





Abstract
This mini-review offers an update on the rare autoimmune polyendocrinopathy (AP) syndrome with a synopsis of recent developments.
Systematic search for studies related to pathogenesis, immunogenetics, screening, diagnosis, clinical spectrum, and epidemiology of AP. AP (orphan code ORPHA 282196) is defined as the autoimmune-induced failure of at least two glands. AP is divided into the rare juvenile type I and the adult types II to IV. The prevalence is 1:100,000 and 1:20,000 for types I and types II to IV, respectively. Whereas type I (ORPHA 3453) is a monogenetic syndrome with an autosomal recessive transmission related to mutations in the autoimmune regulator (AIRE) gene, types II to IV are genetically complex multifactorial syndromes that are strongly associated with certain alleles of HLA genes within the major histocompatibility complex located on chromosome 6, as well as the cytotoxic T lymphocyte antigen 4 and the protein tyrosine phosphatase nonreceptor type 22 genes. Addison disease is the major endocrine component of type II (ORPHA 3143), whereas the coexistence of type 1 diabetes and autoimmune thyroid disease is characteristic for type III (ORPHA 227982). Genetic screening for the AIRE gene is useful in patients with suspected type I, whereas serological screening (i.e., diabetes/adrenal antibodies) is required in patients with monoglandular autoimmunity and suspected AP. If positive, functional endocrine testing of the antibody-positive patients as well as serological screening of their first-degree relatives is recommended.
Timely diagnosis, genetic counseling, and optimal long-term management of AP is best offered in specialized centers.
Autoimmune polyendocrinopathy (AP) is a challenging scientific topic about which there have been several novel genetic and immunological findings in the last few years. Indeed, novel mutations of the responsible autoimmune regulator (AIRE) gene, as well as a proteome analysis in patients with the juvenile AP type I, have been reported. In comparison, in patients with the adult AP type, a genome-wide association study, novel polymorphisms of several susceptibility genes [i.e., protein tyrosine phosphatase nonreceptor type 22 (PTPN22) and cytotoxic T lymphocyte antigen 4 (CTLA-4)], and a sex-dependent alteration of the HLA association have been described. Based on the relevant and achieved progress, this mini-review aims to offer a comprehensive description of the current pathophysiology, immunology, and clinical spectrum of this complex endocrine syndrome, as well as a synopsis of the recent novel and informative reports.
AP is a rare and registered orphan syndrome (orphan code ORPHA 282196) encompassing a wide spectrum of autoimmune diseases. The manifestation of one autoimmune disorder increases the risk of developing others. The time interval between the onsets of the different syndrome manifestations is highly variable; many years may pass between the diagnosis of the first and second endocrine disorder. This may be in part due to the fact that AP and other diseases resulting in autoimmune tissue destruction have a prolonged phase of cellular loss preceding overt autoimmune-induced glandular failure (1–4).
AP or polyglandular failure including thyroid dysfunction was first described in 1904 by Paul Ehrlich in Germany and in 1908 by Claude and Gougerot in France (5). In 1980, the pediatrician Neufeld suggested four types of AP, the very rare juvenile type I and the more frequent adult types II to IV (6). The complex juvenile type I AP, which includes candidiasis and ectodermal dystrophy, was first reported by Thorpe and Handley (7) in 1929 in a 4-year-old child with tetany and mycelial stomatitis. In 1956, Whitaker first described the emblematic triad: primary hypoparathyroidism, adrenal insufficiency, and moniliasis (8); in 1994, a more complete description of the syndrome and the relationship to mutations on chromosome 21 was published (9).
Nomenclature and Definition
Orphan or rare diseases are a group of disorders of very different etiologies, but the common denominator is that they have a very low prevalence and, for most of them, there is no treatment available. An orphan disease is defined as a condition with a prevalence of 1 to 500 in 1,000,000 (10, 11). AP is defined by the autoimmune-induced failure of at least two glands. In all types of AP, females are threefold more frequently affected than males. Onset is most commonly observed in the third and/or fourth decade, although occurrence in childhood can be observed (12). The AP types I to IV are defined as follows:
Type I AP (AP1), also known as AP, candidiasis, and ectodermal dystrophy (APECED), is a juvenile monogenetic syndrome with an autosomal recessive transmission (ORPHA 3453). The male/female sex ratio is 3:4 (13).
Types II to IV are adult forms, with a common manifestation between ages 40 and 60 years. Females are affected more often than males, with a sex ratio of 1:3. Chronic candidiasis is not present in adult AP.
Type II AP (AP2; ORPHA 3143) is characterized by the presence of autoimmune-induced adrenalitis [Addison disease (AD)] with at least one other autoimmune endocrine disorder that can be either autoimmune thyroid disease (AITD) or type 1 diabetes (T1D), or both (14, 15). About half of the AP2 patients initially present with T1D. Further endocrine and nonendocrine component diseases can be present and are shown in Table 1 (16).
Type III AP (AP3; ORPHA 227982) is defined by the presence of AITD and T1D without affecting the adrenal gland, therefore excluding AD. AITD most commonly peaks in the fourth decade in patients with Graves disease (GD) or the fifth to sixth decade in those with autoimmune thyroiditis (AT) (1, 17).
Type IV AP (ORPHA 227990) is a diagnosis of exclusion, with two or more organ-specific autoimmune endocrine syndromes that cannot be assigned to AP2 or AP3 (18).
A common subtype is the immune-dysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome. It is a rare disease linked to the dysfunction of the transcription factor forkhead box P3 (FOXP3), characterized by the dysfunction of regulatory T cells (Tregs) and the subsequent autoimmunity, primarily manifesting as T1D. The thyroid gland is commonly underactive (hypothyroidism) in individuals with this disorder, but it may become overactive (hyperthyroidism) (3). It is inherited primarily in males via an X-linked recessive manner.
