





Abstract
Testotoxicosis is an autosomal-dominant, male-limited disorder. Activating mutations in the luteinizing hormone receptor gene (LHCGR) cause high autonomous testosterone secretion, resulting in early-onset peripheral precocious puberty. Little is known about long-term consequences of testotoxicosis.
We present a rare case of a patient followed for 25 years with two remarkable outcomes: preserved fertility and germ cell neoplasia in situ (GCNIS). He presented with precocious puberty at 10 months of age and was diagnosed with testotoxicosis due to a de novo heterozygous Asp578Tyr mutation in LHCGR. Testicular biopsy in childhood showed Leydig cell hyperplasia with altered cell maturation. From infancy throughout adulthood, elevated testosterone and estradiol, low inhibin B and anti-Müllerian hormone, and completely suppressed follicle-stimulating hormone and luteinizing hormone were noted. Height acceleration and advanced bone age resulted in a reduced final height. Semen analysis revealed ongoing spermatogenesis, and the patient fathered a child by natural conception. Ketoconazole treatment decreased circulating testosterone in childhood, supported by experimental suppression of testosterone production in his adult testis tissue cultured ex vivo. At 25 years of age, ultrasound revealed a testicular tumor, identified as a Leydig cell adenoma, but unexpectedly with GCNIS present in adjacent seminiferous tubules.
The case illustrates that absence of gonadotropins but high intratesticular testosterone concentration is sufficient for spermatogenesis and to allow fatherhood. Our study is also the first description, to our knowledge, of GCNIS in a patient with testotoxicosis. We recommend regular clinical examination and ultrasonic evaluation of the testes in these patients due to potential increased risk of malignancy.
Familial gonadotropin-independent male-limited sexual precocity, also known as testotoxicosis (Online Mendelian Inheritance in Man 176410), is an autosomal-dominant disorder causing early-onset peripheral precocious puberty in boys. This condition is predominantly caused by an activating mutation in the luteinizing hormone receptor (LHR) gene (LHCGR), which causes constitutive activation of the LHR, stimulating testosterone secretion in Leydig cells, despite suppression of luteinizing hormone (LH). Current treatment regimens include aromatase inhibitors and androgen receptor blockade, but little is known about long-term consequences of testotoxicosis, especially in cases with persistent activation of LHR during fetal development. We therefore present data from a 25-year detailed clinical follow-up, from infancy throughout adulthood, in a patient with testotoxicosis. The follow-up included regular measurements of height, reproductive hormones, testicular ultrasound, histological characterization of testicular phenotype, and semen analysis in adult life.
Early Medical History
The index patient was the first-born child of healthy nonconsanguineous parents. At 10 months of age, pubic hair was observed, and growth of genitals, hands, and feet was noticed. Serum testosterone was markedly elevated, reached adult levels of 15.2 nmol/L, and increased further to 35.1 nmol/L at 1.2 years of age. Longitudinal follicle-stimulating hormone (FSH), LH, inhibin B, estradiol, and testosterone levels and testicular volume are shown in Fig. 1. At 7 years of age, linear growth and bone age was ∼15 years (Supplemental Fig. 1). Based upon clinical, biochemical, and histological evaluation of a testicular biopsy performed at 1.5 years of age (detailed later in text), the patient was diagnosed with testotoxicosis. Genetic analyses revealed parents with normal genotypes, but their son with a germline de novo heterozygous G to T mutation at nucleotide 1732, resulting in substitution of Asp578 with Tyr, as previously published (1). In early childhood, he was treated with high-dose ketoconazole, increasing to 500 mg daily with a corresponding 63% decrease in circulating testosterone (from 35.1 nmol/L to 12.9 nmol/L). However, ketoconazole resulted in adrenal insufficiency, which necessitated hydrocortisone treatment, and later in hepatic impairment, demanding alternative antihormonal treatment. During childhood and puberty, the patient was treated with various combinations of available medications, including gonadotropin-releasing hormone (GnRH) agonist, medroxyprogesterone acetate, cyproterone acetate, testolactone, and tamoxifen. When letrozole became available, this was administered also (Fig. 1). Overall, treatment resulted in some regression of the clinical manifestations of precocious puberty. From age 7 to 12 years, the patient was treated with growth hormone in an attempt to increase final adult height.
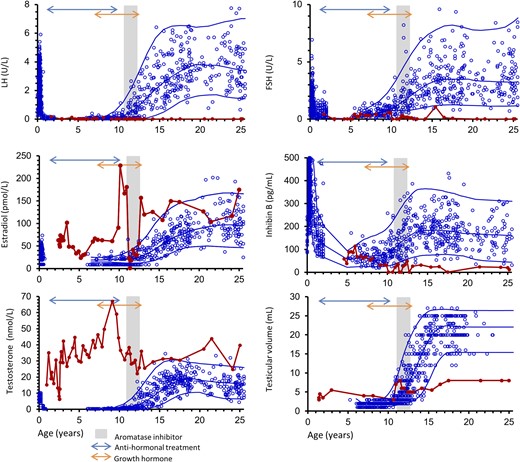
Longitudinal serum concentrations of LH, FSH, estradiol, inhibin B, and testosterone as well as testicular volume measured by orchidometer (red line) are shown according to age and reference ranges based on healthy male children, adolescents, and adults (blue dots). Blue lines represent mean ± 2 standard deviation. Antihormonal and growth hormone treatments are indicated with arrows, and the time period with aromatase inhibition is illustrated by the gray shaded area. Antihormonal treatment included various combinations of GnRH agonist, medroxyprogesterone acetate, cyproterone acetate, testolactone, and tamoxifen.
