





Abstract
Autoimmune thyroid diseases (AITDs) arise from complex interactions among genetic, epigenetic, and environmental factors. Thyroglobulin (TG) is a major susceptibility gene for both Graves disease and Hashimoto thyroiditis. Interferon-α (IFNα), a cytokine secreted during viral infections, has emerged as a key trigger of AITD. We have shown that IFNα upregulates TG transcription; however, how the upregulation of TG transcription by IFNα triggers AITD is still unknown.
To evaluate how IFNα triggers AITD by testing its effects on TG processing.
We exposed human thyroid cells to IFNα and evaluated its effects on TG expression and processing.
Human thyroid cells exposed to INFα had increased levels of TG mRNA but reduced TG protein levels, indicating TG protein degradation. IFNα induced endoplasmic reticulum stress, but surprisingly, neither the use of chemical chaperones nor proteasome inhibitor prevented IFNα-induced TG degradation. IFNα also increased LysoTracker staining and autophagy flux measured by net light chain 3 (LC3)-II and p62 fluxes. In addition, expression of autophagy markers LC3 and autophagy-related gene 5 was higher in thyroid tissues from patients with AITD. Finally, blocking lysosomal degradation prevented IFNα-induced degradation of TG.
We have shown in this study IFNα-induced lysosomal-dependent degradation of TG in human thyroid cells. Our findings suggest that during viral infections, local thyroidal IFNα production can lead to lysosomal TG degradation, releasing pathogenic TG peptides that can trigger AITD.
Autoimmune thyroid diseases (AITDs), including Graves disease (GD) and Hashimoto thyroiditis (HT) are common autoimmune diseases with prevalence in the United States of 1% to 5% (1). Although GD and HT have opposite clinical features, they share common immunopathogenic mechanisms. Both are characterized by infiltration of the thyroid gland by T and B cells reactive to thyroid proteins; this infiltration leads to apoptosis of thyroid cells and hypothyroidism in HT, or production of thyroid-stimulating hormone receptor antibodies and hyperthyroidism in GD (2). AITDs are complex polygenic diseases and they arise from interactions among genetic, epigenetic, and environmental factors (2).
Among the nongenetic factors triggering AITDs (e.g., excess iodine, selenium deficiency, pollutants, infections), interferon-α (IFNα), a critical cytokine secreted by the host immune system in response to viral infections, has recently emerged as a major trigger of AITDs (3–5). Previous data from our group have demonstrated that IFNα upregulates transcription of thyroid antigens [i.e., thyroglobulin (TG), thyroid-stimulating hormone receptor, thyroid peroxidase] and induces thyroid cell necrosis and release of cytokines and chemokines leading to autoimmune thyroiditis by a bystander mechanism (5). Mechanistically, we have mapped a TG promoter variant that was strongly associated with AITDs (−1623 A/G), and that also interacted epigenetically with interferon regulatory factor-1, a major interferon-induced transcription factor. This genetic–epigenetic interaction resulted in upregulation of TG transcription upon exposure of thyroid cells to IFNα (4). However, how the upregulation of TG transcription by IFNα triggers AITDs is still unknown. In the current study, we sought to dissect the downstream molecular mechanisms by which IFNα triggers thyroid autoimmunity.
We found that while IFNα induces TG mRNA overexpression, it also induces degradation of newly formed TG protein within lysosomes in a setting of increased endoplasmic reticulum (ER) stress and autophagy. These downstream effects of IFNα on TG processing could lead to the release of immunogenic peptides and trigger AITDs in genetically susceptible individuals.
Materials and Methods
Cell culture and treatments
Human thyroid ML-1 cells (female origin, a gift from Dr. Johann Schonberger, University of Regensburg, Germany) were maintained in DMEM plus GlutaMAX-1 medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Sigma-Aldrich). We selected ML-1 cells because this human thyroid cell line shows robust expression of TG (6). Cells were grown at 37°C in 95% humidified air with 5% CO2 (pH 7.4). Cells were harvested with trypsin-EDTA (0.02%; ATCC), seeded on six-well plates, 10-cm dishes, or 12-mm–diameter glass coverslips in 24-well plates, and incubated again for 24 hours in the same medium to become partially confluent.
