < 0.0001, respectively.) This was one of the largest studies demonstrating copeptin as a risk factor for CKD (73).
Serum copeptin correlated positively with the albumin/creatinine ratio and negatively with eGFR in a study of 979 patients with diabetes (74). Copeptin correlated positively with GFR in 80 patients with type 1 diabetes (R = 0.86, P = 0.021) and in healthy controls (n = 61, R = 0.61, P = 0.034). Multivariate analysis showed that GFR was associated with copeptin in the patients with type 1 diabetes only (β = 0.23, P = 0.032) (75). Copeptin elevation was associated with creatinine concentration and ESRD in diabetes (76, 77).
Copeptin was associated with the size of the kidneys in a general population study (78). In patients with ADPKD, copeptin was associated with markers of disease severity, including GFR and albuminuria (79, 80). Additionally, the ratio of copeptin to creatinine is associated with kidney size in ADPKD (81). Randomized trial data demonstrate that patients with higher baseline copeptin levels have superior nephroprotection from tolvaptan. A greater copeptin rise at treatment week 3 predicted less eGFR decline at 3 years (82). Copeptin elevation was associated with microalbuminuria and CKD progression in renal transplant patients (83).
Copeptin is also associated with blood pressure. Patients with resistant hypertension had approximately twofold higher copeptin levels than did patients with adequate blood pressure control. This suggests that hypertensives have elevated homeostatic levels of copeptin regarding the osmolarity thresholds leading to AVP release (84). A recent study observed increased AVP levels in women with preeclampsia as early as at 6 weeks of gestation (85). Pregnant C57BL/6J mice receiving AVP infusion become hypertensive and have renal glomerular endotheliosis and pregnancy complications in the absence of placental hypoxia. This suggests that AVP is itself capable of inducing clinical phenotypes of preeclempsia independent from hypoxia-mediated mechanisms (86).
In patients with heart failure (HF), higher copeptin levels are associated with death (63.4% vs 33.0% at 1 year, P = 0.03) after adjusting for confounding factors (87). A study of 340 patients in Denmark who had left ventricular dysfunction found that copeptin levels predicted hospitalization or death at median follow-up of 55 months (hazard ratio, 1.4; 95% CI, 1.1 to 1.9; P < 0.019). However, copeptin did not predict mortality independently of N-terminal pro–B-type natriuretic peptide (88). These studies are in line with prior reports of a significant association of copeptin with HF outcomes (89–92) and of high AVP levels with HF severity (93–95).
Copeptin has also prognostic implications in atherosclerosis-related cardiovascular events, including acute coronary syndromes and stroke. Copeptin plus high-sensitivity cardiac troponin T compared favorably to an algorithm based only on high-sensitivity cardiac troponin T to rule out myocardial infarction (MI) (96). The prognostic value of serum copeptin for a combined endpoint of major cardiovascular events (MACEs) (death, repeat percutaneous coronary intervention, recurrent MI, or coronary artery bypass grafting) was studied retrospectively in 149 patients who successfully received percutaneous coronary intervention after an acute MI. Copeptin was positively associated with MACEs. In multiple logistic regression analyses, higher serum copeptin levels were independently associated with an increased risk of MACEs (OR, 1.6) (97). In other work, copeptin levels are associated with other poor clinical outcomes after acute MI, including HF (98–100).
Another study explored copeptin and renal/cardiovascular morbidity/mortality in two cohorts totaling 4505 patients with type 2 diabetes. As sex-specific tertiles of copeptin increased, the cumulative incidence of cardiovascular events also increased after adjusting for covariates. Pooled cohort analyses including patients with albuminuria found no interaction between copeptin levels and eGFR with cardiovascular events (101).
When HF induces renal failure, or vice versa, this is termed cardiorenal syndrome (102). AVP is implicated in this process. HF is associated with elevated AVP (released nonosmotically), causing fluid retention and peripheral vasoconstriction via V2 and V1 AVP receptors, respectively. HF-derived low cardiac output causes renal ischemia and activation of RAAS to stimulate further AVP, completing the cycle that ultimately leads to cardiorenal syndrome (103). Diuresis-dependent strategies in management of HF are challenged by the inappropriate elevation of AVP. In fact, AVP-directed diuresis-focused strategies have been studied as a result. For example, conivaptan allows dose-dependent diuresis in symptomatic patients with heart failure without changes in BP, heart rate, or electrolyte balance (104, 105). Tolvaptan increases urine volume significantly from a mean of ∼1 L/d to 1.5 L/d (P < 0.01) with stable creatinine levels, giving value in volume-overloaded patients with CKD (106).
