





Abstract
LHX4 encodes a LIM-homeodomain transcription factor that is implicated in early pituitary development. In humans, only 13 heterozygous LHX4 mutations have been associated with congenital hypopituitarism.
The aims of this study were to evaluate the prevalence of LHX4 mutations in patients with hypopituitarism, to define the associated phenotypes, and to characterize the functional impact of the identified variants and the respective role of the 2 LIM domains of LHX4.
We screened 417 unrelated patients with isolated growth hormone deficiency or combined pituitary hormone deficiency associated with ectopic posterior pituitary and/or sella turcica anomalies for LHX4 mutations (Sanger sequencing). In vitro studies were performed to assess the functional consequences of the identified variants.
We identified 7 heterozygous variations, including p.(Tyr131*), p.(Arg48Thrfs*104), c.606+1G>T, p.Arg65Val, p.Thr163Pro, p.Arg221Gln, and p.Arg235Gln), that were associated with variable expressivity; 5 of the 7 were also associated with incomplete penetrance. The p.(Tyr131*), p.(Arg48Thrfs*104), p.Ala65Val, p.Thr163Pro, and p.Arg221Gln LHX4 variants are unable to transactivate the POU1F1 and GH promoters. As suggested by transactivation, subcellular localization, and protein-protein interaction studies, p.Arg235Gln is probably a rare polymorphism. Coimmunoprecipitation studies identified LHX3 as a potential protein partner of LHX4. As revealed by functional studies of LIM-defective recombinant LHX4 proteins, the LIM1 and LIM2 domains are not redundant.
This study, performed in the largest cohort of patients screened so far for LHX4 mutations, describes 6 disease-causing mutations that are responsible for congenital hypopituitarism. LHX4 mutations were found to be associated with variable expressivity, and most of them with incomplete penetrance; their contribution to pituitary deficits that are associated with an ectopic posterior pituitary and/or a sella turcica defect is ∼1.4% in the 417 probands tested.
Molecular defects in genes that are involved in pituitary development and function are responsible for congenital forms of isolated growth hormone deficiency (IGHD) or combined pituitary hormone deficiency (CPHD) that may be syndromic (1–7). Among them is LHX4, a member of the LIM-homeodomain (LIM-HD) family. Correct pituitary organogenesis requires the coordinated expression in both space and time of several transcription factors including LHX4.
The anterior pituitary originates from Rathke’s pouch, an invagination of the oral ectoderm that derives from the most anterior part of the neural ridge. In the mouse, Lhx4 acts with its close paralog Lhx3 to regulate Rathke’s pouch maturation through cellular proliferation and differentiation of pituitary lineages (somatotroph, lactotroph, tyrotroph, gonadotroph, and corticotroph). Indeed, Rathke’s pouch in mice with a homozygous invalidation of Lhx4 and Lhx3 remains primitive (i.e., pouch rudiment), whereas 1 wild-type allele of either Lhx4 or Lhx3 is sufficient to form a definitive pouch (8). Mice with a homozygous Lhx4 invalidation present anterior pituitary hypoplasia and die shortly after birth, whereas heterozygous mutants are asymptomatic (9). Further studies of these knockout mice described a morphological anomaly of the sphenoid bone cartilage unable to close properly underneath the pituitary gland (10). This latter feature was in fact initially reported in the first patient identified with an LHX4 heterozygous mutation, who displayed a CPHD associated with a poorly developed sella turcica (7).
From a more general viewpoint, little is known about the contribution of LHX4 molecular defects to hypopituitarism or about the disease phenotype associated with those defects. To date, only 13 heterozygous LHX4 sequence variations have been reported in humans, 9 of which were assessed functionally (11–20). The patients displayed IGHD or CPHD associated with an ectopic posterior pituitary (EPP), an anterior pituitary hypoplasia, a poorly developed sella turcica, and/or a Chiari malformation. Five of these mutations have also been associated with incomplete penetrance (13, 14, 17, 18), which impedes interpretation of the variant pathogenicity. At the structural level, as a member of the LIM-HD protein family, LHX4 possesses a homeodomain and 2 double zinc-finger LIM domains. Molecular studies have shown that through its homeodomain, LHX4 can regulate pituitary-specific genes, including POU1F1, GH, PRL, αGSU, FSHβ, and TSHβ (12,13, 21–23). As for the LIM domains involved in protein-protein interactions, their function is still unclear. In the case of LHX4, the LIM domains are known to interact with at least 3 partners containing a LIM-interacting domain (LID): IFT172 (24), LDB1 (25), and ISL2 (25, 26).
The current study was performed with the aim to assess the contribution of LHX4 defects in a large cohort of unrelated patients (n = 417) with syndromic hypopituitarism and to describe the associated phenotypic spectrum. Our objective was also to evaluate the functional consequences of the identified LHX4 variants and to better characterize the potential role of the LIM domains in LHX4 biological activity.
Patients and Methods
Patients
A total of 417 unrelated patients presenting a congenital hypopituitarism associated with EPP and/or sella turcica malformations and whose samples had been referred to our laboratory from the years 2000 to 2015 for molecular investigation were studied. Detailed phenotypic data, including hormonal data and magnetic resonance imaging (MRI) of the hypothalamic-pituitary area, were collected through a standardized form. Among the 417 patients, 324 presented with an EPP, 23 had a sella turcica anomaly, and 70 presented with both defects. MRI scans were made and assessed in the Pediatric Endocrinology Departments of university hospitals; all available MRI data were provided to our laboratory. Patients or parents of patients younger than 18 years of age gave their written informed consent for genetic studies to be performed. Blood samples were collected from patients and, whenever possible, from their first-degree relatives.