IFNα (Millipore) was added to cells to a final concentration of 1000 U/mL, 5000 U/mL, and 10,000 U/mL for 24, 48, or 72 hours. Cells were pretreated for 12 hours with 500 μM of 4-phenylbutyric acid (PBA; Sigma-Aldrich), 100 μM of tauroursodeoxycholic acid (TUDCA; Calbiochem), 0.8 μM of MG132 (Sigma-Aldrich) or with medium only, and then cultured in the presence of IFNα 5000 U/mL for 48 hours. As a positive control for ER stress, 0.5 μM of thapsigargin (THAP; Sigma-Aldrich) was added to cells overnight. For cycloheximide assays, ML-1 cells were treated with cycloheximide (50 μg/mL) for 4, 8, 12, and 24 hours before collecting the cells and cultured in the presence of IFNα 5000 U/mL for 48 hours.
Human thyroid specimens
Studies were approved as exempt (deidentified tissues) by the institutional review boards of Albert Einstein College of Medicine and Icahn School of Medicine at Mount Sinai. A total of eight deidentified human thyroid specimens were obtained (five from patients with GD and three from patients with HT) from the Department of Pathology. Normal adjacent thyroid tissues (also deidentified; n = 5) obtained at surgeries for thyroid nodules were used as controls.
RNA analyses
Total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific) followed by DNase treatment (Thermo Fisher Scientific) according to the manufacturer’s instructions. Total RNA (1 μg) was reverse transcribed using the Superscript III kit (Thermo Fisher Scientific) following the manufacturer's instructions. Real-time RT-PCR analyses were performed in a fluorescent temperature cycler (AbiPRISM 7300; Applied Biosystems). After an initial incubation at 50°C for 2 minutes and 95°C for 10 minutes, the reactions were cycled 40 times using the following parameters for all genes studied: 95°C for 15 seconds, 60°C for 30 seconds, and 72°C for 45 seconds. SYBR Green (Applied Biosystems) fluorescence was detected at the end of each cycle. The expression of each gene of interest was normalized to that of glyceraldehyde-3-phosphate dehydrogenase and the relative mRNA levels were determined using the comparative threshold cycle method.
The sequences for the forward and reverse primers (Integrated DNA Technologies) are shown in Supplemental Table 1. The assays were repeated at least three times, and the data were merged after normalization. The results are expressed relative to the values of the control group, which were considered to be equal to 1.
Western blot analyses
ML-1 cells were lysed in ice-cold RIPA lysis buffer supplemented with a broad-spectrum protease inhibitor cocktail (Roche). Lysates were centrifuged at 13,000 rpm for 30 minutes and supernatants were collected. Total protein concentration was determined using bicinchoninic acid protein assay kit (Thermo Fisher Scientific) and 30 to 50 μg of sample was resolved by 4% to 15% gradient or 12% Tris-glycine gels (BioRad). Protein was transferred on nitrocellulose or polyvinylidene difluoride membranes (BioRad) and blocked for 1 hour at room temperature (RT) in Odyssey blocking buffer (LI-COR). Membranes were blotted overnight at 4°C with the primary antibodies listed in Supplemental Table 2. Membranes were washed with Tris-buffered saline containing 0.1% Tween 20, followed by incubation for 1 hour at RT with infrared-labeled anti-rabbit or anti-mouse IgG (1:20,000; LI-COR) secondary antibodies. Immunolabeled proteins were visualized using the LI-COR Odyssey system. Quantitative densitometric analysis was performed using Odyssey software. The blots were repeated at least three times and protein levels were normalized to those of β-actin.
Autophagic measurements
Autophagic flux was estimated by immunoblot for light chain 3 (LC3)-II and sequestosome 1 (also called p62) in cells untreated or treated with the lysosomal protease inhibitor mixture 20 mM NH4Cl/200 μM leupeptin (Thermo Fisher Scientific) for the last 12 hours of the IFNα treatment. The level of LC3 lipidation was quantified as the ratio of the measured LC3-II to β-actin levels. LC3 flux and p62 degradation was quantified as the relative ratio of LC3-II or p62 to β-actin values between samples with or without the lysosomal proteolysis inhibitors (net flux).
Small-interfering RNA–induced mRNA knockdown
ML-1 cells were transfected with ON-TARGETplus SMARTpool (Dharmacon) against human autophagy-related gene 7 (Atg7) to a final concentration of 25 nM, using DharmaFECT 1 transfection reagent. Cells transfected with ON-TARGETplus nontargeting control small-interfering RNA were used as negative controls. Cells were harvested 48 hours after transfection and incubation with IFNα and mRNA expression of ATg7 and Western blot analyses of TG were performed.