Studies on copeptin and stroke have not been concordant. In a case-control study that included 119 healthy controls and 119 patients with stroke, copeptin was associated with ischemic stroke, and unexpectedly negatively associated with hemorrhagic stroke. Stroke prevalence increased with copeptin (5% ischemic, 8% hemorrhagic) (107) in one study whereas ischemic stroke did not increase with copeptin in another study (108), and in yet another study, the association occurred only in patients with diabetes (109).
Copeptin may have prognostic value in pulmonary embolism. Within the first 30 days, 843 normotensive patients with pulmonary embolism demonstrated a 7.6-fold increased risk of death (P = 0.001) with elevated copeptin levels (110).
All of the above association data suggest that copeptin is associated with outcomes in various pathologic states related to kidney and cardiovascular disease, and this list is increasing. However, the leap from associations and biomarker potential of copeptin to causality and, thus, therapeutic target potential of AVP requires interventional studies that so far have only been performed successfully, outside the context of hyponatremia, for ADPKD (111).
AVP Pathophysiology Beyond Osmolarity Control
Although it is a very “old” hormone, the full extent of AVP pathophysiological implications is only now being recognized. Besides its traditional role to regulate water homeostasis (Fig. 2) and systemic vasoconstrictive effects, there are several pathophysiological mechanisms that directly link AVP to kidney disease (Fig. 4).
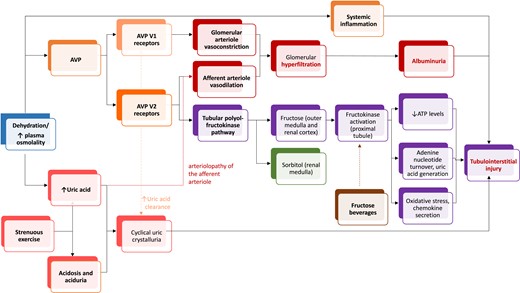
Postulated mechanisms for kidney injury triggered by dehydration. In the renal medulla, the presence of sorbitol and other osmolytes protects from hyperosmolarity (green).
In rats, AVP causes glomerular hypertension and kidney hypertrophy, and the reverse occurs when AVP is decreased with water intake (112), with similar findings in healthy humans (113). In this regard, AVP increases urinary albumin excretion in a dose-dependent manner (69). In rats with subtotal nephrectomy, tripling water intake reduced AVP secretion, osmolairy, proteinuria, renal hypertrophy, and mortality (55). These findings were reproduced in diabetic animal studies (114, 115). AVP infusion also accelerated CKD progression in subtotally nephrectomized AVP-deficient rats (116). In patients with ADPKD, the AVP V2 receptor blocker tolvaptan reversibly decreased hyperfiltration and also decreased albuminuria in randomized clinical trials (111, 117). Thus, AVP may be involved in glomerular hyperfiltration and may help increase urinary albumin excretion for humans and rats (114). However, further human studies are needed to demonstrate that these effects are also evident in disease conditions other than ADPKD. There are several potential mechanisms linking vasopressin to hyperfiltration, a driver of albuminuria and CKD progression. Vasopressin-induced hyperfiltration may be related to macula densa sodium concentration reducing feedback from the tubule to the glomerulus via complex AVP-dependent intrarenal handling of urea, whereby there is more urea in the loop of Henle (57, 118). Additionally, AVP V2 receptors (those blocked by tolvaptan) induce vasodilation in rabbit afferent arterioles (119).
There is preclinical evidence that AVP, likely via a V2 receptor–dependent pathway, may directly cause kidney injury (3). Thus, dehydration-induced hyperosmolarity and chronic stimulation of the V2 receptor may lead to kidney damage (54, 118). Experimental heat stress nephropathy in male mice induced by recurrent heat exposure with limited water availability was characterized by evidence of glomerular (albuminuria, mesangiolysis, matrix expansion) and tubulointerstitial injury (elevated urinary neutrophil gelatinase-associated protein, fibrosis, inflammation). Desmopressin, a V2 receptor agonist, twice daily for 5 weeks was associated with albuminuria and tubulointerstitial fibrosis in mice, which was worse in heat-stress mice.