Demographic, Clinical, Serological, and Genetic Data of the Various Types of AP
| AP (ORPHA: 282196) Subtypes | Juvenile I (ORPHA: 3453) | Adult | ||
|---|---|---|---|---|
| II (ORPHA:3143) | III (ORPHA:227982) | IV (ORPHA:227990) | ||
| Prevalence | 1:100,000 | 1:20,000 | ||
| Sex ratio (male/female) | 3:4 | 1:3 | ||
| Onset | Childhood | Adulthood | ||
| Endocrine disorders / manifestations | Hypoparathyroidism | AD | AITD | Hypogonadism |
| Hypogonadism | Hypoparathyroidism | T1D | Hypoparathyroidism | |
| Adrenal failure | AITD | Hypogonadism | Hypopituitarism | |
| T1D | AITD | |||
| T1D | ||||
| Non - endocrine disorders / manifestations | Candidiasis | Autoimmune gastritis | Autoimmune gastritis | Autoimmune gastritis |
| Moniliasis | Celiac disease | Pernicious anemia | Pernicious anemia | |
| Ectodermal dystrophy | Inflammatory bowel disease | Celiac disease | Celiac disease | |
| Enamel hypoplasia | Autoimmune pancreatitis | Inflammatory bowel disease | Inflammatory bowel disease | |
| Keratitis | Vitiligo | Autoimmune pancreatitis | Autoimmune pancreatitis | |
| Hyposplenism | Alopecia | Autoimmune hepatitis | Primary biliary cirrhosis | |
| Tubulointerstitial nephritis | Urticaria | Primary biliary cirrhosis | Vitiligo | |
| Psoriasis | Vitiligo | Alopecia | ||
| Rheumatoid arthritis | Alopecia | Urticaria | ||
| Urticaria | Pemphigus | |||
| Psoriasis | Psoriasis | |||
| Neurodermitis | Neurodermitis | |||
| Rheumatoid arthritis | Myasthenia gravis | |||
| Systemic lupus erythematosus | Sicca/Sjögren syndrome | |||
| Myasthenia gravis | ||||
| Sicca/Sjögren-syndrome | ||||
| Inheritance | Monogenic autosomal recessive transmission | Polygenic | ||
| HLA haplotypes / AIRE gene | AIRE gene, autosomal recessive and dominant X-linked, FOXP3 | DRB1*04:04-DQA1*03:01-DQB1*03:02 | DRB1*04:01-DQA1*03:01-DQB1*03:02 | HLA-DRB1*03:01-DQA1*05:01-DQB1*02:01 |
| Single nucleotide polymorphisms | PTPN22+1858 C/T (rs2476601), CTLA-4 C/T60 (rs3087243), Bsm I (rs1544410), Aps I (rs7975232), Taq I (rs731236), IL2-Ra CD25 (rs10795791), TNF-α -308 (rs1800629) | |||
| Auto-antigens | CaSR | 21-OH | TSH-R | 17-OH |
| 17-OH | CaSR | TPO | CaSR | |
| 21-OH | TSH-R | Tg | TSH-R | |
| TPO | GAD | TPO | ||
| Tg | Insulin | Tg | ||
| GAD | IA-2 | GAD | ||
| Insulin | Islet cell | Insulin | ||
| IA-2 | ZnT8 | IA-2 | ||
| Islet cell | Islet cell | |||
| ZnT8 | ZnT8 | |||
| AP (ORPHA: 282196) Subtypes | Juvenile I (ORPHA: 3453) | Adult | ||
|---|---|---|---|---|
| II (ORPHA:3143) | III (ORPHA:227982) | IV (ORPHA:227990) | ||
| Prevalence | 1:100,000 | 1:20,000 | ||
| Sex ratio (male/female) | 3:4 | 1:3 | ||
| Onset | Childhood | Adulthood | ||
| Endocrine disorders / manifestations | Hypoparathyroidism | AD | AITD | Hypogonadism |
| Hypogonadism | Hypoparathyroidism | T1D | Hypoparathyroidism | |
| Adrenal failure | AITD | Hypogonadism | Hypopituitarism | |
| T1D | AITD | |||
| T1D | ||||
| Non - endocrine disorders / manifestations | Candidiasis | Autoimmune gastritis | Autoimmune gastritis | Autoimmune gastritis |
| Moniliasis | Celiac disease | Pernicious anemia | Pernicious anemia | |
| Ectodermal dystrophy | Inflammatory bowel disease | Celiac disease | Celiac disease | |
| Enamel hypoplasia | Autoimmune pancreatitis | Inflammatory bowel disease | Inflammatory bowel disease | |
| Keratitis | Vitiligo | Autoimmune pancreatitis | Autoimmune pancreatitis | |
| Hyposplenism | Alopecia | Autoimmune hepatitis | Primary biliary cirrhosis | |
| Tubulointerstitial nephritis | Urticaria | Primary biliary cirrhosis | Vitiligo | |
| Psoriasis | Vitiligo | Alopecia | ||
| Rheumatoid arthritis | Alopecia | Urticaria | ||
| Urticaria | Pemphigus | |||
| Psoriasis | Psoriasis | |||
| Neurodermitis | Neurodermitis | |||
| Rheumatoid arthritis | Myasthenia gravis | |||
| Systemic lupus erythematosus | Sicca/Sjögren syndrome | |||
| Myasthenia gravis | ||||
| Sicca/Sjögren-syndrome | ||||
| Inheritance | Monogenic autosomal recessive transmission | Polygenic | ||
| HLA haplotypes / AIRE gene | AIRE gene, autosomal recessive and dominant X-linked, FOXP3 | DRB1*04:04-DQA1*03:01-DQB1*03:02 | DRB1*04:01-DQA1*03:01-DQB1*03:02 | HLA-DRB1*03:01-DQA1*05:01-DQB1*02:01 |
| Single nucleotide polymorphisms | PTPN22+1858 C/T (rs2476601), CTLA-4 C/T60 (rs3087243), Bsm I (rs1544410), Aps I (rs7975232), Taq I (rs731236), IL2-Ra CD25 (rs10795791), TNF-α -308 (rs1800629) | |||
| Auto-antigens | CaSR | 21-OH | TSH-R | 17-OH |
| 17-OH | CaSR | TPO | CaSR | |
| 21-OH | TSH-R | Tg | TSH-R | |
| TPO | GAD | TPO | ||
| Tg | Insulin | Tg | ||
| GAD | IA-2 | GAD | ||
| Insulin | Islet cell | Insulin | ||
| IA-2 | ZnT8 | IA-2 | ||
| Islet cell | Islet cell | |||
| ZnT8 | ZnT8 | |||
Abbreviations: 17-OH, 17-hydroxylase; IA-2, tyrosine-phosphatase; Tg, thyroglobulin; TSH-R, TSH receptor; ZnT8, zinc transporter 8.
Demographic, Clinical, Serological, and Genetic Data of the Various Types of AP
| AP (ORPHA: 282196) Subtypes | Juvenile I (ORPHA: 3453) | Adult | ||
|---|---|---|---|---|
| II (ORPHA:3143) | III (ORPHA:227982) | IV (ORPHA:227990) | ||
| Prevalence | 1:100,000 | 1:20,000 | ||
| Sex ratio (male/female) | 3:4 | 1:3 | ||
| Onset | Childhood | Adulthood | ||
| Endocrine disorders / manifestations | Hypoparathyroidism | AD | AITD | Hypogonadism |
| Hypogonadism | Hypoparathyroidism | T1D | Hypoparathyroidism | |
| Adrenal failure | AITD | Hypogonadism | Hypopituitarism | |
| T1D | AITD | |||
| T1D | ||||
| Non - endocrine disorders / manifestations | Candidiasis | Autoimmune gastritis | Autoimmune gastritis | Autoimmune gastritis |
| Moniliasis | Celiac disease | Pernicious anemia | Pernicious anemia | |
| Ectodermal dystrophy | Inflammatory bowel disease | Celiac disease | Celiac disease | |
| Enamel hypoplasia | Autoimmune pancreatitis | Inflammatory bowel disease | Inflammatory bowel disease | |
| Keratitis | Vitiligo | Autoimmune pancreatitis | Autoimmune pancreatitis | |
| Hyposplenism | Alopecia | Autoimmune hepatitis | Primary biliary cirrhosis | |
| Tubulointerstitial nephritis | Urticaria | Primary biliary cirrhosis | Vitiligo | |
| Psoriasis | Vitiligo | Alopecia | ||
| Rheumatoid arthritis | Alopecia | Urticaria | ||
| Urticaria | Pemphigus | |||
| Psoriasis | Psoriasis | |||
| Neurodermitis | Neurodermitis | |||
| Rheumatoid arthritis | Myasthenia gravis | |||
| Systemic lupus erythematosus | Sicca/Sjögren syndrome | |||
| Myasthenia gravis | ||||
| Sicca/Sjögren-syndrome | ||||
| Inheritance | Monogenic autosomal recessive transmission | Polygenic | ||
| HLA haplotypes / AIRE gene | AIRE gene, autosomal recessive and dominant X-linked, FOXP3 | DRB1*04:04-DQA1*03:01-DQB1*03:02 | DRB1*04:01-DQA1*03:01-DQB1*03:02 | HLA-DRB1*03:01-DQA1*05:01-DQB1*02:01 |
| Single nucleotide polymorphisms | PTPN22+1858 C/T (rs2476601), CTLA-4 C/T60 (rs3087243), Bsm I (rs1544410), Aps I (rs7975232), Taq I (rs731236), IL2-Ra CD25 (rs10795791), TNF-α -308 (rs1800629) | |||
| Auto-antigens | CaSR | 21-OH | TSH-R | 17-OH |
| 17-OH | CaSR | TPO | CaSR | |
| 21-OH | TSH-R | Tg | TSH-R | |
| TPO | GAD | TPO | ||
| Tg | Insulin | Tg | ||
| GAD | IA-2 | GAD | ||
| Insulin | Islet cell | Insulin | ||
| IA-2 | ZnT8 | IA-2 | ||
| Islet cell | Islet cell | |||
| ZnT8 | ZnT8 | |||
| AP (ORPHA: 282196) Subtypes | Juvenile I (ORPHA: 3453) | Adult | ||
|---|---|---|---|---|
| II (ORPHA:3143) | III (ORPHA:227982) | IV (ORPHA:227990) | ||
| Prevalence | 1:100,000 | 1:20,000 | ||
| Sex ratio (male/female) | 3:4 | 1:3 | ||
| Onset | Childhood | Adulthood | ||
| Endocrine disorders / manifestations | Hypoparathyroidism | AD | AITD | Hypogonadism |
| Hypogonadism | Hypoparathyroidism | T1D | Hypoparathyroidism | |