Adult Auxological, Body Composition, and Endocrine Parameters
At 18 years of age, adult height reached 164 cm (target height 172.6 cm), weight was 52 kg, and body mass index 19.3 kg/m2. Bone mineral density (measured by dual-energy x-ray absorptiometry) was normal (T-score L1–L4; −0.8 standard deviation), whereas serum levels of testosterone were 39.8 nmol/L, estradiol 175 pmol/L, sex hormone–binding globulin 25 nmol/L, inhibin B 12 pg/mL, anti-Müllerian hormone 23 pmol/L, FSH <0.05 IU/L, and LH <0.05 IU/L. Other endocrine and metabolic parameters were normal, illustrated by serum concentration of: prolactin, 191 IU/L; insulin-like growth factor-I, 175 ng/mL; IGFBP-3, 3657 ng/mL; cholesterol, 3.8 mmol/L; high-density lipoprotein, 0.98 mmol/L; low-density lipoprotein, 2.3 mmol/L; triglyceride, 1.81 mmol/L; and hemoglobin A1c, 37 mmol/mol.
Testicular Function in Adulthood
At the age of 18 years, testicular volume was 8 mL, and semen quality was impaired, with sperm concentration 3.4 million/mL, semen volume 2.2 mL, progressive motility 14%, and total motility 21%. Therefore, cryopreservation of semen was performed. However, at the age of 23.8 years, the patient fathered a son conceived by natural conception. The son also presented with signs of virilization in infancy, and investigations revealed advanced bone age, elevated serum testosterone of 18.4 nmol/L, and undetectable LH (<0.05 U/L). An identical LHCGR mutation was demonstrated, and he was successfully treated with antiandrogen bicalutamide and aromatase inhibitor letrozole.
The index patient was followed with testicular ultrasonography at regular intervals, and, at the age of 25 years, a suspicious mass in the left testis was detected. The tumor was excised, and histological evaluation showed a Leydig cell adenoma. Unexpectedly, in the testicular tissue adjacent to the adenoma, a preinvasive stage of testicular cancer, germ cell neoplasia in situ (GCNIS; detailed later in text), was detected, and orchiectomy was subsequently performed. A contralateral biopsy was taken at the same time, which showed Leydig cell hyperplasia but no signs of GCNIS.
Histopathological Findings in the Testes
Bilateral testicular biopsies taken at the age of 1.5 years showed extensive Leydig cell hyperplasia with fully differentiated Leydig cells, seminiferous tubules with partially differentiated Sertoli cells, spermatogonia, and a few primary spermatocytes that had entered meiosis, but no spermatids were present. A detailed immunohistochemical profiling of Leydig cell markers (2) was conducted later on these archived specimens. Accelerated maturation of Leydig cells was evident compared with age-matched control, illustrated by marked expression of INSL3 and CYP11A1, although presence of immature Leydig cells characterized by DLK1 expression and possibly persisting fetal Leydig cells (SULT2A1 expression) was also found (Fig. 2). Sertoli cell maturation was also more advanced, illustrated by the early loss of anti-Müllerian hormone expression, absence of podoplanin (D2-40), and early expression of the androgen receptor known to be expressed in mature Sertoli cells (Supplemental Table 1).

Histology and selected immunohistochemical staining of testicular biopsies in the patient with testotoxicosis at 1.5 years of age as well as in the orchiectomy specimen and contralateral biopsy at 25 years of age, note the difference when compared with matched controls. Top panels show testis biopsies from patient as a child and adult compared with matched controls (adult specimen: contralateral biopsy with unaffected tissue). Green circles mark intertubular areas with Leydig cells; blue circles mark tubuli with Sertoli and germ cells. Note a remarkable expansion of Leydig cells in patient. In childhood, the patient had advanced Leydig cell maturation compared with control (marked by strong expression of INSL3) but in adulthood, the Leydig cells were immature compared with control (marked by strong expression of DLK1 and low expression of INSL3). Bottom panels show tissue from patient at 25 years of age in an area adjacent to a Leydig cell tumor with GCNIS. In addition to typical tubules containing GCNIS cells (positive for D2-40 and OCT4; left panels), an atypical pattern was observed in a subpopulation: GCNIS cells with smaller nuclei and retained high positivity for MAGE-A4 (right panel, marked by red arrows). CYP11A1, marker of steroidogenesis in Leydig cells; D2-40, marker of immature Sertoli cells and GCNIS cells; DLK1, marker of immature Leydig cells; INSL3, marker of mature Leydig cells; MAGE-A4, spermatogonia marker; OCT4: marker of GCNIS cells; PLAP, marker of GCNIS cells. *Control tissue from adult with seminoma; shown is the unaffected area adjacent to the tumor.