Immunofluorescence
ML-1 cells (n = 1.5 × 105) were seeded on poly-d-lysine–coated, 12-mm glass coverslips in 24-well plates and allowed to attach for 24 hours at 37°C. Cells were then treated with vehicle or with the indicated drugs (see Results) at the indicated experimental settings and fixed in 4% paraformaldehyde (Sigma-Aldrich) in PBS for 15 minutes at RT. Cells were then permeabilized by 0.5% Triton X-100 (Thermo Fisher Scientific) in PBS for 10 minutes at RT, and blocked for 1 hour at RT with 1% normal goat serum (Cell Signaling) in PBS. After the blocking step, cells were incubated overnight at 4°C with the primary antibodies diluted in blocking buffer. Indirect immunofluorescence was performed using rabbit monoclonal anti phospho-eIF2α (1:100; Cell Signaling), mouse monoclonal anti-CHOP (CCAAT/enhancer-binding protein homologous protein; 1:500; Cell Signaling). Secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 568 (1:500; Thermo Scientific) were used for 1 hour at RT. We added 4′,6-diamidino-2-phenylindole (Thermo Fisher Scientific) for nuclear stain. After final washes with PBS, the coverslips were mounted on microscope slides with Vectashield mounting media (Vector Laboratories). The stained slides were observed with a Leica DM6000 microscope.
LysoTracker staining
LysoTracker Deep Red (Thermo Fisher Scientific) was used to label lysosomes according to the manufacturer’s instructions. Briefly, ML-1 cells plated in a glass eight-chamber slide were incubated with 50 nM of LysoTracker for 1 hour at 37°C. Loading solution was replaced with fresh medium before analysis and cells were observed under a Zeiss LSM 880 Airyscan microscope.
Statistics
Results are shown as mean ± SE and represent data from a minimum of three independent experiments. One-way ANOVA followed by Student-Newman-Keuls multiple comparison test or Student t test for unpaired data were used for the assessment of differences between the treatment groups. A P value of < 0.05 was considered significant.
Results
IFNα upregulated TG mRNA levels but reduced protein levels in human thyroid cells
To investigate how IFNα could affect TG processing in human thyroid cells, we exposed ML-1 cells to increasing doses of IFNα (1000, 5000, 10,000 U/mL) for 48 hours. A time-course analysis from 12 hours to 72 hours revealed significant changes in TG expression after 48 hours (data not shown). As shown in Fig. 1A, TG mRNA levels, as assessed by quantitative RT-PCR, were significantly increased by 5000 U/mL and 10,000 U/mL of IFNα. Surprisingly, despite the increase in TG mRNA levels triggered by IFNα, TG protein levels were reduced after all IFNα doses tested (Fig. 1B and 1C), suggesting that IFNα could be stimulating TG protein degradation.

IFNα upregulates TG mRNA levels but reduces protein levels in human thyroid cells. (A) Comparison of TG mRNA and protein levels upon exposure to different doses of IFNα (1000, 5000, and 10,000 U/mL) for 48 h. The data were normalized for each mRNA level and protein level relative to CTR group (cells not treated with IFNα). (B) Representative immunoblot of TG (330 kDa) and β-actin (45 kDa) of ML-1 cells treated with the same increasing doses of IFNα as in (A) for 48 h and (C) a respective analysis by densitometry. Data are shown as mean ± SE of at least three independent experiments. Statistical analysis was performed using one-way ANOVA followed by Student-Newman-Keuls multiple comparison test. *P < 0.05, **P < 0.01 vs CTR cells. Conc, concentration; CTR, control.
IFNα activated unfolded protein response in human thyroid cells
The first potential mechanism for IFNα-mediated TG degradation we tested was endoplasmic reticulum–associated protein degradation (ERAD). Thyroid follicular cells, which are professional secretory cells because of their heavy engagement in TG production and secretion, are very sensitive to disruptions in ER homeostasis (7). To test whether IFNα induces ER stress in human thyroid cells, we exposed ML-1 cells to increasing doses of IFNα (1000, 5000, 10,000 U/ml) for 48 hours and checked the activation of different branches of the unfolded protein response (UPR).
IFNα induced all major signaling branches of the UPR, including activating transcription factor 6, inositol-requiring enzyme 1, and double-stranded RNA activated protein kinase–like ER kinase (PERK), as demonstrated by the upregulation of binding immunoglobulin protein (BIP), X-box binding protein 1 (XBP-1), spliced XBP-1 (sXBP-1), activating transcription factor 4, and CHOP mRNA levels (Fig. 2A). Protein levels of BIP, a protein chaperone and specific ER stress marker, were stimulated by 5000 and 10,000 U/mL of IFNα (Fig. 2B and 2C). The upregulation of the UPR by IFNα was also confirmed by immunofluorescence, showing that IFNα induced both phosphorylated eIF2α (Fig. 2D and 2E) and CHOP (Fig. 2F and 2G) levels, confirming the activation of PERK signaling pathway. In turn, UPR activation by the ER stressor THAP increased intrathyroidal mRNA expression of IFNα2 and its target gene MHC I (Supplemental Fig. 1), indicating a vicious cycle of augmented IFNα production and ER stress.