| Adrenal failure | AITD | Hypogonadism | Hypopituitarism | |
| T1D | AITD | |||
| T1D | ||||
| Non - endocrine disorders / manifestations | Candidiasis | Autoimmune gastritis | Autoimmune gastritis | Autoimmune gastritis |
| Moniliasis | Celiac disease | Pernicious anemia | Pernicious anemia | |
| Ectodermal dystrophy | Inflammatory bowel disease | Celiac disease | Celiac disease | |
| Enamel hypoplasia | Autoimmune pancreatitis | Inflammatory bowel disease | Inflammatory bowel disease | |
| Keratitis | Vitiligo | Autoimmune pancreatitis | Autoimmune pancreatitis | |
| Hyposplenism | Alopecia | Autoimmune hepatitis | Primary biliary cirrhosis | |
| Tubulointerstitial nephritis | Urticaria | Primary biliary cirrhosis | Vitiligo | |
| Psoriasis | Vitiligo | Alopecia | ||
| Rheumatoid arthritis | Alopecia | Urticaria | ||
| Urticaria | Pemphigus | |||
| Psoriasis | Psoriasis | |||
| Neurodermitis | Neurodermitis | |||
| Rheumatoid arthritis | Myasthenia gravis | |||
| Systemic lupus erythematosus | Sicca/Sjögren syndrome | |||
| Myasthenia gravis | ||||
| Sicca/Sjögren-syndrome | ||||
| Inheritance | Monogenic autosomal recessive transmission | Polygenic | ||
| HLA haplotypes / AIRE gene | AIRE gene, autosomal recessive and dominant X-linked, FOXP3 | DRB1*04:04-DQA1*03:01-DQB1*03:02 | DRB1*04:01-DQA1*03:01-DQB1*03:02 | HLA-DRB1*03:01-DQA1*05:01-DQB1*02:01 |
| Single nucleotide polymorphisms | PTPN22+1858 C/T (rs2476601), CTLA-4 C/T60 (rs3087243), Bsm I (rs1544410), Aps I (rs7975232), Taq I (rs731236), IL2-Ra CD25 (rs10795791), TNF-α -308 (rs1800629) | |||
| Auto-antigens | CaSR | 21-OH | TSH-R | 17-OH |
| 17-OH | CaSR | TPO | CaSR | |
| 21-OH | TSH-R | Tg | TSH-R | |
| TPO | GAD | TPO | ||
| Tg | Insulin | Tg | ||
| GAD | IA-2 | GAD | ||
| Insulin | Islet cell | Insulin | ||
| IA-2 | ZnT8 | IA-2 | ||
| Islet cell | Islet cell | |||
| ZnT8 | ZnT8 | |||
Abbreviations: 17-OH, 17-hydroxylase; IA-2, tyrosine-phosphatase; Tg, thyroglobulin; TSH-R, TSH receptor; ZnT8, zinc transporter 8.
Epidemiology
AP is a rare syndrome with an approximate prevalence of 1:100,000 for type I and 1:20,000 for types II to IV in the United States (19). Type I has been studied far more thoroughly than the adult types II to IV, and still little is known about the precise prevalence and incidence of AP in different countries and ethnic groups around the world. The highest prevalence of AP1 has been found in populations with a high degree of kindredship or descendants of small founder populations such as Iranian Jews (1:600 to 1:9000) and Finns (1:25,000) (19). Compared with the AP–candidiasis–ectodermal dystrophy syndrome, type II is the more common syndrome, with a prevalence of 1.4 to 2 per 100,000, without any apparent preference of ethnic groups (20). The incidence of types II to IV varies between 1.4 and 4.5 per 100,000 depending on the published source. However, owing to the heterogeneous expression pattern, it is assumed that there are higher numbers of unreported cases. The actual incidence is therefore estimated at 1:20,000 (12, 21). What particularly makes AP a rare syndrome is the combination of AITD with a prevalence of 27 to 448 per 100,000 subjects per year for AT and 21 to 120 per 100,000 subjects per year for GD (22), together with other infrequent autoimmune-mediated diseases, with prevalence between 0.0002% and 8.5%.
Pathogenesis and Immunogenetics
AP1 is related to the AIRE gene, located on the long arm of chromosome 21, involving 14 exons. The AIRE gene codes for the AIRE protein. The AIRE protein is a transcription factor, which interferes with immune regulation, and contributes to the negative selection of autoreactive thymocytes. Therefore, its impairment allows for the appearance and spread of autoreactive lymphocytes. AIRE also regulates reactions against microbial agents, especially against mycosis. AIRE deficiency contributes to an alteration in the intracellular communication between monocytes and T helper (Th) cells (13).
Autoaggression in AP types II to IV is considered to be multifactorial. The antigen-specific immune response is initiated by antigen-presenting cells. These ubiquitous, immature dendritic cells pick up antigen molecules in nonlymphoid organs. After fragmenting the antigens, they migrate to the secondary lymphoid organs presenting their HLA class I or II associated antigen fragments. By using different cytokines, antigen presentation stimulates the cellular immune response via cytotoxic T lymphocytes (Th1) and/or the humoral immune response via activation of antigen-specific Th cells (Th2) and stimulation of B lymphocytes. Activation of mononuclear phagocytes also occurs during the Th1 response because Th1 cytokines comprise proinflammatory mediators. T suppressor cells regulate the immune responses, as autoaggression occurs when immune tolerance is lost. These T suppressor cells regulate self-reactive T and B cells and may therefore have profound influence on the control of human autoimmune diseases (23).
The influence of genetic factors on multiple autoimmune disorders has been demonstrated by the significant clustering of AITD within families and twin studies for both AT and GD, but mostly through recent genome-wide association studies, which suggest a common genetic susceptibility (24, 25). The inheritance pattern seems to be autosomal dominant with incomplete penetrance in some patients. Several genetic loci may interact with environmental factors. However, the exact underlying pathogenic mechanisms are not yet completely characterized. Environmental factors such as high iodine intake, exposure to irradiation, and deficiency of vitamin D and selenium all seem to be of particular importance for the occurrence of autoimmune diseases in susceptible subjects. Also, chemical contaminants such as polychlorinated biphenyls, their metabolites, and polybrominated diethyl ethers that bind to thyroid transport proteins and disrupt the thyroid function by displacing thyroxine may be involved (26). Smoking plays a dual role: increasing the risk for GD-associated orbitopathy (27) while reducing the occurrence of AT (28).
The adult AP types are strongly associated with certain alleles of HLA genes within the major histocompatibility complex (MHC). Several HLA class I and II alleles have been identified to influence AP. The HLA gene complex is located on chromosome 6. It encodes the MHC proteins. HLA class I presents peptides from inside the cell, whereas HLA class II presents antigens from outside of the cell to T lymphocytes (29).
Class I genes encode for the HLA antigens A, B, and C, and class II genes encode for the α- and β-chains of the heterodimer HLA class II antigens DR, DP, or DQ. There are two possible mechanisms of how HLA class II could lend susceptibility to both T1D and AITD. The first mechanism relates to the HLA binding groove structure. The second mechanism is directly related to the peptide binding (29, 30). In AP, the haplotypes HLA-DRB1*03:01-DQA1*05:01-DQB1*02:01 and HLA-DRB1*04:01-DQA1*03:01-DQB1*03:02 are significantly overrepresented, whereas the HLA-DRB1*15:01-DQA1*01:02-DQB1*06:02 haplotype is underrepresented and therefore protective of AP. The DRB1*04-DQ8 haplotype differentiates between AP1 and AP2. The DRB1*04:04-DQA1*03:01-DQB1*03:02 haplotype is associated with AD and, therefore AP2, whereas the DRB1*04:01-DQA1*03:01-DQB1*03:02 haplotype is susceptible for AP3, including T1D. A shared common proinflammatory genetic background as well as a defect in immune regulation could explain the coexistence of different organ-specific and non–organ-specific autoimmune diseases in patients (29–32).