Screening for LHX4 mutations
For all patients, genomic DNA was isolated from peripheral blood, and all 6 coding exons of LHX4 (NM_033343) and their flanking intronic regions were analyzed by Sanger sequencing. The amplified samples were purified by Exo-SAP (Affymetrix, Santa Clara, CA). Internal primers were used to sequence the corresponding polymerase chain reaction (PCR) products, using an ABI 3130XL automated capillary DNA sequencer (Applied Biosystems, Foster City, CA). Other genes known to be responsible for pituitary deficits with an EPP had been previously analyzed by PCR-based sequencing, and no potential mutations were found. HESX1 has been sequenced in all probands, and SOX3 has been sequenced only in male patients because this gene is located on chromosome X.
Plasmid constructs
An HA-tag was introduced at the 5′ end of the human LHX4 cDNA expression vector (pTracer-LHX4_WT-HA) (7), which was then subjected to site-directed mutagenesis using the Quick-Change kit (Stratagene; Agilent, Santa Clara, CA) to generate an empty vector (pTracer-Mock) and the following plasmids encoding the different mutated forms of LHX4: pTracer-LHX4_A65V-HA, pTracer-LHX4_T163P-HA, pTracer-LHX4_R221Q-HA, pTracer-LHX4_R235Q-HA, pTracer-LHX4_Y131*-HA, pTracer-LHX4_R48Tfs*104-HA, pTracer-LHX4_ΔLIM1-HA, pTracer-LHX4_ΔLIM2-HA, and pTracer-LHX4_C57-60A-HA, in which alanine residues substituted for zinc-associated Cys57 and Cys60. Reporter vectors encompassing sequences of the 5′-proximal promoter of the human GH (−446 to +20; NM_000515.4; +1 designates the transcription start site) and PRL (−163 to +469; NM_000948.5) genes were obtained as previously described for the POU1F1 promoter reporter vector (21). The ISL2 cDNA was generated by real-time PCR using human fetal brain RNA as a template (Clontech, Palo Alto, CA) and primers containing a FLAG-tag at the 5′ end. The amplified product was cloned into the expression vector pcDNA3.1/V5-his-TOPO (Thermo Fisher Scientific, Waltham, MA) to generate pcDNA3.1-ISL2_WT. A FLAG-tagged pcDNA3.1-LHX3_WT plasmid was also used to evaluate potential interactions with LHX4.
Cell culture and transfection
Chinese hamster ovary (CHO) cells, obtained from ATCC (Manassas, VA), and human embryonic kidney (HEK293T) cells were grown in Dulbecco’s modified Eagle medium (Gibco, Life Technologies) supplemented with 10% fetal bovine serum, penicillin, and streptomycin at 37°C under 5% CO2. CHO cells were used for luciferase activation assays because they yielded stronger signals than HEK293T cells. For other experiments, HEK293T cells were chosen because they presented higher transfection efficiency. CHO cells were transfected at approximately 80% confluence using lipofectamine reagent (Thermo Fisher Scientific) in OptiMEM medium (Gibco), according to the manufacturer’s standard protocol. In HEK293T cells, transfections were performed at approximately 80% confluence by the Fugene HD transfection reagent (Promega, Madison, WI), as recommended by the manufacturer, with a 3:1 Fugene/DNA ratio.
Western blot analyses
HEK293T cells were transfected with 3 µg of the pTracer expression vectors encoding HA-tagged LHX4 proteins. Proteins extracted from the cellular lysates were separated on 12% sodium dodecyl sulfate polyacrylamide gels, then wet-transferred to polyvinylidene difluoride membranes (Hybond, Amersham, Buckinghamshire, UK). Peroxidase-conjugated mouse anti-HA monoclonal antibody 3F10 (Roche Life Science, Indianapolis, IN) and horseradish peroxidase–conjugated glyceraldehyde 3-phosphate dehydrogenase antibody (14C10; Cell Signaling Technology, Danvers, MA) were used at 1:1,000 dilution. Results were visualized using SuperSignal West Dura substrate (Pierce Biotechnology, Rockford, IL), a chemiluminescence camera, and Quantity One software (Biorad, Hercules, CA).
Immunofluorescence analyses
pTracer-LHX4_WT-HA in amounts of 0.4 µg was transfected in HEK293T cells that were cultured for 24 hours in glass coverslips coated with poly-l-lysin (Sigma-Aldrich, St Louis, MO) in 24-well culture plates. Cells were subsequently fixed in 4% (v/v) paraformaldehyde/ phosphate-buffered saline and permeabilized in 0.25% Igepal/phosphate-buffered saline (Sigma-Aldrich). Mouse anti-HA monoclonal antibody clone H-7 (Sigma-Aldrich) was used at 1:1000 to detect HA-tagged LHX4 proteins. The secondary antibody was goat antimouse immunoglobulin G, Alexa Fluor 488 (Thermo Fisher Scientific) at 1:2000. Nuclei were stained using 0.004% DAPI (Sigma-Aldrich)/SSC 2× (Thermo Fisher Scientific) and Prolong Antifade Gold Reagent (Thermo Fisher Scientific). Cells were examined with a Nikon 80i fluorescence microscope (Nikon, Tokyo, Japan).
Luciferase activity assays
CHO cells were seeded onto 24-well plates coated with poly-l-lysin (Sigma-Aldrich) and cotransfected with 100 ng of the plasmids with the luciferase reporter driven by the POU1F1, GH, or PRL proximal promoters, and either 30 ng of empty pTracer expression vector and/or LHX4 expression vectors per well. Additional assays, assessing a potential dominant negative effect of the identified LHX4 variants, involved cotransfection of equal amount of pTracer-LHX4_WT expression vector with other LHX4-derived pTracer constructs (15 ng each), and 100 ng of the reporter plasmid. After 24 hours, cell lysates were assayed for luciferase activity using the Luciferase Assay System (Promega) on a TriStar LB 941 luminometer (Berthold Technologies, Munich, Germany). The total amount of proteins, as determined through the Pierce Coomassie Plus Protein Assay Reagent (Thermo Fisher Scientific), was used to normalize luciferase activity. Results are shown as means ± standard deviations (SDs) of 3 independent experiments performed in triplicate.