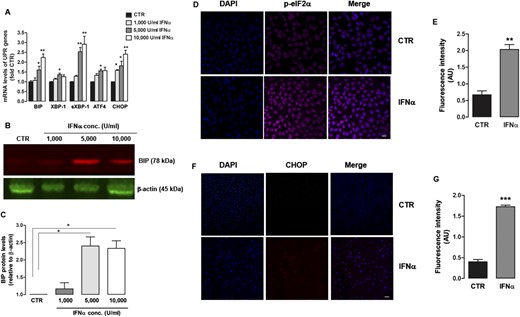
IFNα induces UPR activation in human thyroid cells. (A) mRNA expression of UPR genes BIP, XBP-1, sXBP-1, ATF4, CHOP measured by quantitative RT-PCR in ML-1 cells exposed to increasing doses of IFNα (1000, 5000, and 10,000 U/mL) for 48 h. The data were normalized for each mRNA level relative to the CTR group. (B) Representative immunoblot of BIP (78 kDa) and β-actin (45 kDa) of ML-1 cells treated with the same increasing doses of IFNα as in (A) for 48 h and (C) a respective analysis by densitometry. (D) Immunofluorescence staining for DAPI (blue) and pelF2α (purple) in ML-1 cells treated with IFNα (5000 U/mL) for 48 h and (E) a respective quantification of fluorescence signal intensity. (F) Immunofluorescence staining for DAPI (blue) and CHOP (red) in cells treated with IFNα (5000 U/mL) for 48 h and (G) a respective quantification of fluorescence signal intensity. Scale bars, 10 μm. The data are shown as mean ± SE of at least three independent experiments. Statistical analysis was performed using one-way ANOVA followed by Student-Newman-Keuls multiple comparison test. *P < 0.05, **P < 0.01, *** P < 0.001 vs CTR cells. Conc, concentration; CTR, control; DAPI, 4′,6-diamidino-2-phenylindole.
IFNα-induced TG degradation was not mediated by ubiquitin-proteasome system
The canonical ERAD pathway targets unfolded or misfolded proteins retained in the ER for ubiquitination and subsequent degradation by the ubiquitin-proteasome system (8). To examine the contribution of ERAD to IFNα-induced TG degradation, we treated ML-1 cells with two chemical chaperones, PBA and TUDCA, that facilitate protein folding and reduce ER stress. As a positive control for ER stress, we used THAP, a noncompetitive inhibitor of the Sarco/ER Ca2+ ATPase 2. As expected, THAP induced expression of UPR markers BIP (Supplemental Fig. 2A), sXBP-1 (Supplemental Fig. 2B), and CHOP (Supplemental Fig. 2C). Treatment with 5000 U/mL IFNα for 48 hours increased BIP, sXBP-1, and CHOP expression, whereas PBA and TUDCA completely prevented the effects of IFNα on these UPR markers (Supplemental Fig. 2). PBA and TUDCA, however, did not prevent IFNα-induced TG degradation; as shown in Supplemental Fig. 2D and 2E, the protein levels of TG after cotreatment with chemical chaperones plus IFNα were similar to those in cells exposed to only IFNα. Moreover, neither the selective proteasome inhibitor MG132 alone nor the combination of MG132 and PBA or TUDCA could reverse the decrease in TG levels triggered by IFNα. These data suggested that although IFNα triggered ER stress and UPR in human thyroid cells, it did not trigger protein degradation of TG through ERAD.
IFNα stimulated autophagy in human thyroid cells
Macroautophagy (hereafter referred to as autophagy) is a highly conserved, lysosomal dependent-catabolic process that contributes to quality control and maintenance of the cellular energetic balance through degradation of organelles, proteins, and other macromolecules in lysosomes (9). Because bidirectional interaction among these two proteolytic systems—ubiquitin-proteasome system and autophagy—has been shown in many different cell systems, we investigated whether IFNα also affects autophagy in human thyroid cells, as a potential mechanism for TG degradation. Because lysosomes are key organelles in the autophagic process, we tested lysosomal acidification by LysoTracker staining and found that IFNα (5000 U/mL) increased the staining at all the time points studied (24 to 72 hours), indicating that treatment with IFNα resulted in an expansion of autophagic/lysosomal compartments (Fig. 3A and 3B), similar to that observed during induction of autophagy in response to serum deprivation for 16 to 18 hours (Fig. 3A and 3B).