Single-nucleotide polymorphisms (SNPs) in various genes, that is, PTPN22, CTLA-4, vitamin D receptor (VDR), BsmI, ApaI, and TaqI, IL-2 receptor α (IL-2Ra), and TNF-α, were reported as susceptibility risk factors for AP (25, 31, 33–37). Other possible risk genes include FOXP3, the MHC class I chain–related gene A (MICA), and variable number of tandem repeats (VNTR).
The PTPN22 gene is located on chromosome 1p13 and encodes the lymphoid tyrosine phosphatase, the strongest inhibitor of T cell activation. It is expressed in mature and immature T and B lymphocytes. By binding to protein kinase, it inhibits the T lymphocyte antigen receptor signaling pathway and thereby limits the response to antigens. AITD, T1D, and AP3 were observed to be associated with the minor T allele of the +1858 (rs2476601) C/T substitutions. The association seems to be stronger for GD than for AT. The mutation causes a tryptophan-to-arginine substitution in the LYP protein at codon 620 (38, 39). This R620W amino acid substitution, located in a polyproline motif, PLPXR, within the Lyp phosphatase protein is thought to be involved in binding to SH3 domains during protein–protein interactions (40, 41).
The CTLA-4 gene on chromosome 2, location 2q33, is a major susceptibility gene for AP3. CTLA-4 is a negative regulator of the T cell activation. Two SNPs, CT60 (rs3087243) and AG49 (rs231775), are associated with a lower level of soluble CTLA-4 and therefore less regulation of the T cells, promoting the development of autoimmunity, that is, T1D and/or AITD. The A/G49 SNP results in a threonine-to-alanine substitution in the signal peptide of the CTLA-4 protein. This leads to less efficient glycosylation in the endoplasmic reticulum and reduced surface expression of the CTLA-4 protein, which affects CTLA-4 function or expression, resulting in increased T cell activation (38, 42, 43).
The VDR is expressed in immune cells and reduces the production of proinflammatory cytokine-activated T cells by directly inhibiting them. Vitamin D3 suppresses activated T cells, resulting in improved phagocytosis as well as the suppression of γ-interferon production, which may therefore reduce the occurrence of T1D in humans. Three SNPs, BsmI (rs1544410), ApaI (rs7975232), and TaqI (rs731236), have been associated with T1D (44–46).
The IL-2Ra gene is located on chromosome 10p15 and is a differentiation factor via the protein IL-2Ra chain (CD25; rs10795791). It actively suppresses autoreactive T cells in the periphery, regulating the function of natural killer cells, B cells, and especially Tregs. Therefore, polymorphisms in the CD25 gene region could influence the development of autoimmune diseases. The CD25 SNP (rs10795791) is associated with both T1D and AITD, and especially GD (47).
The TNF-α gene is located on chromosome 6p21, within the HLA class III region of the MHC, specifically between HLA-B loci of class I and HLA-D loci of class II. Both the C/C genotype and the A allele of the G/A genotype of the −308 (rs1800629) SNP in the promoter region of the gene are associated with increased transcription and production of the TNF-α protein and confer susceptibility to both AITD and T1D. The endogenous production of TNF-α is influenced by TNF-α promoter polymorphisms, thereby affecting mRNA and protein expression levels. Higher levels of TNF-α transcription may facilitate the inflammatory response in autoimmunity (48–50).
The FOXP3 gene is located on the p arm of the X chromosome (Xp11.23), containing 11 coding exons and belonging to the forkhead/winged-helix family of transcriptional regulators. The FOXP3 transcription factor occupies the promoters for genes involved in Treg function; it therefore controls Treg differentiation and is considered the master regulator of Treg development and function. Mutations of the FOXP3 regulatory pathway affect thymocytes developing within the thymus that during thymopoiesis are transformed into mature Tregs, which can lead to autoimmune glandular diseases (33, 51, 52).
MICA is an associated locus within the MHC region, encoding a highly polymorphic cell surface glycoprotein. It is located on the p arm of chromosome 6 (6p21.33), and the protein is expressed in two isoforms formed by alternative splicing: MICA1 and MICA2, which is lacking exon 3. The MICA protein is expressed in epithelial and intestinal cells and is a ligand for receptors on the surface of natural killer cells. MICA acts as a possibly stress-induced antigen that is broadly recognized by intestinal epithelial T cells, binding to CD8 T cells carrying the integral membrane protein receptor natural killer group 2, member D (NKG2D) as well as natural killer cells. When engaged, the NKG2D–MICA complex results in the activation of T cell responses and natural killer cells against epithelial stressor cells expressing MICA on their surface. Therefore, it is also connected to organ-specific autoimmune diseases such as T1D (33, 51, 53, 54).
A penta-allelic 86-bp tandem repeat of a VNTR occurs in intron 2, of which allele 2 (IL1RN*2) is associated with autoimmune conditions and of a 600-bp tandem repeat polymorphism, ∼50 bp of which are located in the insulin gene, leading to susceptibility to T1D (33, 51). Table 2 offers a synopsis of the susceptibility genes for AP (31, 38, 39, 44, 51, 55–57).
Susceptibility Genes for AP
| Gene | Function | Chromosome Location | Mutations/Polymorphisms | Mutation/Polymorphism Phenotype |
|---|---|---|---|---|
| AIRE | • Contains 14 exons encoding a 545–amino acid protein | 21q22.3 | p.R257 in SAND | • Juvenile AP1 |
| • Plays key role in negative selection of T effector cells in thymus by promoting transcription of tissue-specific proteins | p.P252L in SAND | |||
| • Instrumental in generating subset of Tregs | p.G228W in SAND | |||
| p.G305S in PHD1 | ||||
| c.463G>A | ||||
| c.653-1G>A | ||||
| PTPN22 | • Encodes lymphoid tyrosine phosphatase, a strong inhibitor of T cell activation, expressed in B and T lymphocytes | 1p13 | +1858 C/T (rs2476601) | • Polymorphisms lead to increased T cell activation |
| • Inhibits T lymphocyte antigen receptor signaling pathway | • Increased susceptibility for AP3, AITD, especially Hashimoto thyroiditis, T1D | |||
| CTLA-4 | • Encodes a receptor that is expressed on T cells and is a negative regulator of T cell activation | 2q33 | C/T60 (rs3087243) A/G49 (rs231775) | • The threonine-to-alanine substitution in the signal peptide of the CTLA-4 protein leads to less efficient glycosylation in endoplasmic reticulum and reduced surface expression of CTLA-4 protein, which affects CTLA-4 function or expression |
| • Increased susceptibility for AITD, T1D, and AP3 | ||||
| VDR | • Expressed on immune cells | 12q13.11 | BsmI (rs1544410) | • Polymorphisms lead to increased T cell activation |
| • Directly inhibits activated T cells | ApsI (rs7975232) | • Increased susceptibility for T1D | ||
| • Reduces production of proinflammatory cytokines (IFNγ) | TaqI (rs731236) | |||
| IL2-Ra | • As differentiation factor and via CD25, actively suppresses autoreactive T cells and regulates function of natural killer cells, B cells, and especially Tregs | 10p15 | CD25 (rs10795791) | • Polymorphisms in the CD25 gene region might affect function of Tregs |
| • Increased susceptibility for T1D and AITD, especially GD | ||||
| TNF-α | • Located on chromosome 6p21, within HLA class III region of MHC. | 6p21, within HLA class III region of MHC | −308 (rs1800629) | • Polymorphisms in promoter region of the gene associated with increased transcription and production of TNF-α protein |