Coimmunoprecipitation assays
HEK293T cells were transfected with 3 µg of pTracer-LHX4-HA and 3 µg of either pcDNA3.1-ISL2_WT-FLAG or pcDNA3.1-LHX3_WT-FLAG in 100-mm dishes. Cells were harvested 24 hours after transfection, and protein extracts were divided into 2 equal parts, 1 part for immunoprecipitation with a mouse anti-HA monoclonal antibody (Sigma-Aldrich) or a mouse anti-LHX4 monoclonal antibody (E10; Santa Cruz Biotechnology, Santa Cruz, CA), and the other part for immunoprecipitation with a mouse anti-FLAG monoclonal antibody (Sigma-Aldrich). For each part, 10% of protein extracts were kept as input for quantification. Immunoprecipitation was achieved overnight at 4°C, and protein complexes were collected using protein G-coated magnetic beads from the Universal Magnetic Co-IP kit (Active Motif, Carlsbad, CA). Protein samples were then resolved by western blotting, as mentioned previously.
Results
Spectrum of LHX4 variants in the cohort of 417 patients with hypopituitarism associated with ectopic posterior pituitary and/or sella turcica anomalies
The screening of the 6 coding exons of LHX4 and their flanking intronic sequences led to the identification of previously unreported LHX4 variants in 7 of the 417 patients (1.7%; Figs. 1 and 2).
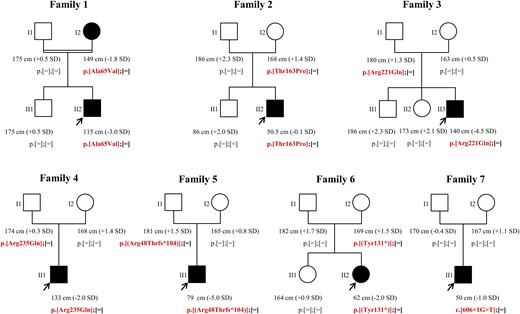
Pedigree, phenotype, and genotype of the 7 families in which an LHX4 variation was identified. Filled symbols represent subjects with pituitary deficits, and empty symbols represent healthy subjects. The proband heights were before recombinant human GH treatment. Arrows represent index cases.
![Identified LHX4 variants and in silico analysis. (A) Representation of LHX4 variants. Missense (in black) and putative truncated (in gray) variants identified in our cohort. (B) LIM1 domain is schematically represented with its 2 zinc-finger motifs. Cysteines 57 and 60 are represented in dark gray. (C) Alignments from Clustal Omega [online tool from The European Bioinformatics Institute (EMBL-EBI), http://www.ebi.ac.uk/Tools/msa/clustalo/] show highly conserved amino acid sequence of LHX4 between species (from Homo sapiens to Drosophila melanogaster). Missense variations (framed in red) target evolutionarily conserved amino acids. Fully conserved residue (*); strongly similar properties (:); weakly similar properties (.). LIM, LIM domain; NLS, putative nuclear localization signal.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jcem/102/1/10.1210_jc.2016-3158/3/m_jc.2016-3158f2.jpeg?Expires=1747902231&Signature=bgu2~eHFayb6T~MvEIFESfMnVxfeeGV45ddLkNQtfDuHEqSCVfCvLaYMbGNSOiRt28zy6iyxugZeZs-oPeXWAzs~uUjYOxJAiLDh8p3huf6GuWFp1p4WsWX2PTZtBUJ-QT1s13Rc6d5xLKFkViktkN0zlNYTyT4REX55bbc8wIfZZMcKoC7e--F8mMRojjnkpE7vn-YjvwQgWaMgLUgpdLz9Hs4DrQbWc4-dEH58vedBz9z1niFv~J2SHFq3FsDTcr1Y6EdyDs1EDnBEh-6wOkmJk-1-4Wi-23oNgk-tuF-Vt6AR2xU8~wO6J-9N3UNCwwH2n4FPz9P8l1nXEVHFeQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Identified LHX4 variants and in silico analysis. (A) Representation of LHX4 variants. Missense (in black) and putative truncated (in gray) variants identified in our cohort. (B) LIM1 domain is schematically represented with its 2 zinc-finger motifs. Cysteines 57 and 60 are represented in dark gray. (C) Alignments from Clustal Omega [online tool from The European Bioinformatics Institute (EMBL-EBI), http://www.ebi.ac.uk/Tools/msa/clustalo/] show highly conserved amino acid sequence of LHX4 between species (from Homo sapiens to Drosophila melanogaster). Missense variations (framed in red) target evolutionarily conserved amino acids. Fully conserved residue (*); strongly similar properties (:); weakly similar properties (.). LIM, LIM domain; NLS, putative nuclear localization signal.
Phenotype of patients with identified LHX4 allelic variations
Family 1
The propositus (II2), born to a consanguineous union, was referred to an endocrinologist for short stature (−3 SD). He was diagnosed with a transient somatotroph deficiency, as already described in patients with inherited IGHD (27). Pituitary MRI revealed an EPP and no anomaly of the sella turcica (Table 1). Sanger sequencing of LHX4 revealed that he carried the c.194C>T mutation in the heterozygous state, predicting an amino acid substitution at codon 65 (p.Ala65Val), within the first LIM domain of LHX4 [Fig. 1 and Fig. 2(A) and 2(B)] The patient’s mother, who also carries the mutation in the heterozygous state, presented a normal height but close to −2 SD (149 cm; −1.8 SD) and low insulin-like growth factor 1 levels (110 ng/mL; normal range, 100 to 295).