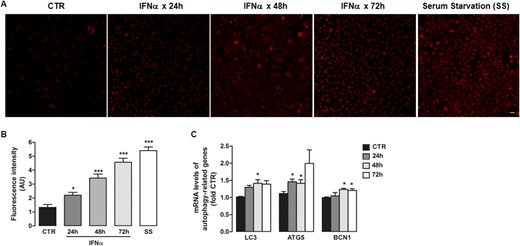
IFNα induces autophagy markers. (A) Representative immunofluorescence images after LysoTracker staining for lysosomes in ML-1 cells treated with IFNα (5000 U/mL) for 24, 48, and 72 h, and cells under serum starvation for 16 to 18 h. Scale bar, 10 μm. (B) Respective quantification of fluorescence signal intensity. (C) mRNA expression of autophagy genes LC3, ATG5, and beclin 1 measured by quantitative RT-PCR in ML-1 cells exposed to 5000 U/mL IFNα for 24, 48, and 72 h. Data are shown as mean ± SE of at least three independent experiments. Statistical analysis was performed using one-way ANOVA followed by Student-Newman-Keuls multiple comparison test. *P < 0.05, *** P < 0.001 vs CTR cells. AU, arbitrary units; CTR, control.
In addition, exposure of ML-1 cells to IFNα (5000 U/mL) for 48 hours upregulated mRNA levels of LC3, autophagy-related gene 5 (ATG5) and beclin 1, all key genes in autophagic pathways (Fig. 3C). In turn, autophagy induction by serum starvation induced intrathyroidal mRNA levels of IFNα2 and its target gene 2'-5′-oligoadenylate synthetase (Supplemental Fig. 3), suggesting a self-perpetuating cycle of increased IFNα and autophagy.
Analysis of steady-state levels of LC3 and p62 revealed a significant 30% reduction of LC3-II (P < 0.05; Fig. 4A and 4B) in IFNα-treated cells and a trend toward a decrease in the intracellular levels of p62 (Fig. 4C and 4D) compatible with autophagic activation. We next directly measured the autophagic flux, detected as the increase in LC3-II and p62 protein levels upon blockage of lysosomal degradation (by lysosomal protease inhibitors). Treatment with IFNα increased LC3-II (Fig. 4A) and p62 (Fig. 4C) lysosomal degradation over the levels detected in untreated cells. IFNα exposure increased net LC3-II flux (net differences in p62 levels on addition of inhibitors) by 35% (P < 0.05; Fig. 4B) and net p62 flux by 112% (P < 0.05; Fig. 4D) compared with control cells. These results confirmed that treatment with IFNα increased autophagic flux.
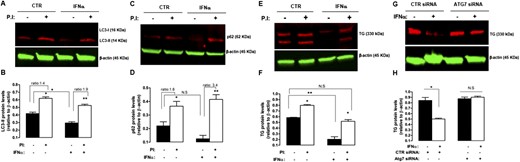
Blockage of lysosomal degradation and autophagy prevented IFNα-induced protein degradation of TG. (A) Representative immunoblot of p62 (62 kDa) and β-actin (45 kDa) and (B) corresponding densitometric analysis showing increased p62 protein expression levels in ML-1 cells treated with IFNα (5000 U/mL) for 48 h, followed by protease inhibitors (to block lysosomal degradation). (C) Representative immunoblot of LC3-II (14 kDa) and β-actin (45 kDa) and (D) corresponding densitometric analysis showing increased LC3-II expression levels in ML-1 cells treated with IFNα (5000 U/mL) for 48 h, followed by PIs (to block lysosomal degradation). Note that LC3-I (16 kDa) levels were not detected. (E) Representative immunoblot of TG (330 kDa) and β-actin (45 kDa) and (F) corresponding densitometric analysis showing blockage of TG degradation in ML-1 cells treated with IFNα (5000 U/mL) for 48 h, followed by blockage of lysosomal degradation by PIs. Where indicated, a combination of lysosomal protease inhibitors (20 mM NH4Cl and 200 µM leupeptin) was added 12 h before collecting the cells (see Methods). (G) Representative immunoblot of TG (330 kDa) and β-actin (45 kDa) and (H) corresponding densitometric analysis showing blockage of TG degradation in ML-1 cells treated with IFNα (5000 U/mL) for 48 h after small-interfering RNA–mediated Atg7 knockdown. Data are shown as mean ± SE of at least three independent experiments. Statistical analysis was performed using Student t test. *P < 0.05, **P < 0.01. CTR, control; N.S, nonsignificant; PI, protease inhibitor.