| • Higher levels of TNF-α transcription facilitate inflammatory response in autoimmunity | ||||
| • Increased susceptibility for AITD and T1D | ||||
| FOXP3 | • Contains 11 coding exons, belongs to the forkhead/winged-helix family of transcriptional regulators. | p arm of X chromosome (Xp11.23) | rs3761549 (−2383C/T) | • Polymorphisms might affect function of Tregs |
| • FOXP3 transcription factor occupies the promoters for genes involved in Treg function | • Increased susceptibility for autoimmune glandular diseases | |||
| • Controls Treg differentiation and is considered the master regulator of Treg development and function | ||||
| MICA | • Encodes highly polymorphic cell surface glycoprotein | p arm of chromosome 6 (6p21.33) | MICA*A5 variant | • Polymorphisms increase susceptibility for organ-specific autoimmune diseases, that is, T1D |
| • Protein expressed in two isoforms formed by alternative splicing: MICA1 and MICA2 | ||||
| • MICA protein is a ligand for receptors on natural killer cells. | ||||
| • Stress-induced antigen broadly recognized by intestinal epithelial T cells | ||||
| INS-VNTR | • Encompasses 1430 bp | 11p15 | Short class I (26–63 repeats) | • Polymorphisms might affect translation of preproinsulin and production of mature insulin |
| • Comprises three exons and two introns interspersed with several polymorphisms in linkage disequilibrium | Intermediate class II (64–139 repeats) | • Increased susceptibility for T1D | ||
| • Translation of preproinsulin, precursor of mature insulin. | Larger class III (140–210 repeats) | |||
| • VNTR region of 14- to 15-bp consensus sequence upstream of insulin gene in insulin promoter |
| Gene | Function | Chromosome Location | Mutations/Polymorphisms | Mutation/Polymorphism Phenotype |
|---|---|---|---|---|
| AIRE | • Contains 14 exons encoding a 545–amino acid protein | 21q22.3 | p.R257 in SAND | • Juvenile AP1 |
| • Plays key role in negative selection of T effector cells in thymus by promoting transcription of tissue-specific proteins | p.P252L in SAND | |||
| • Instrumental in generating subset of Tregs | p.G228W in SAND | |||
| p.G305S in PHD1 | ||||
| c.463G>A | ||||
| c.653-1G>A | ||||
| PTPN22 | • Encodes lymphoid tyrosine phosphatase, a strong inhibitor of T cell activation, expressed in B and T lymphocytes | 1p13 | +1858 C/T (rs2476601) | • Polymorphisms lead to increased T cell activation |
| • Inhibits T lymphocyte antigen receptor signaling pathway | • Increased susceptibility for AP3, AITD, especially Hashimoto thyroiditis, T1D | |||
| CTLA-4 | • Encodes a receptor that is expressed on T cells and is a negative regulator of T cell activation | 2q33 | C/T60 (rs3087243) A/G49 (rs231775) | • The threonine-to-alanine substitution in the signal peptide of the CTLA-4 protein leads to less efficient glycosylation in endoplasmic reticulum and reduced surface expression of CTLA-4 protein, which affects CTLA-4 function or expression |
| • Increased susceptibility for AITD, T1D, and AP3 | ||||
| VDR | • Expressed on immune cells | 12q13.11 | BsmI (rs1544410) | • Polymorphisms lead to increased T cell activation |
| • Directly inhibits activated T cells | ApsI (rs7975232) | • Increased susceptibility for T1D | ||
| • Reduces production of proinflammatory cytokines (IFNγ) | TaqI (rs731236) | |||
| IL2-Ra | • As differentiation factor and via CD25, actively suppresses autoreactive T cells and regulates function of natural killer cells, B cells, and especially Tregs | 10p15 | CD25 (rs10795791) | • Polymorphisms in the CD25 gene region might affect function of Tregs |
| • Increased susceptibility for T1D and AITD, especially GD | ||||
| TNF-α | • Located on chromosome 6p21, within HLA class III region of MHC. | 6p21, within HLA class III region of MHC | −308 (rs1800629) | • Polymorphisms in promoter region of the gene associated with increased transcription and production of TNF-α protein |
| • Higher levels of TNF-α transcription facilitate inflammatory response in autoimmunity | ||||
| • Increased susceptibility for AITD and T1D | ||||
| FOXP3 | • Contains 11 coding exons, belongs to the forkhead/winged-helix family of transcriptional regulators. | p arm of X chromosome (Xp11.23) | rs3761549 (−2383C/T) | • Polymorphisms might affect function of Tregs |
| • FOXP3 transcription factor occupies the promoters for genes involved in Treg function | • Increased susceptibility for autoimmune glandular diseases | |||
| • Controls Treg differentiation and is considered the master regulator of Treg development and function | ||||
| MICA | • Encodes highly polymorphic cell surface glycoprotein | p arm of chromosome 6 (6p21.33) | MICA*A5 variant | • Polymorphisms increase susceptibility for organ-specific autoimmune diseases, that is, T1D |
| • Protein expressed in two isoforms formed by alternative splicing: MICA1 and MICA2 | ||||
| • MICA protein is a ligand for receptors on natural killer cells. | ||||
| • Stress-induced antigen broadly recognized by intestinal epithelial T cells | ||||
| INS-VNTR | • Encompasses 1430 bp | 11p15 | Short class I (26–63 repeats) | • Polymorphisms might affect translation of preproinsulin and production of mature insulin |
| • Comprises three exons and two introns interspersed with several polymorphisms in linkage disequilibrium | Intermediate class II (64–139 repeats) | • Increased susceptibility for T1D | ||
| • Translation of preproinsulin, precursor of mature insulin. | Larger class III (140–210 repeats) | |||
| • VNTR region of 14- to 15-bp consensus sequence upstream of insulin gene in insulin promoter |
Susceptibility Genes for AP
| Gene | Function | Chromosome Location | Mutations/Polymorphisms | Mutation/Polymorphism Phenotype |
|---|---|---|---|---|
| AIRE | • Contains 14 exons encoding a 545–amino acid protein | 21q22.3 | p.R257 in SAND | • Juvenile AP1 |
| • Plays key role in negative selection of T effector cells in thymus by promoting transcription of tissue-specific proteins | p.P252L in SAND | |||
| • Instrumental in generating subset of Tregs | p.G228W in SAND | |||
| p.G305S in PHD1 | ||||
| c.463G>A | ||||
| c.653-1G>A | ||||
| PTPN22 | • Encodes lymphoid tyrosine phosphatase, a strong inhibitor of T cell activation, expressed in B and T lymphocytes | 1p13 | +1858 C/T (rs2476601) | • Polymorphisms lead to increased T cell activation |
| • Inhibits T lymphocyte antigen receptor signaling pathway | • Increased susceptibility for AP3, AITD, especially Hashimoto thyroiditis, T1D | |||
| CTLA-4 | • Encodes a receptor that is expressed on T cells and is a negative regulator of T cell activation | 2q33 | C/T60 (rs3087243) A/G49 (rs231775) | • The threonine-to-alanine substitution in the signal peptide of the CTLA-4 protein leads to less efficient glycosylation in endoplasmic reticulum and reduced surface expression of CTLA-4 protein, which affects CTLA-4 function or expression |
| • Increased susceptibility for AITD, T1D, and AP3 | ||||
| VDR | • Expressed on immune cells | 12q13.11 | BsmI (rs1544410) | • Polymorphisms lead to increased T cell activation |
| • Directly inhibits activated T cells | ApsI (rs7975232) | • Increased susceptibility for T1D | ||
| • Reduces production of proinflammatory cytokines (IFNγ) | TaqI (rs731236) | |||
| IL2-Ra | • As differentiation factor and via CD25, actively suppresses autoreactive T cells and regulates function of natural killer cells, B cells, and especially Tregs | 10p15 | CD25 (rs10795791) | • Polymorphisms in the CD25 gene region might affect function of Tregs |
| • Increased susceptibility for T1D and AITD, especially GD | ||||