Phenotype and Genotype Data of the Index Cases From the 7 Families With Identified LHX4 Variations
| Family 1, II2 | Family 2, II2 | Family 3, II1 | Family 4, II1 | Family 5, II1 | Family 6, II2 | Family 7, II1 | |
|---|---|---|---|---|---|---|---|
| Clinical phenotype | |||||||
| Height at diagnosis in SD (cm; age) | −3 SD (118; 9 y, 6 mo) | −0.1 SD (50.5; 9 d) | −4.5 SD (140.5; 15 y, 2 mo) | −1.85 SD (133; 2 y, 6 mo) | −5 SD (79; 2 y, 6 mo) | −2 SD (62; 8 mo) | −1 SD (50; 1 d) |
| Pituitary deficit(s) | GH (transient) | GH, TSH, FSH/LH, ACTH | GH, FSH/LHa | GH, TSH | GH, TSH, FSH/LH, ACTH | GH, PRL, TSH, FSH/LH, ACTH | GH, TSH, ACTH |
| Other feature(s) | Micropenis | Aortic coarctation, pyelocaliceal duplicity, micropenis | Arched palate | Bilateral papillary coloboma | None | Epilepsy | Type I Chiari, rounded forehead, micropenis |
| Pituitary imaging | |||||||
| Anterior pituitary | Normal | Hypoplastic | Normal | NA | Hypoplastic | Aplastic | Hypoplastic |
| Posterior pituitary | Ectopic | Ectopic | Ectopic (double) | Ectopic | Eutopic | Eutopic | Ectopic (double) |
| Sella turcica | Normal | Shallow | Shallow | Sphenoid bone dehiscence | Hypoplastic | Normal | Shallow |
| Endocrine explorations | |||||||
| GH/insulin, ng/mL (basal and +60 min) | NA and 1.5 | 5.74 and NA | NA | NA | NA | < 2 and 0.95 | < 0.1b |
| GH/glucagon, mIU/L (basal and +60 min) | NA and 10 | NA | < 0.5 and 0.7 | NA | 0.36 and 0.72 | NA and 3 | NA |
| GH/ornithine, ng/mL (basal and +60 min) | NA | NA | NA | 0.3 and 2.2 | NA | NA | NA |
| GH/L-Dopa, ng/mL (basal and +60 min) | NA | NA | NA | < 0.1 and 0.2 | NA | NA | NA |
| IGF-I, ng/mL (normal values) | 42 (> 100) | 31 (20–120) | 23 (140–496) | 10 (62–308) | 53 (24–144) | 11 (103–391) | NA |
| TSH, mIU/L (normal values) | 1.4 (0.5–6) | 7.92 (1–10) | 1.81 (0.6–4) | 0.8 (0.4–4) | 4.43 (0.3–3.8) | 0.01 (0.1–4) | 0.06 (0.1–4) |
| FT4, pmol/L (normal values) | 19.3 (> 10) | 7.45 (10–22) | 10.07 (9–15) | 11.2 (12–21.9) | 3.3 (6–15.5) | 19 (10–25) | 3.8 (7–20) |
| FSH, mIU/mL (normal values; age) | 0.8 (< 2; 9 y, 6 mo) | 5.8 (0.4–5; 1 mo, 10 d) | 0.4 (0.2–5.6; 15 y, 2 mo) | < 0.4 (NA; 3 y, 9 mo) | 1.19 (1–10.5; 2 y, 6 mo) | 1.1 (2–9; 21 y, 7 mo) | < 0.5 (NA; 1 d) |
| LH, mIU/mL (normal values; age) | 0.1 (< 0.1; 9 y, 6 mo) | 2.1 (0.5–6; 1 mo, 10 d) | 0.2 (0.2–2; 15 y, 2 mo) | < 0.2 (NA; 3 y, 9 mo) | 0.25 (1–8.4; 2 y, 6 mo) | 0.7 (1–5; 21 y, 7 mo) | < 0.5 (NA; 1 d) |
| Testosterone, ng/mL (normal values; age) | 0.6 (< 0.1; 9 y, 6 mo) | 0.15 (0.5–3.5; 1 mo, 10 d) | < 0.07 (< 0.5; 15 y, 2 mo) | < 0.14 (0.05–0.09; 8 y, 9 mo) | 0.1 (> 12; 14 y, 6 mo) | — | 0.03 (NA; 1 d) |
| Cortisol/synacthen, ng/mL: basal at 8 am and +60 min (normal values at +60 min) | 152 and 282 (> 150) | 27 and 125 (> 180) | 142.3 and 199.2 (> 180) | 135 and 269c (> 200) | 53 and 169 (>200) | 83 and 98b (> 180) | < 11b |
| Molecular data | |||||||
| Variation (nucleotide) | c.194C>T | c.487A>C | c.662G>A | c.704G>A | c.143_144delGA | c.393C>G | c.606+1G>T |
| Variation (protein) | p.Ala65Val | p.Thr163Pro | p.Arg221Gln | p.Arg235Gln | p.(Arg48Thrfs*104) | p.(Tyr131*) | — |
| Genotype | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous |
| Consanguinity | Yes | No | No | No | No | No | No |
| Conservation | Vertebrates | Invariant | Invariant | Mammals | — | — | — |
| Location | LIM1 | Homeodomain | — | — | — | — | — |
| Allele frequency | |||||||
| 1000 Genomes | 0 | 0 | 2 × 10−4 | 0 | 0 | 0 | 0 |