Stress and desmopressin activate the polyol–fructokinase pathway in the renal cortex, likely via interstitial osmolarity, which is another potential driver of kidney injury (1, 3). Hyperosmolarity activates the enzyme aldose reductase, which is normally active in the hyperosmolar renal medulla where it metabolizes glucose into sorbitol (120), an intracellular osmolyte that protects medullary cells from hypertonicity-induced injury (121). Although sorbitol is the end product of the polyol pathway in the renal medulla, sorbitol dehydrogenase in the outer medulla and renal cortex sorbitol can further degrade sorbitol to fructose, a substrate for the proximal tubular enzyme fructokinase. Fructokinase activation may lead to a fall in ATP levels and increases in adenine nucleotide turnover, uric acid production, oxidative stress, and chemokine (monocyte chemoattractant protein-1) release, processes that can cause local renal injury (122, 123). Sodas may worsen the effect.
Uric acid is another potential link between AVP and kidney injury. Heat stress causing nephropathy has been theorized since the 1970s (124). Hyperuricemia is associated with effective circulating volume depletion in heat-stress nephropathy. Hard physical work in hot environments with inadequate hydration results in increased serum osmolarity and in increased serum uric acid levels derived from muscle turnover. Additionally, concentrated urines in this setting may exceed the uric acid insolubility threshold, a process further facilitated by urine acidity. Serum uric acid may cause glomerular hypertension, whereas urinary uric acid may cause injury in tubules through crystalline and noncrystalline effects. In a study of male sugarcane workers, the daily work shift was associated with increased serum creatinine and uric acid, urine concentration and acidification, and uric acid crystalluria, supporting the concept that repetitive subclinical kidney injury through recurrent exposure to heat, exercise, and dehydration may favor the development and progression of CKD (125).
In spontaneously hypertensive rats, chronic recurrent dehydration induced by restricting drinking to a 2-hour period each day for 4 weeks exacerbated hypertension and promoted renal damage, as assessed by decreased GFR, increased urinary neutrophil gelatinase-associated lipocalcin, and more severe renal fibrosis and an increased Th1/Th2 cell ratio. Thus, recurrent dehydration was associated with kidney damage even in the absence of heat exposure or strenuous physical exercise (126). Several previous studies had reported a protective effect of increasing water intake to reduce urine concentration, or administering AVP receptor antagonists, on CKD progression in rats (127, 128). The amount of water used for this purpose was roughly equivalent to 2 to 2.5 L/d in humans.
CKD progression via renal injury and inflammation is associated with recurrent dehydration and hyperosmolarity (129). Regarding osmolytes, in a cohort of 227 patients with CKD, urine metabolomics identified increased urine concentrations of the osmolytes betaine and myo-inositol to predict CKD progression (130). Interestingly, kidney transcriptomics identified decreased expression of osmolyte transporters Slc6a12 and Slc5a11 mRNA in injured renal tissue, consistent with a role of osmolyte disturbances in CKD progression (130).
Hyperosmolarity promotes CKD (131) via cytokines from monocytes (132, 133) as seen in a prospective study showing that dialysate sodium reduction reduced cytokine levels (134). There is also evidence that a high sodium intake is associated with a proinflammatory state (135). AVP secretion is also closely related with immune activity (136, 137). The relationship between AVP and the immune response may be bidirectional because nuclei that make AVP also respond to inflammation (138), and AVP receptors are found on immune cells (139). To make things more complicated, both AVP and the gut microbiota regulate and are potentially regulated by immune-related pathways (140, 141). Therefore, the interaction between gut microbiota and AVP expression may be in part mediated by the immune system.
Other potential deleterious mechanisms of increased osmolarity include activation of the sympathetic (142) and intracerebral angiotensin II activation (143, 144). Additionally, AVP causes mitochondrial dysfunction (145).