| TNF-α | • Located on chromosome 6p21, within HLA class III region of MHC. | 6p21, within HLA class III region of MHC | −308 (rs1800629) | • Polymorphisms in promoter region of the gene associated with increased transcription and production of TNF-α protein |
| • Higher levels of TNF-α transcription facilitate inflammatory response in autoimmunity | ||||
| • Increased susceptibility for AITD and T1D | ||||
| FOXP3 | • Contains 11 coding exons, belongs to the forkhead/winged-helix family of transcriptional regulators. | p arm of X chromosome (Xp11.23) | rs3761549 (−2383C/T) | • Polymorphisms might affect function of Tregs |
| • FOXP3 transcription factor occupies the promoters for genes involved in Treg function | • Increased susceptibility for autoimmune glandular diseases | |||
| • Controls Treg differentiation and is considered the master regulator of Treg development and function | ||||
| MICA | • Encodes highly polymorphic cell surface glycoprotein | p arm of chromosome 6 (6p21.33) | MICA*A5 variant | • Polymorphisms increase susceptibility for organ-specific autoimmune diseases, that is, T1D |
| • Protein expressed in two isoforms formed by alternative splicing: MICA1 and MICA2 | ||||
| • MICA protein is a ligand for receptors on natural killer cells. | ||||
| • Stress-induced antigen broadly recognized by intestinal epithelial T cells | ||||
| INS-VNTR | • Encompasses 1430 bp | 11p15 | Short class I (26–63 repeats) | • Polymorphisms might affect translation of preproinsulin and production of mature insulin |
| • Comprises three exons and two introns interspersed with several polymorphisms in linkage disequilibrium | Intermediate class II (64–139 repeats) | • Increased susceptibility for T1D | ||
| • Translation of preproinsulin, precursor of mature insulin. | Larger class III (140–210 repeats) | |||
| • VNTR region of 14- to 15-bp consensus sequence upstream of insulin gene in insulin promoter |
| Gene | Function | Chromosome Location | Mutations/Polymorphisms | Mutation/Polymorphism Phenotype |
|---|---|---|---|---|
| AIRE | • Contains 14 exons encoding a 545–amino acid protein | 21q22.3 | p.R257 in SAND | • Juvenile AP1 |
| • Plays key role in negative selection of T effector cells in thymus by promoting transcription of tissue-specific proteins | p.P252L in SAND | |||
| • Instrumental in generating subset of Tregs | p.G228W in SAND | |||
| p.G305S in PHD1 | ||||
| c.463G>A | ||||
| c.653-1G>A | ||||
| PTPN22 | • Encodes lymphoid tyrosine phosphatase, a strong inhibitor of T cell activation, expressed in B and T lymphocytes | 1p13 | +1858 C/T (rs2476601) | • Polymorphisms lead to increased T cell activation |
| • Inhibits T lymphocyte antigen receptor signaling pathway | • Increased susceptibility for AP3, AITD, especially Hashimoto thyroiditis, T1D | |||
| CTLA-4 | • Encodes a receptor that is expressed on T cells and is a negative regulator of T cell activation | 2q33 | C/T60 (rs3087243) A/G49 (rs231775) | • The threonine-to-alanine substitution in the signal peptide of the CTLA-4 protein leads to less efficient glycosylation in endoplasmic reticulum and reduced surface expression of CTLA-4 protein, which affects CTLA-4 function or expression |
| • Increased susceptibility for AITD, T1D, and AP3 | ||||
| VDR | • Expressed on immune cells | 12q13.11 | BsmI (rs1544410) | • Polymorphisms lead to increased T cell activation |
| • Directly inhibits activated T cells | ApsI (rs7975232) | • Increased susceptibility for T1D | ||
| • Reduces production of proinflammatory cytokines (IFNγ) | TaqI (rs731236) | |||
| IL2-Ra | • As differentiation factor and via CD25, actively suppresses autoreactive T cells and regulates function of natural killer cells, B cells, and especially Tregs | 10p15 | CD25 (rs10795791) | • Polymorphisms in the CD25 gene region might affect function of Tregs |
| • Increased susceptibility for T1D and AITD, especially GD | ||||
| TNF-α | • Located on chromosome 6p21, within HLA class III region of MHC. | 6p21, within HLA class III region of MHC | −308 (rs1800629) | • Polymorphisms in promoter region of the gene associated with increased transcription and production of TNF-α protein |
| • Higher levels of TNF-α transcription facilitate inflammatory response in autoimmunity | ||||
| • Increased susceptibility for AITD and T1D | ||||
| FOXP3 | • Contains 11 coding exons, belongs to the forkhead/winged-helix family of transcriptional regulators. | p arm of X chromosome (Xp11.23) | rs3761549 (−2383C/T) | • Polymorphisms might affect function of Tregs |
| • FOXP3 transcription factor occupies the promoters for genes involved in Treg function | • Increased susceptibility for autoimmune glandular diseases | |||
| • Controls Treg differentiation and is considered the master regulator of Treg development and function | ||||
| MICA | • Encodes highly polymorphic cell surface glycoprotein | p arm of chromosome 6 (6p21.33) | MICA*A5 variant | • Polymorphisms increase susceptibility for organ-specific autoimmune diseases, that is, T1D |
| • Protein expressed in two isoforms formed by alternative splicing: MICA1 and MICA2 | ||||
| • MICA protein is a ligand for receptors on natural killer cells. | ||||
| • Stress-induced antigen broadly recognized by intestinal epithelial T cells | ||||
| INS-VNTR | • Encompasses 1430 bp | 11p15 | Short class I (26–63 repeats) | • Polymorphisms might affect translation of preproinsulin and production of mature insulin |
| • Comprises three exons and two introns interspersed with several polymorphisms in linkage disequilibrium | Intermediate class II (64–139 repeats) | • Increased susceptibility for T1D | ||
| • Translation of preproinsulin, precursor of mature insulin. | Larger class III (140–210 repeats) | |||
| • VNTR region of 14- to 15-bp consensus sequence upstream of insulin gene in insulin promoter |
Clinical Spectrum
Endocrine manifestations
Endocrine autoimmune diseases comprise AD, T1D, AT, GD, primary hypogonadism, primary hypoparathyroidism, and hypopituitarism (Fig. 1). AD is an autoimmune disorder of the adrenal gland, characterized by primary adrenal failure and hypocortisolism (58). T1D is characterized by an autoimmune reaction against the β cells of the pancreas, resulting in the loss of the ability to produce insulin and an associated insulin deficiency. In autoimmune AT, the thyroid gland is gradually destroyed by cytotoxic T cells, natural killer cells, and/or antibody (Ab)-dependent cellular cytotoxicity. Symptoms may include hypothyroidism, weight gain, and depression. Autoimmune-induced GD results in hyperthyroidism with or without goiter (59). GD is caused by long-term thyrocyte stimulation by functional thyroid-stimulating immunoglobulins (27, 60–67). Primary hypogonadism is defined as gonadal failure to respond to gonadotropin stimulation and sex hormone biosynthesis (68). Primary hypoparathyroidism is characterized by low levels of PTH, hypocalcemia, and hyperphosphatemia (69). Hypopituitarism is due to a failure of function of the pituitary gland or hypothalamus (70). Silent autoantibodies are prevalent long before the disorder is diagnosed. AP has been based on the presence of organ-specific Abs in the serum, lymphocyte infiltration in the affected gland, cellular immune defects, and an association with the HLA genes or immune response genes. Autoantibodies to the various endocrine and nonendocrine tissues not only offer a diagnostic clue to the autoimmune nature of diseases, but they also can be used to identify asymptomatic individuals who are at risk for developing other disease manifestations of the syndrome (Table 1).
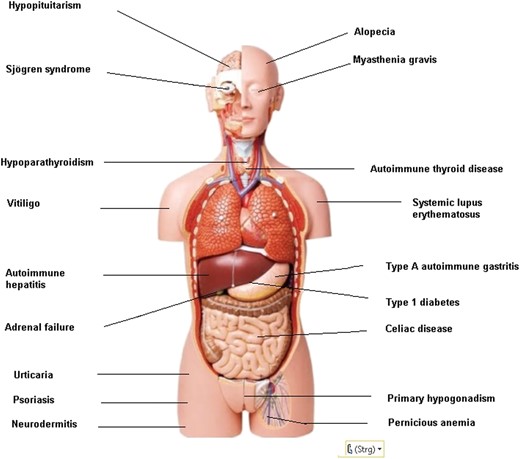
Autoimmune endocrine and nonendocrine disorders in AP.