| ExAC | 8 × 10−6 | 0 | 10−5 | 5 × 10−5 | 0 | 0 | 0 |
| Family 1, II2 | Family 2, II2 | Family 3, II1 | Family 4, II1 | Family 5, II1 | Family 6, II2 | Family 7, II1 | |
|---|---|---|---|---|---|---|---|
| Clinical phenotype | |||||||
| Height at diagnosis in SD (cm; age) | −3 SD (118; 9 y, 6 mo) | −0.1 SD (50.5; 9 d) | −4.5 SD (140.5; 15 y, 2 mo) | −1.85 SD (133; 2 y, 6 mo) | −5 SD (79; 2 y, 6 mo) | −2 SD (62; 8 mo) | −1 SD (50; 1 d) |
| Pituitary deficit(s) | GH (transient) | GH, TSH, FSH/LH, ACTH | GH, FSH/LHa | GH, TSH | GH, TSH, FSH/LH, ACTH | GH, PRL, TSH, FSH/LH, ACTH | GH, TSH, ACTH |
| Other feature(s) | Micropenis | Aortic coarctation, pyelocaliceal duplicity, micropenis | Arched palate | Bilateral papillary coloboma | None | Epilepsy | Type I Chiari, rounded forehead, micropenis |
| Pituitary imaging | |||||||
| Anterior pituitary | Normal | Hypoplastic | Normal | NA | Hypoplastic | Aplastic | Hypoplastic |
| Posterior pituitary | Ectopic | Ectopic | Ectopic (double) | Ectopic | Eutopic | Eutopic | Ectopic (double) |
| Sella turcica | Normal | Shallow | Shallow | Sphenoid bone dehiscence | Hypoplastic | Normal | Shallow |
| Endocrine explorations | |||||||
| GH/insulin, ng/mL (basal and +60 min) | NA and 1.5 | 5.74 and NA | NA | NA | NA | < 2 and 0.95 | < 0.1b |
| GH/glucagon, mIU/L (basal and +60 min) | NA and 10 | NA | < 0.5 and 0.7 | NA | 0.36 and 0.72 | NA and 3 | NA |
| GH/ornithine, ng/mL (basal and +60 min) | NA | NA | NA | 0.3 and 2.2 | NA | NA | NA |
| GH/L-Dopa, ng/mL (basal and +60 min) | NA | NA | NA | < 0.1 and 0.2 | NA | NA | NA |
| IGF-I, ng/mL (normal values) | 42 (> 100) | 31 (20–120) | 23 (140–496) | 10 (62–308) | 53 (24–144) | 11 (103–391) | NA |
| TSH, mIU/L (normal values) | 1.4 (0.5–6) | 7.92 (1–10) | 1.81 (0.6–4) | 0.8 (0.4–4) | 4.43 (0.3–3.8) | 0.01 (0.1–4) | 0.06 (0.1–4) |
| FT4, pmol/L (normal values) | 19.3 (> 10) | 7.45 (10–22) | 10.07 (9–15) | 11.2 (12–21.9) | 3.3 (6–15.5) | 19 (10–25) | 3.8 (7–20) |
| FSH, mIU/mL (normal values; age) | 0.8 (< 2; 9 y, 6 mo) | 5.8 (0.4–5; 1 mo, 10 d) | 0.4 (0.2–5.6; 15 y, 2 mo) | < 0.4 (NA; 3 y, 9 mo) | 1.19 (1–10.5; 2 y, 6 mo) | 1.1 (2–9; 21 y, 7 mo) | < 0.5 (NA; 1 d) |
| LH, mIU/mL (normal values; age) | 0.1 (< 0.1; 9 y, 6 mo) | 2.1 (0.5–6; 1 mo, 10 d) | 0.2 (0.2–2; 15 y, 2 mo) | < 0.2 (NA; 3 y, 9 mo) | 0.25 (1–8.4; 2 y, 6 mo) | 0.7 (1–5; 21 y, 7 mo) | < 0.5 (NA; 1 d) |
| Testosterone, ng/mL (normal values; age) | 0.6 (< 0.1; 9 y, 6 mo) | 0.15 (0.5–3.5; 1 mo, 10 d) | < 0.07 (< 0.5; 15 y, 2 mo) | < 0.14 (0.05–0.09; 8 y, 9 mo) | 0.1 (> 12; 14 y, 6 mo) | — | 0.03 (NA; 1 d) |
| Cortisol/synacthen, ng/mL: basal at 8 am and +60 min (normal values at +60 min) | 152 and 282 (> 150) | 27 and 125 (> 180) | 142.3 and 199.2 (> 180) | 135 and 269c (> 200) | 53 and 169 (>200) | 83 and 98b (> 180) | < 11b |
| Molecular data | |||||||
| Variation (nucleotide) | c.194C>T | c.487A>C | c.662G>A | c.704G>A | c.143_144delGA | c.393C>G | c.606+1G>T |
| Variation (protein) | p.Ala65Val | p.Thr163Pro | p.Arg221Gln | p.Arg235Gln | p.(Arg48Thrfs*104) | p.(Tyr131*) | — |
| Genotype | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous |
| Consanguinity | Yes | No | No | No | No | No | No |
| Conservation | Vertebrates | Invariant | Invariant | Mammals | — | — | — |
| Location | LIM1 | Homeodomain | — | — | — | — | — |
| Allele frequency | |||||||
| 1000 Genomes | 0 | 0 | 2 × 10−4 | 0 | 0 | 0 | 0 |
| ExAC | 8 × 10−6 | 0 | 10−5 | 5 × 10−5 | 0 | 0 | 0 |
The em dash (—) indicates irrelevant.