Paradoxical Impact of Sodium Intake on Water Balance
In classical physiology, ingestion of a salty meal leads to hypertonicity, which stimulates fluid intake with resultant expansion of extracellular fluid volume (146). However, this concept has been challenged by recent findings. In contrast to traditional belief, a long-term high-salt intake decreased fluid intake in humans. Although the mechanisms are unclear, this may be partly attributed to reduced urinary osmolyte-free water (147). In mice, increased salt intake stimulated catabolism, resulting in urea production that was partly mediated by increased plasma cortisol levels. Furthermore, mice with high dietary salt intake chose to eat more overall, possibly to counter catabolism. High salt intake increases renal medullary urea transporters, and a higher production of urea and reduced urea excretion results in medullary urea concentration with the ability to concentrate the urine. However, surprisingly, urine volume did not increase with salt intake. Thus, increased urine water excretion likely depends on additional water sources, such as increased endogenous water production. Taken together, decreased free water excretion in urine and elevated endogenous water production were associated with lower fluid intake without a rise in tonicity (148). Increasing salt intake for 7 days in healthy men was not associated with changes in fluid intake or urine volume (149). When dietary sodium is increased to 15 g/d, healthy male subjects demonstrated net negative fluid balance with stable extracellular fluid balance. This means that water was recruited from intracellular spaces, protected from insensible losses, or protected from metabolic losses (150). Potential explanations for these diverse findings include patient characteristics, endocrine and other physiologic perturbations, and the amount and chronological pattern of ingested salt (151). An improved understanding of these systems may help tackle the contribution of hyperosmolarity and AVP to disease by designing approaches that do not have to rely on difficult-to-implement dietary or drinking habit modifications.
Conclusion and Research Ahead
Recent evidence suggests that the regulation and impact of AVP and hyperosmolarity are more complex than previously thought and include regulation of AVP secretion by acid-sensing taste receptor cells, median preoptic nucleus neurons in the brain, pharyngeal, esophageal, and/or gastric sensors, and gut microbiota. Surprisingly, and contrary to prior literature, long-term salt intake decreased fluid consumption in humans. Copeptin and AVP levels are associated with chronic disease outcomes. There is also recent data on the direct relationship between sodium, hyperosmolarity, and AVP with the immune system. Several underlying mechanisms may be involved in the novel observations regarding salt intake, hyperosmolarity, and AVP secretion and actions, and it is important to clarify them to develop novel therapeutic approaches. Some controversy exists regarding fluid intake and progression of CKD. Whereas some studies suggested an association between higher fluid intake and slower CKD progression, others did not. The nature of fluid intake in response to hypertonicity driven by urinary concentrating defects, or as a proactive action leading to lower AVP levels, may underlie the controversial results. In this regard, although fluid intake may be of the same magnitude in both scenarios, the relationship to AVP levels and tonicity would be the opposite. Definitive proof may come from clinical trials of increased water intake or other approaches. However, novel imaginative ways to effortlessly increase electrolyte-free water intake are required. In any case, the recent demonstration of nephroprotection by tolvaptan in ADPKD and the surprising finding that this is associated with decreased hyperfiltration and albuminuria (a potential universal nephroprotective mechanism) on top of direct effects on cyst size (a disease-specific mechanism) emphasize the need to further understand the AVP system and its therapeutic implications for chronic cardiovascular and renal diseases.
Acknowledgments
Financial Support: M.K. gratefully acknowledges use of the services and facilities of the Koç University Research Center for Translational Medicine (KUTTAM), funded by the Presidency of Turkey, Presidency of Strategy and Budget. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Presidency of Strategy and Budget. A.O. was supported by Instituto de Salud Carlos III (ISCIII) and Fonds Européen de Développement Économique et Régional (FEDER) Grant PI16/02057, Sociedad Española de Nefrologia, Fundacion Renal Iñigo Alvarez de Toledo (FRIAT), and by ISCIII Red de Investigacion Renal (REDinREN) Grant RD016/009.
Disclosure Summary: The authors have nothing to disclose.
Data Availability: Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations:
- ADPKD
autosomal-dominant type of polycystic kidney disease
- AQP
aquaporin
- AVP
arginine vasopressin
- CKD
chronic kidney disease
- eGFR
estimated glomerular filtration rate
- ESRD
end-stage renal disease
- GFR
glomerular filtration rate
- HF
heart failure
- MACE
major cardiovascular event
- MI
myocardial infarction
- PKD
polycystic kidney disease
- RAAS
renin-angiotensin-aldosterone system

{kind=link}
{kind=link}
{kind=link}
{kind=link}