Nonendocrine diseases
Nonendocrine organ system diseases associated with AP include autoimmune-induced skin diseases, that is, vitiligo or loss of melanocytes, alopecia (characterized by nonscarring hair loss), chronic urticaria, the formation of itchy wheals, pemphigus (characterized by autoimmune blistering), psoriasis (an autoimmune inflammatory skin disease), dermatomyositis, and neurodermitis, with a prevalence ranging from 0.1% to 8.5% depending on region and age (71). Other nonendocrine disorders include autoimmune gastritis, which causes malabsorption of essential elements and pernicious or macrocytic anemia, generally asymptomatic up to an advanced stage, and a predisposing factor to carcinoid tumor and gastric adenocarcinoma (72). Celiac disease is caused by gluten intolerance, leading to immune-mediated enteropathy and malabsorption of most nutrients and vitamins, with a prevalence of 1:270 in Finland to 1:5000 in North America (73). Inflammatory bowel diseases (Crohn disease and ulcerative colitis) are characterized by an imbalanced production of proinflammatory mediators (i.e., TNF-α) and increased recruitment of leukocytes to the site of inflammation (74). Autoimmune pancreatitis belongs to the spectrum of immunoglobulin G, subclass 4–related diseases, which typically presents with obstructive jaundice (75). Primary biliary cirrhosis is characterized by destruction, cholestasis, and cirrhosis, and it is accompanied by the occurrence of specific antimitochondrial Abs and autoreactive T cells (76).
Rheumatoid arthritis is characterized by progressive joint destruction (77) and ankylosing spondylitis, a progressive form of arthritis of the axial skeleton (78). Systemic lupus erythematosus is characterized by autoantibodies directed against nuclear antigens and affects every organ in the body. Common manifestations include rash, arthritis, and fatigue. Prevalence varies with ethnicity, but it is estimated to be 1 per 1000 overall, with a male-to-female ratio of 1:10 (79). Myasthenia gravis may occur. It is a rare autoimmune neuromuscular junction disorder initially with ocular weakness, asymmetric ptosis, and binocular diplopia later with painless, fluctuating, fatigable weakness involving specific muscle groups. The approximate prevalence rate is 1 out of 5000 subjects (80). Scleroderma, a systemic disorder with extensive accumulation of extracellular matrix proteins and small vessel vasculopathy, is also observed (81). Autoimmune hepatitis, leading to progressive inflammatory fibrotic liver disease and cirrhosis, may occur (82). The sicca (dry eye)/Sjögren syndrome is characterized by xerostomia and keratoconjunctivitis through a progressive lymphocytic and plasma cell infiltration of the salivary and lachrymal glands, and Ab production (83–85) is quite frequent in AP3. Patients affected by one autoimmune disorder are more frequently affected by others. The prevalence of the associated nonendocrine autoimmune diseases varies between the four types of AP (16).
Screening and Diagnosis
Serological screening in patients with monoglandular autoimmune syndrome is predictive for the development of future autoimmune endocrine diseases. Although target tissues or glands differ, several common threads link the syndromes of AP. Autoantibody screening, especially for T1D and AD, is the most important diagnostic tool and can give precise insight into the exact disease constellation. Alternatively, given that a significant percentage of the population is thyroperoxidase (TPO) Ab positive (∼15%), serological testing of the less specific Abs, that is, TPO Ab and/or thyroglobulin Ab, is limited, particularly for screening purposes in the absence of functional abnormalities of the target endocrine organ. Serological screening for T1D includes anti–glutamate decarboxylase (GAD), anti-insulin, anti–IA-2, anti–islet cell, and anti–zinc transporter Abs. Furthermore, the 21-hydroxylase (21-OH) Ab and the calcium-sensitive receptor (CaSR) Ab are present in AD and primary hypoparathyroidism, respectively (19, 27, 60, 61, 86). If serological screening is positive, functional screening (i.e., ACTH stimulation test or oral glucose tolerance test) is required to confirm the glandular dysfunction. Finally, owing to the frequent involvement of family members, serological screening of first-degree relatives is recommended (Fig. 2) (87). For juvenile AP, serological tests include 17-hydroxylase/21-OH Ab (cytochrome 450) and CaSR Ab for early testing of autoimmune-induced primary hypogonadism, AD, and primary hypoparathyroidism, respectively. Definitive diagnosis of glandular failure within AP1 is obtained by functional testing and measurement of serum levels of gonadotropins, testosterone or estradiol, baseline cortisol, parathormone, and electrolytes.
Algorithm for diagnosis of AP.
Genetic testing for mutations in the AIRE gene associated with juvenile AP is recommended when there is a likely presumption of AP1. Genetic testing identifies AP1 and permits early and accurate diagnosis of AP1 after only one of the primary component diseases of AP1 has developed. It informs patients and physicians of the risk of developing other manifestations of AP1 and supports the need for genetic counseling. Also, mutational analysis of the AIRE gene helps to identify patients with atypical phenotypes that can resemble AP1. For family members of AP1 patients, genetic diagnosis allows the avoidance of unnecessary follow-up of family members when AIRE mutations are not present. Genetic testing for mutations in the AIRE gene identifies AP1-associated mutations in relatives of AP1 patients prior to the occurrence of symptoms and signs, allowing early intervention and helping prevent potentially fatal complications from untreated endocrine insufficiencies. Children below the age of 10 years with monoglandular autoimmune disease (e.g., AD, autoimmune hypoparathyroidism, and autoimmune primary hypogonadism) should be tested for AIRE mutations. In the presence of a positive AIRE gene mutation, the risk for developing polyglandular failure is extremely high. The relative risks should be discussed during genetic counseling of the families at risk.
Management and Counseling
The treatment of AP1 is based on the organ systems involved and is specifically directed at replacing the various hormones that are deficient and treating yeast infections. Immunosuppressive therapy may be indicated in severe cases of AP1. However, there is no current cure for AP1. Prognosis depends on whether infections can be successfully controlled, and whether the critical hormone deficiencies are remedied. Each of the component disorders of the adult AP types is characterized by several stages, beginning with active organ-specific autoimmunity and followed by metabolic abnormalities with clinically manifest or overt organ dysfunction. Circulating organ-specific autoantibodies are observed in the various component diseases of AP2/AP3. The presence of such Abs precedes clinical overt disease. Because these autoantibodies are predictive for the development of future AP, siblings and kindred of affected AP patients should be screened (88, 89). Indeed, progression to T1D at 10-year follow-up after islet autoantibody seroconversion in children with multiple islet autoantibodies was 70% (95% CI, 65 to 74), and in children with a single islet autoantibody was only 14.5% (10–19). Risk of T1D in children who had no islet autoantibodies was 0.4% (0.2% to 0.6% by the age of 15 years) (88). In the presence of 21-OH Ab, cumulative incidence, cumulative rate, and annual incidence of AD within AP1 were 82.8%, 94.2%, and 18.5%, respectively (89). Several endocrine component disorders can be adequately treated with hormonal replacement therapy when the disease is recognized early. Regular follow-up of patients with monoglandular autoimmune syndrome, most especially those with AD and T1D, and to a lesser degree AT, is warranted, because a second autoimmune glandular disease may occur between 1 and 20 years after the manifestation of the first glandular failure (14). Furthermore, serological screening of the first-degree relatives of patients with AP2/AP3 is recommended owing to the high prevalence of various autoantibodies in their kindred (90). The presence of organ autoantibodies in these relatives should be followed by functional organ testing to diagnosis possible glandular dysfunction (1).