Blockage of lysosomal degradation and autophagy prevented IFNα-induced degradation of TG
Lysosomes play an essential role in normal physiological TG processing; as a prohormone, TG is hydrolyzed by proteases within these organelles to release the active thyroid hormones triiodothyronine and thyroxine (10). Therefore, we tested the lysosomal-dependent degradation system as another potential proteolytic system that could be responsible for IFNα-induced TG degradation. When a combination of lysosomal protease inhibitors was added to ML-1 cells during the last 12 hours of their 48-hour exposure to IFNα, TG protein levels were twofold higher than in absence of protease inhibitors, and the TG protein levels were similar to those in control cells incubated without protease inhibitors (Fig. 4E and 4F). These data demonstrated that blockage of lysosomal degradation almost completely rescued TG levels. Interestingly, treatment of ML-1 cells with inhibitors of lysosomal proteolysis led to increased TG levels in control cells, consistent with the notion that TG is processed within lysosomes of thyrocytes (Fig. 4E).
Because autophagy is one of the major pathways for degrading cytoplasmic proteins, we tested next whether blocking Atg7, an essential molecule for autophagy induction, would prevent IFNα-induced TG degradation. After inhibition of autophagy by knockdown of Atg7 using small-interfering RNA, the abundance of TG protein levels in IFNα-treated cells were similar to those in control-treated cells (Fig. 4G and 4H), indicating that autophagy blockage completely rescued TG protein levels.
Finally, we checked if IFNα affects TG translation in addition to promoting its lysosomal degradation. As shown in Supplemental Fig. 4, TG protein degradation was still present after protein translation inhibition by cycloheximide, suggesting that IFNα only triggers TG degradation but does not significantly suppress TG translation.
Thyroid tissues from patients with AITD expressed increased levels of UPR and autophagy markers
In view of our data showing that IFNα increased TG degradation within lysosomes and triggered ER stress and autophagy in thyroid cells, we evaluated the role of autophagy and ER stress in the etiology of AITD. We tested thyroid tissues from patients with AITD (GD tissues, n = 5; HT tissues, n = 3) for the expression of autophagy-related genes (i.e., LC3, ATG5, and beclin1) and UPR markers (i.e., BIP, CHOP, XBP-1, sXBP-1, and activating transcription factor 4) by quantitative RT-PCR (Fig. 5A and 5B). There was a substantial increase in mRNA levels of all UPR markers analyzed and ATG5 and LC3 levels in thyroid tissues from patients with AITD compared with normal thyroid tissues, supporting our data that suggested a role for both ER stress and autophagy in the pathogenesis of thyroid autoimmunity. However, this association does not prove causation.

GD and HT tissues express increased mRNA levels of UPR and autophagy markers. (A) mRNA expression levels of UPR genes BIP, CHOP, XBP-1, sXBP-1, and ATF4 and (B) autophagy genes LC3, ATG5, and BCN1 measured by quantitative RT-PCR in thyroid tissues of eight patients with AITD (empty squares) and five normal adjacent thyroid tissues obtained at surgeries for thyroid nodules (black triangles). The data were normalized for each mRNA level relative to the CTR group. Data are shown as mean ± SE. Statistical analysis was performed using one-way ANOVA followed by Student-Newman-Keuls multiple comparison test. *P < 0.05, **P < 0.01 vs CTR cells. CTR, control.
Discussion
TG is a 660-kDa glycoprotein homodimer that accounts for ∼80% of total thyroid protein content and serves as a precursor for thyroid hormones thyroxine and triiodothyronine (11). Whole-genome linkage screens and association studies have mapped the TG gene as a key susceptibility gene for both forms of AITD: GD and HT (12–14). Indeed, TG is the key autoantigen in thyroid autoimmunity as evidenced by the fact that IgG anti-TG antibodies are found in 75% of patients with AITD (2). Moreover, autoimmune response to TG is the earliest event in the development of AITD in mice (15), and the classical model of human autoimmune thyroiditis is induced by immunizing mice with TG protein (16). Several single nucleotide polymorphisms (SNPs) in the TG locus are associated with AITD in humans and in mice (4, 14, 17), but the mechanisms by which these SNPs trigger disease are still unclear.