Abbreviations: ACTH, adrenocorticotropic hormone; ExAC, Exome Aggregation Consortium; FSH, follicle-stimulating hormone; FT4, free thryoxine; IGF-I, insulin-like growth factor I; LH, luteinizing hormone; NA, not available; PRL, prolactin; TSH, thyrotropin.
aIn patient 3, low FSH, LH, and testosterone levels at the age of 15 years, 2 months may be linked to a pubertal delay rather than to hypogonadotropic hypogonadism. This patient indeed showed normal testosterone levels at 19 years of age (5 ng/mL; normal range, 3.5 to 11.5 ng/mL), more than 1 year after testosterone treatment cessation.
bTest was performed when patient had severe spontaneous hypoglycemia.
cDynamic test was performed with ornithine.
Phenotype and Genotype Data of the Index Cases From the 7 Families With Identified LHX4 Variations
| Family 1, II2 | Family 2, II2 | Family 3, II1 | Family 4, II1 | Family 5, II1 | Family 6, II2 | Family 7, II1 | |
|---|---|---|---|---|---|---|---|
| Clinical phenotype | |||||||
| Height at diagnosis in SD (cm; age) | −3 SD (118; 9 y, 6 mo) | −0.1 SD (50.5; 9 d) | −4.5 SD (140.5; 15 y, 2 mo) | −1.85 SD (133; 2 y, 6 mo) | −5 SD (79; 2 y, 6 mo) | −2 SD (62; 8 mo) | −1 SD (50; 1 d) |
| Pituitary deficit(s) | GH (transient) | GH, TSH, FSH/LH, ACTH | GH, FSH/LHa | GH, TSH | GH, TSH, FSH/LH, ACTH | GH, PRL, TSH, FSH/LH, ACTH | GH, TSH, ACTH |
| Other feature(s) | Micropenis | Aortic coarctation, pyelocaliceal duplicity, micropenis | Arched palate | Bilateral papillary coloboma | None | Epilepsy | Type I Chiari, rounded forehead, micropenis |
| Pituitary imaging | |||||||
| Anterior pituitary | Normal | Hypoplastic | Normal | NA | Hypoplastic | Aplastic | Hypoplastic |
| Posterior pituitary | Ectopic | Ectopic | Ectopic (double) | Ectopic | Eutopic | Eutopic | Ectopic (double) |
| Sella turcica | Normal | Shallow | Shallow | Sphenoid bone dehiscence | Hypoplastic | Normal | Shallow |
| Endocrine explorations | |||||||
| GH/insulin, ng/mL (basal and +60 min) | NA and 1.5 | 5.74 and NA | NA | NA | NA | < 2 and 0.95 | < 0.1b |
| GH/glucagon, mIU/L (basal and +60 min) | NA and 10 | NA | < 0.5 and 0.7 | NA | 0.36 and 0.72 | NA and 3 | NA |
| GH/ornithine, ng/mL (basal and +60 min) | NA | NA | NA | 0.3 and 2.2 | NA | NA | NA |
| GH/L-Dopa, ng/mL (basal and +60 min) | NA | NA | NA | < 0.1 and 0.2 | NA | NA | NA |
| IGF-I, ng/mL (normal values) | 42 (> 100) | 31 (20–120) | 23 (140–496) | 10 (62–308) | 53 (24–144) | 11 (103–391) | NA |
| TSH, mIU/L (normal values) | 1.4 (0.5–6) | 7.92 (1–10) | 1.81 (0.6–4) | 0.8 (0.4–4) | 4.43 (0.3–3.8) | 0.01 (0.1–4) | 0.06 (0.1–4) |
| FT4, pmol/L (normal values) | 19.3 (> 10) | 7.45 (10–22) | 10.07 (9–15) | 11.2 (12–21.9) | 3.3 (6–15.5) | 19 (10–25) | 3.8 (7–20) |
| FSH, mIU/mL (normal values; age) | 0.8 (< 2; 9 y, 6 mo) | 5.8 (0.4–5; 1 mo, 10 d) | 0.4 (0.2–5.6; 15 y, 2 mo) | < 0.4 (NA; 3 y, 9 mo) | 1.19 (1–10.5; 2 y, 6 mo) | 1.1 (2–9; 21 y, 7 mo) | < 0.5 (NA; 1 d) |
| LH, mIU/mL (normal values; age) | 0.1 (< 0.1; 9 y, 6 mo) | 2.1 (0.5–6; 1 mo, 10 d) | 0.2 (0.2–2; 15 y, 2 mo) | < 0.2 (NA; 3 y, 9 mo) | 0.25 (1–8.4; 2 y, 6 mo) | 0.7 (1–5; 21 y, 7 mo) | < 0.5 (NA; 1 d) |
| Testosterone, ng/mL (normal values; age) | 0.6 (< 0.1; 9 y, 6 mo) | 0.15 (0.5–3.5; 1 mo, 10 d) | < 0.07 (< 0.5; 15 y, 2 mo) | < 0.14 (0.05–0.09; 8 y, 9 mo) | 0.1 (> 12; 14 y, 6 mo) | — | 0.03 (NA; 1 d) |
| Cortisol/synacthen, ng/mL: basal at 8 am and +60 min (normal values at +60 min) | 152 and 282 (> 150) | 27 and 125 (> 180) | 142.3 and 199.2 (> 180) | 135 and 269c (> 200) | 53 and 169 (>200) | 83 and 98b (> 180) | < 11b |
| Molecular data | |||||||
| Variation (nucleotide) | c.194C>T | c.487A>C | c.662G>A | c.704G>A | c.143_144delGA | c.393C>G | c.606+1G>T |
| Variation (protein) | p.Ala65Val | p.Thr163Pro | p.Arg221Gln | p.Arg235Gln | p.(Arg48Thrfs*104) | p.(Tyr131*) | — |