Genetic counseling is recommended for families and kindred of patients with AP1 because this is a monogenetic disease. Counseling should be done before performing genetic testing of the AIRE gene. For the adult AP type encompassing AP2/AP3, genetic counseling is optional, as several genes and environmental factors may be involved in the pathogenesis of the disease related to the loss of immune self-tolerance. Based on a genetic predisposition, external factors such as pathogens, that is, viral or bacterial infections (91), and psychosocial factors might induce autoimmune reactions. Counseling for those with the adult type should emphasize the rationale for a regular follow-up of any kindred and especially first-degree relatives with positive endocrine and nonendocrine Abs.
Recent Developments
The AIRE gene, by facilitating negative selection of T cells in the thymus, building the thymic microarchitecture, and inducing a specific subset of Tregs, shapes central immunological tolerance. Recent data suggest that certain mutations located in the SAND and PHD1 domains exert a dominant-negative effect on the wild-type AIRE gene, resulting in milder forms of autoimmune diseases. These findings also indicate that the AIRE gene contributes to autoimmunity in more common organ-specific autoimmune disorders as well (55).
Phenotyping 112 Russian patients with AP1 revealed several additional and uncommon phenotypes. The major Finnish mutation c.769C.T (p.R257*) was found in 72% of the alleles and was the most frequent in Russia. Furthermore, nine unknown mutations were reported: c.38T.C (p.L13P), c.173C.T (p.A58V), c.280C.T (p.Q94*), c.554C.G (p.S185*), c.661A.T (p.K221*), c.821del (p.Gly274Afs*104), c.1195G.C (p.A399P), c.1302C.A (p.C434*), and c.1497del (p.A500Pfs*21). This study demonstrated the clinical utility of measuring interferon Abs as a screening tool for AP1 (92).
Proteome arrays were used to search for autoimmune targets in AP1. Using established autoantigens revealed a highly reliable detection of autoantibodies. Melanoma-associated antigen B2 (MAGEB2) and protein disulfide isomerase-like, testis-expressed (PDILT) antigen were identified as novel autoantigens in AP1. These findings suggest that only a very limited portion of the proteome is targeted by the immune system, in contrast to the broad defect of thymic presentation associated with AIRE deficiency (93).
The HLA-DR binding pocket has a unique amino acid signature. Islet and thyroid peptides were screened for their ability to bind to the HLA-DR binding pocket. Four peptides (Tg.1571, GAD.492, TPO.758, TPO.338) were identified that were presented by antigen-presenting cells and elicited a T cell response, which led to the conclusion that both thyroid and islet peptides can bind to this flexible binding pocket and induce thyroid and islet-specific T cell responses, thus triggering T1D and AITD in the same subject (94).
A genome-wide approach has been used to identify new genes and mechanisms causing the co-occurrence of T1D and AITD. In one study, 346 whites with AP3 were analyzed along with 727 sex- and ethnicity-matched healthy controls, and multiple signals within the HLA region were identified. Conditioning studies suggested that a few of them contributed independently to the strong association of the HLA locus with AP. Furthermore, a locus on 1p13 containing the PTPN22 gene showed genome-wide significant associations. The major pathways contributing to the pathogenesis of AP are B cell development, CD40, and CTLA-4 signaling, suggesting that complex mechanisms involving T cell and B cell pathways are involved in the strong genetic association between AITD and T1D (25).
The HLA-DR variant containing arginine at position 74 (DR_1-Arg74) as specific HLA class II variant increases the risk for AITD. Blocking thyroglobulin peptides, especially Tg.2098, bound to DR_1-Arg74 could block continuous T cell activation, ending the autoimmune response in AT. To identify small molecules that can block T cell activation, a large number of compounds were screened. Cepharanthine was able to block T cell activation by preventing peptide binding, particularly of Tg.2098 to DR_1-Arg74 (95). Risk alleles for AP include HLA-DR and HLA-DQ with Ala in HLA-DQB1 at position 57 (Ala57), whereas amino acid Asp57 is protective. Two hundred seventy-eight patients with AP2 and 1373 patients with isolated endocrinopathies were compared with healthy controls in relationship to sex to differentiate the effects of HLA-DQB1 amino acid variants at position 57, finding that HLA-DQB1 confers strong susceptibility by Ala57 homozygosity and protection by non-Ala57, both in monoglandular and polyglandular syndromes. Frequencies of HLA-DQB1 amino acids also differentiated between AP types. HLA-DQB1 Ala57 heterozygous women have an increased risk for AD or AITD, whereas males have an increased risk for T1D, revealing sex-dependent increased susceptibility (96).
Single-base extension in a large group of adult AP showed that PTPN22 and CTLA-4 polymorphisms are associated with AP and differentiate between polyglandular and monoglandular autoimmunity (38). Also, a cross-sectional immunogenic study demonstrated that sex alters the MHC class I HLA-A association with polyglandular autoimmunity (97). Furthermore, patients with AP, monoglandular autoimmunity, and celiac disease and their first-degree relatives share a common genetic background of HLA antigens DQ2 (DQA1*0501-DQB1*0201) and DQ8 (DQA1*0301-DQB1*0302), as well as overlapping functional single-nucleotide polymorphisms of various susceptibility genes involved in the immune regulation (84, 98).
Activating Abs directed at the extracellular CaSR were described in monoglandular autoimmune hypoparathyroidism and AP (99). CaSR Abs of the IgG1 subtype stimulate phosphorylation of ERK1/2 and inositol phosphate accumulation. Treatment of a patient diagnosed with hypoparathyroidism, hypocalcemia, and hypomagnesemia with prednisone and azathioprine led to normalization of her serum calcium and magnesium, CaSR Ab titer, and Ab‐mediated stimulation of ERK1/2 phosphorylation and inositol phosphate accumulation. This demonstrates, to our knowledge for the first time, the response of CaSR Ab–mediated hypoparathyroidism to immunosuppressive therapy (100).
Conclusions
The overall prevalence of AP is below the threshold for rarity in Europe. Each of its subtypes and clinical variants, however, has a very low prevalence and therefore qualifies as a rare or ORPHAN condition. The clinical presentation of AP variants is often preceded by an asymptomatic latent period of up to 10 years characterized by the presence of autoantibodies, which are useful markers for the prediction of AP. Still, the absence of these Abs does not exclude the disease. In view of the possible long interval between the first manifestation of AP and the subsequent development of further autoimmune endocrinopathies, regular and long-term observation of patients with endocrine autoimmune disorders is warranted. Furthermore, screening for autoimmune endocrine diseases of their first-degree relatives is recommended, especially for the offspring of patients with AP and/or T1D. Although genetic screening is useful in patients with AP1, the influence of genetic and environmental factors combined may lead to the onset of autoimmune disorders in different organs of the same subject.
Acknowledgments
We are most grateful to Tanja Diana (Endocrine and Thyroid Research Laboratory, Johannes Gutenberg University Medical Center) and Paul D. Olivo (Department of Molecular Microbiology, Washington University School of Medicine, St. Louis, MO) for their constructive and helpful comments.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations:
- 21-OH
21-hydroxylase
- Ab
antibody
- AD
Addison disease
- AIRE
autoimmune regulator
- AITD
autoimmune thyroid disease
- AP
autoimmune polyendocrinopathy
- AP1
type I AP
- AP2
type II AP
- AP3
type III AP
- AT
autoimmune thyroiditis
- CaSR
calcium-sensitive receptor
- CTLA-4
cytotoxic T lymphocyte antigen 4
- FOXP3
forkhead box P3
- GAD
glutamate decarboxylase
- GD
Graves disease
- IL-2Ra
IL-2 receptor α
- MHC
major histocompatibility complex
- MICA
MHC class I chain–related gene A
- NKG2D
natural killer group 2
- member D
PTPN22
- protein tyrosine phosphatase nonreceptor type 22
SNP
- single-nucleotide polymorphism
T1D
- type 1 diabetes
Th
- T helper
TPO
- thyroperoxidase
Treg
- regulatory T cell
VDR
- vitamin D receptor
VNTR, variable number of tandem repeats

{kind=link}
{kind=link}