Our study demonstrated that exposing human thyroid cells to INFα induced an increase in TG mRNA levels but a reduction in TG protein levels, indicating TG protein degradation. Furthermore, we demonstrated that IFNα induced degradation of TG via the autophagy/lysosomal system. Taken together, our findings suggest an attractive mechanism for the development of AITD. The mechanism we propose is that during viral infections, local thyroidal IFNα production upregulates TG synthesis coupled with triggering TG degradation within lysosomes, resulting in the release of pathogenic TG peptides that can trigger thyroid autoimmunity. Moreover, we have demonstrated that IFNα induced ER stress in thyroid cells, which can lead to thyroid cell apoptosis that can further trigger immune cell recruitment and autoimmune responses targeting thyroid antigens (18).
IFNα plays a key role in the development of several autoimmune diseases, most notably systemic lupus erythematosus (SLE) and type 1 diabetes (T1D). Most patients with SLE have an increased IFNα signature and many risk gene variants for SLE are located in loci linked to IFNα (19). In addition, increased IFNα levels were found in the islets of deceased diabetic individuals (20) and β cells of patients with T1D (21). Finally, IFNα has been shown to initiate T1D in in NOD mice (22). Likewise, there are abundant data showing that IFNα treatment of hepatitis C and other conditions can trigger thyroiditis in up to 40% of patients (23). Taken together, these data and our data support a general role of IFNα in autoimmunity and point to a new mechanism by which IFNα can trigger autoimmunity. It remains to be determined whether the mechanism we identified is unique to AITD or is relevant to other autoimmune diseases. Moreover, we have recently shown that IFNα can trigger ER stress affecting insulin processing in β cells (24), supporting a similar role for IFNα in other autoimmune diseases.
We have shown that IFNα exposure stimulated autophagic flux in thyroid cells. Our data are consistent with those of Schmeisser et al. (25), who reported that IFNα induces autophagy through JAK1/STAT1 in different cell types. Autophagy plays pivotal roles in innate and adaptive immunity, including antigen processing and presentation, pathogen recognition, and removal of intracellular microorganisms (26). Recently, autophagy has been implicated as an important mechanism in autoimmunity. Specifically, several SNPs in autophagy-related genes were reported to be associated with autoimmune disorders (26), such as Crohn disease (27), SLE (28), and rheumatoid arthritis (29). Moreover, IFNα-induced autophagy in B cells is important in the pathogenesis of SLE (30). Similarly, autophagy has also been implicated in the pathogenesis of AITD. Yoon et al. (31) reported that expression levels of LC3 and p62 and the number of autophagic vacuoles were higher in Graves orbitopathy cells than in control cells. In addition, HT thyroid tissues expressed higher protein levels of LC3B-II (32). Moreover, studies have shown increased lysosomal degradation of TG in thyroid tissues from patients with GD (33) (34), supporting our hypothesis that IFNα-induced lysosomal degradation of TG into pathogenic peptides may play a key role in the initiation of AITD. Consistent with these findings, we detected increased expression levels of ATG5 and LC3 in thyroid tissues from patients with AITD. However, how perturbations in autophagy trigger autoimmunity remains to be elucidated.
Autophagy is dynamically intertwined with ER stress and acts as a prosurvival mechanism under stress conditions (35). For example, PERK and IRE1 pathways that are triggered in response to ER stress regulate several autophagy-related genes (36). Interestingly, our study showed that IFNα exposure triggered autophagy and ER stress in thyrocytes. As highly secretory cells, thyrocytes are inherently susceptible to disruptions in ER homeostasis (37) (38), even under normal physiological conditions, and IFNα likely induces additional ER stress in thyrocytes. ER stress that usually results from the accumulation of misfolded or unfolded proteins within the ER lumen activates an adaptive coordinated program to alleviate ER stress and restore protein homeostasis (39). If the adaptive response fails, cells undergo apoptosis and this has been proposed to produce neo-self antigens and trigger an autoimmune response in genetically susceptible individuals (39). Impaired ER stress responses have been linked to several autoimmune diseases, such as T1D (40), rheumatoid arthritis (41), and SLE (42), and our study may suggest a link to AITD as well. Taking into account that induction of ER stress and autophagy stimulates intrathyroidal expression of IFNα and its target genes, such as MHC I and 2'-5′-oligoadenylate synthetase, one can propose that IFNα exposure initiates a vicious cycle of stressed thyrocytes that provoke an autoimmune attack.