| Genotype | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous |
| Consanguinity | Yes | No | No | No | No | No | No |
| Conservation | Vertebrates | Invariant | Invariant | Mammals | — | — | — |
| Location | LIM1 | Homeodomain | — | — | — | — | — |
| Allele frequency | |||||||
| 1000 Genomes | 0 | 0 | 2 × 10−4 | 0 | 0 | 0 | 0 |
| ExAC | 8 × 10−6 | 0 | 10−5 | 5 × 10−5 | 0 | 0 | 0 |
| Family 1, II2 | Family 2, II2 | Family 3, II1 | Family 4, II1 | Family 5, II1 | Family 6, II2 | Family 7, II1 | |
|---|---|---|---|---|---|---|---|
| Clinical phenotype | |||||||
| Height at diagnosis in SD (cm; age) | −3 SD (118; 9 y, 6 mo) | −0.1 SD (50.5; 9 d) | −4.5 SD (140.5; 15 y, 2 mo) | −1.85 SD (133; 2 y, 6 mo) | −5 SD (79; 2 y, 6 mo) | −2 SD (62; 8 mo) | −1 SD (50; 1 d) |
| Pituitary deficit(s) | GH (transient) | GH, TSH, FSH/LH, ACTH | GH, FSH/LHa | GH, TSH | GH, TSH, FSH/LH, ACTH | GH, PRL, TSH, FSH/LH, ACTH | GH, TSH, ACTH |
| Other feature(s) | Micropenis | Aortic coarctation, pyelocaliceal duplicity, micropenis | Arched palate | Bilateral papillary coloboma | None | Epilepsy | Type I Chiari, rounded forehead, micropenis |
| Pituitary imaging | |||||||
| Anterior pituitary | Normal | Hypoplastic | Normal | NA | Hypoplastic | Aplastic | Hypoplastic |
| Posterior pituitary | Ectopic | Ectopic | Ectopic (double) | Ectopic | Eutopic | Eutopic | Ectopic (double) |
| Sella turcica | Normal | Shallow | Shallow | Sphenoid bone dehiscence | Hypoplastic | Normal | Shallow |
| Endocrine explorations | |||||||
| GH/insulin, ng/mL (basal and +60 min) | NA and 1.5 | 5.74 and NA | NA | NA | NA | < 2 and 0.95 | < 0.1b |
| GH/glucagon, mIU/L (basal and +60 min) | NA and 10 | NA | < 0.5 and 0.7 | NA | 0.36 and 0.72 | NA and 3 | NA |
| GH/ornithine, ng/mL (basal and +60 min) | NA | NA | NA | 0.3 and 2.2 | NA | NA | NA |
| GH/L-Dopa, ng/mL (basal and +60 min) | NA | NA | NA | < 0.1 and 0.2 | NA | NA | NA |
| IGF-I, ng/mL (normal values) | 42 (> 100) | 31 (20–120) | 23 (140–496) | 10 (62–308) | 53 (24–144) | 11 (103–391) | NA |
| TSH, mIU/L (normal values) | 1.4 (0.5–6) | 7.92 (1–10) | 1.81 (0.6–4) | 0.8 (0.4–4) | 4.43 (0.3–3.8) | 0.01 (0.1–4) | 0.06 (0.1–4) |
| FT4, pmol/L (normal values) | 19.3 (> 10) | 7.45 (10–22) | 10.07 (9–15) | 11.2 (12–21.9) | 3.3 (6–15.5) | 19 (10–25) | 3.8 (7–20) |
| FSH, mIU/mL (normal values; age) | 0.8 (< 2; 9 y, 6 mo) | 5.8 (0.4–5; 1 mo, 10 d) | 0.4 (0.2–5.6; 15 y, 2 mo) | < 0.4 (NA; 3 y, 9 mo) | 1.19 (1–10.5; 2 y, 6 mo) | 1.1 (2–9; 21 y, 7 mo) | < 0.5 (NA; 1 d) |
| LH, mIU/mL (normal values; age) | 0.1 (< 0.1; 9 y, 6 mo) | 2.1 (0.5–6; 1 mo, 10 d) | 0.2 (0.2–2; 15 y, 2 mo) | < 0.2 (NA; 3 y, 9 mo) | 0.25 (1–8.4; 2 y, 6 mo) | 0.7 (1–5; 21 y, 7 mo) | < 0.5 (NA; 1 d) |
| Testosterone, ng/mL (normal values; age) | 0.6 (< 0.1; 9 y, 6 mo) | 0.15 (0.5–3.5; 1 mo, 10 d) | < 0.07 (< 0.5; 15 y, 2 mo) | < 0.14 (0.05–0.09; 8 y, 9 mo) | 0.1 (> 12; 14 y, 6 mo) | — | 0.03 (NA; 1 d) |
| Cortisol/synacthen, ng/mL: basal at 8 am and +60 min (normal values at +60 min) | 152 and 282 (> 150) | 27 and 125 (> 180) | 142.3 and 199.2 (> 180) | 135 and 269c (> 200) | 53 and 169 (>200) | 83 and 98b (> 180) | < 11b |
| Molecular data | |||||||
| Variation (nucleotide) | c.194C>T | c.487A>C | c.662G>A | c.704G>A | c.143_144delGA | c.393C>G | c.606+1G>T |
| Variation (protein) | p.Ala65Val | p.Thr163Pro | p.Arg221Gln | p.Arg235Gln | p.(Arg48Thrfs*104) | p.(Tyr131*) | — |
| Genotype | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous |
| Consanguinity | Yes | No | No | No | No | No | No |
| Conservation | Vertebrates | Invariant | Invariant | Mammals | — | — | — |
| Location | LIM1 | Homeodomain | — | — | — | — | — |
| Allele frequency | |||||||
| 1000 Genomes | 0 | 0 | 2 × 10−4 | 0 | 0 | 0 | 0 |
| ExAC | 8 × 10−6 | 0 | 10−5 | 5 × 10−5 | 0 | 0 | 0 |
The em dash (—) indicates irrelevant.