The histopathological analysis of the testis in adulthood revealed persisting clusters of immature Leydig cells characterized by expression of DLK1 (Fig. 2) compared with adult control.
Unexpectedly, within the mass of Leydig cell adenoma, seminiferous tubules with abnormal germ cells were recognized. All cells displayed the expression profile pathognomonic of GCNIS (3), and most of these cells had large irregular nuclei and were negative for MAGE-A4. However, a subset of tubules contained OCT4+/PLAP+ germ cells with morphological characteristics comparable with normal spermatogonia and a strong MAGE-A4+ staining (Fig. 2; Supplemental Table 1).
Experimental Confirmation of Testosterone Suppression by Ex Vivo Ketoconazole Treatment
Testis tissue from the patient was retrieved after orchiectomy and cultured using an ex vivo hanging-drop approach for 5 days [methodology described in detail previously (4)] and treated with ketoconazole 10 µmol/L for 72 hours, alongside control tissue from another patient undergoing orchiectomy due to testicular cancer, in which only healthy tissue adjacent to the tumorous area was cultured. A marked decrease in production of testosterone, determined by liquid chromatography–tandem mass spectrometry (5), was observed in both control (Δ testosterone −271%) and index patient (Δ testosterone −44%) after ketoconazole treatment.
Discussion
Activating mutations in LHCGR leads to early virilization, but less focus has been on the consequences during adulthood. This case describes an unusual and protracted outcome of testotoxicosis. The findings in infancy with Leydig cell hyperplasia, premature Sertoli cell maturation, and start of meiosis have previously been described in boys with precocious puberty due to activating LHR germline mutations (6, 7). However, unlike just one case with Leydig cell adenoma in a patient with somatic LHR mutation, the adult presentation of GCNIS has, to our knowledge, never been reported (8). The presence of GCNIS was surprising, but a literature search revealed one study reporting seminoma in a patient with an activating LHR mutation (9). Seminomas and nonseminomas in young adults originate from GCNIS (10). Interestingly, our patient harbored an atypical GCNIS, with the presence of both regular GCNIS cells and a subpopulation of GCNIS cells with smaller nuclei and also expressing the spermatogonial marker MAGE-A4, suggestive of a less invasive or transitional stage of malignancy (11). Immunohistological evaluation strongly indicated an accelerated differentiation of Leydig cells, although different populations were detected, illustrated by concomitant presence of mature-, immature-, and fetal-like Leydig cells. Regular ultrasonic evaluation revealed what might have been a slow transformation from Leydig cell hyperplasia to Leydig cell adenoma in our patient, but whether such a transformation is part of the etiology of testotoxicosis remains unknown.
The hallmark of testotoxicosis is excessive testosterone production. The index patient was treated with ketoconazole to successfully inhibit circulating testosterone in vivo during childhood. Using testicular specimens from this patient in an ex vivo model, we have confirmed the effect of this treatment in testis with constitutive activation of LHR. However, in the postpubertal male, intratesticular testosterone concentration is >100 times higher than in serum, and our patient may therefore have been exposed to abnormally increased testosterone levels peripherally as well as locally in the testes, most likely already during fetal development. The consequences of this balance for germ cell function are largely unknown, but an imbalance in steroidogenesis and accelerated puberty could potentially disrupt gonocyte differentiation and promote neoplastic changes in germ cells. Conversely, the high intratesticular testosterone has most likely maintained spermatogenesis in light of lifelong levels of gonadotropins below levels of detection. Our clinical observations are in line with experimental studies in mice showing that androgen alone can stimulate spermatogenesis in GnRH and LHR knockout animals presenting with low testosterone and azoospermia, in which testosterone replacement was able to restore spermatogenesis to produce offspring (12).
Conclusion
This case illustrates that spermatogenesis is maintained at a level to produce offspring by natural conception despite lif-long suppression of gonadotropins in patients with testotoxicosis. Our findings support data from animal models indicating that high intratesticular testosterone concentrations may induce spermatogenesis even in the absence of gonadotropins. Moreover, we show in this report a patient with testotoxicosis in whom precancerous lesions (GCNIS) were detected by routine follow-up before overt testicular cancer had developed. The exact mechanism for this link remains to be determined, but we suggest regular clinical examination and ultrasonic evaluation of the testes in such patients due to a potential increased risk of testicular malignancy.
Abbreviations:
- FSH
follicle-stimulating hormone
- GCNIS
germ cell neoplasia in situ
- GnRH
gonadotropin-releasing hormone
- LH
luteinizing hormone
- LHCGR
luteinizing hormone receptor gene
- LHR
luteinizing hormone receptor.
Acknowledgments
Financial Support: This work was supported by the International Center for Research and Research Training in Endocrine Disruption of Male Reproduction and Child Health.
Disclosure Summary: The authors have nothing to disclose.
References

{kind=link}
{kind=link}