In summary, we have shown that in human thyroid cells, IFNα induces lysosomal degradation of TG and upregulation of ER stress and autophagy. Taken together, findings of the current study and previous studies suggest that IFNα (e.g., during viral infections or in individuals receiving therapeutic IFNα) disrupts thyrocytes and can trigger AITD by the integration of three effects: (1) stimulating proinflammatory cytokines and chemokines, inducing expression of HLA class I and activating cytotoxic T cells (5); (2) upregulating TG mRNA expression (4) (5); and (3) inducing ER stress and autophagy that trigger TG degradation by lysosomal hydrolases possibly into immunogenic peptides that could be presented to T cells, leading to activation of self-reactive T cells within the thyroid (Fig. 6).
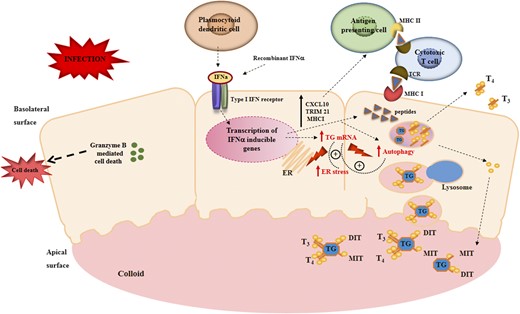
Potential mechanisms by which IFNα disrupts thyrocytes to trigger AITDs. Thyroid follicles are composed of a monolayer of polarized thyroid follicular cells with the apical surface facing the interior of the follicle containing the colloid and the basolateral surface facing the blood vessels. TG is stored in the colloid; upon TSH stimulation of the thyroid-stimulating hormone receptor, TG is endocytosed and processed in lysosomes to release T3 and T4 into the blood. In response to infection, plasmocytoid dendritic cells and other cells produce locally IFNα, which binds to its receptor and activates the JAK/STAT pathway (this would also happen in individuals receiving therapeutic IFNα). The activation of the JAK/STAT pathway results in the stimulation of proinflammatory cytokines, increased expression of HLA class I on thyrocytes, and activation of cytotoxic T cells. In addition, IFNα also triggers TG mRNA upregulation as well as ER stress and autophagic proteolysis, leading to processing of TG by lysosomal hydrolases into immunogenic peptides. Induction of ER stress and autophagy stimulates intrathyroidal expression of IFNα and its target genes, initiating a vicious cycle of stressed thyrocytes that provoke an autoimmune attack. In genetically susceptible individuals (e.g., individuals carrying the TG SNP (−1623A→G), these peptides generated from TG will be presented to T cells and activate self-reactive T cells, triggering thyroid autoimmunity. CXCL10, C-X-C motif chemokine ligand 10; DIT, diodotyrosine; MHC I, major histocompatibility complex I; MHC II, major histocompatibility complex II; MIT, monoiodotyrosine; T3, triiodothyronine; T4, thyroxine; TCR, T-cell receptor; TRIM21, tripartite motif containing 21.
Abbreviations:
- AITD
autoimmune thyroid disease
- ATG5
autophagy-related gene 5
- BIP
binding immunoglobulin protein
- CHOP
CCAAT/enhancer-binding protein homologous protein
- ER
endoplasmic reticulum
- ERAD
endoplasmic reticulum–associated protein degradation
- GD
Graves disease
- HT
Hashimoto thyroiditis
- IFNα
interferon-α
- LC3
light chain 3
- PBA
phenylbutyric acid
- PERK
protein kinase–like endoplasmic reticulum kinase
- RT
room temperature
- SLE
systemic lupus erythematosus
- SNP
single nucleotide polymorphism
- sXBP-1
spliced X-box binding protein 1
- T1D
type 1 diabetes
- TG
thyroglobulin
- THAP
thapsigargin
- TUDCA
tauroursodeoxycholic acid
- UPR
unfolded protein response
- XBP-1
X-box binding protein 1
Acknowledgments
We thank Dr. Ana Maria Cuervo (Albert Einstein College of Medicine) for her helpful advice and discussions and for critically reading the manuscript; Dr. Terry Davies (Icahn School of Medicine at Mount Sinai) for providing us thyroid tissues from patients with Graves disease; and Dr. Mihaela Stefan (Albert Einstein College of Medicine) for her valuable comments.
Financial Support: This work was supported in part by the National Institute of Diabetes and Digestive and Kidney Diseases (Grants DK061659, DK067555, and DK073681 to Y.T.). J.M.-M. is supported by a postdoctoral fellowship from the American Diabetes Association (Grant 1-15-MI-03) and by the Leducq Foundation Transatlantic Network of Excellence.
Disclosure Summary: The authors have nothing to disclose.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}