Abbreviations: ACTH, adrenocorticotropic hormone; ExAC, Exome Aggregation Consortium; FSH, follicle-stimulating hormone; FT4, free thryoxine; IGF-I, insulin-like growth factor I; LH, luteinizing hormone; NA, not available; PRL, prolactin; TSH, thyrotropin.
aIn patient 3, low FSH, LH, and testosterone levels at the age of 15 years, 2 months may be linked to a pubertal delay rather than to hypogonadotropic hypogonadism. This patient indeed showed normal testosterone levels at 19 years of age (5 ng/mL; normal range, 3.5 to 11.5 ng/mL), more than 1 year after testosterone treatment cessation.
bTest was performed when patient had severe spontaneous hypoglycemia.
cDynamic test was performed with ornithine.
Family 2
The propositus (II2) was referred to a physician for CPHD involving the somatotroph, thyrotroph, gonadotroph, and corticotroph axes. He also presented the following features: aortic coarctation, pyelocaliceal duplicity, and micropenis (Table 1). MRI of the pituitary area showed a hypoplastic anterior pituitary and an EPP together with a shallow sella turcica (Fig. 3). Analysis of the LHX4 gene showed the presence of the p.Thr163Pro allelic variant (c.487A>C) in the heterozygous state [Figs. 1 and 2(A)]. Threonine located at position 163 is part of the homeodomain of LHX4, next to the putative nuclear localization signal of the protein [Fig. 2(A)].
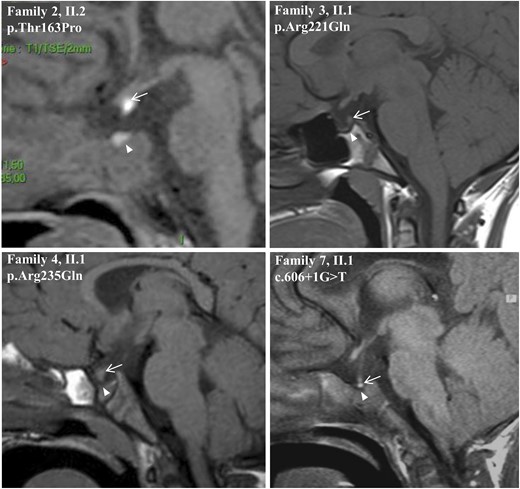
Sagittal MRI views of the index patients from families 2, 3, 4, and 7. Images show EPP and sella turcica anomalies. White arrows show the posterior pituitary and white arrowheads indicate the sella turcica.
Family 3
The propositus (II1) was referred to a pediatrician for severe growth retardation at 15 years of age (−4.5 SD). He also featured an arched palate. Hormonal data showed somatotroph and gonadotroph deficiencies (Table 1). Pituitary MRI revealed an EPP and an interrupted pituitary stalk associated with a shallow sella turcica (Fig. 3). LHX4 sequencing showed that the propositus carried the p.Arg221Gln (c.662G>A) allelic variant in the heterozygous state [Figs. 1 and 2(A)].
Family 4
The propositus (II1) was diagnosed with short stature (−1.85 SD) and a pituitary deficit involving growth hormone (GH) and thyrotropin. MRI investigations showed an EPP and a severe bone malformation of the sella turcica (Table 1). The sphenoid bone at the origin of the sella turcica was dehiscent (Fig. 3). The propositus also displayed bilateral papillary coloboma. LHX4 analysis revealed the presence of the c.704G>A mutation in the heterozygous state, predicting the p.Arg235Gln missense variation [Figs. 1 and 2(A)].
Family 5
The propositus (II1) was referred to an endocrinologist for severe short stature (−5 SD) and pituitary hormone deficiency at the age of 2.5 years. Hormonal investigations showed a GH, thyrotropin, follicle-stimulating hormone/luteinizing hormone, and adrenocorticotropic hormone deficit. MRI of the pituitary area revealed a severe hypoplasia of the anterior pituitary associated with a normal location of the posterior pituitary and a poorly delineated sella turcica (Table 1). LHX4 gene analysis identified a heterozygous 2-bp deletion (c.143_144delGA). This frameshift mutation, p.(Arg48Thrfs*104), leads to a stop codon at position 152 [Figs. 1 and 2(A)].
Family 6
The propositus (II2) was referred to an endocrinologist at the age of 2 years for a mild short stature (−2 SD) and epilepsy crisis. Hormonal assessments revealed a panhypopituitarism, which, as shown by pituitary MRI, was secondary to an aplastic anterior pituitary. Posterior pituitary was originally described as ectopic when referred to us, but further MRI studies revealed a normal location of the posterior pituitary (Table 1). LHX4 analysis showed the presence of the p.(Tyr131*) mutation (c.393C>G) in the heterozygous state (Fig. 1). This mutation predicts a premature stop codon in the LIM2 domain of LHX4 [Fig. 2(A)].
Family 7
The propositus (II1) was referred to a pediatrician for pituitary deficits and macrocrania with hydrocephaly. Hormonal deficiency affected somatotroph, thyrotroph, and corticotroph lineages. MRI and radiography of the pituitary region revealed a shallow sella turcica and a type I Chiari malformation (Table 1). The anterior pituitary was found to be hypoplastic, and the posterior pituitary had 2 distinct signals (Fig. 3). Sequencing of LHX4 exon-intron boundaries led to the identification of the de novo c.606+1G>T mutation in the heterozygous state. This mutation, which involves the invariant dinucleotide of the splice-donor site of intron 4, is therefore deleterious. Kinship was confirmed by microsatellite analysis.
In silico analyses of the identified LHX4 allelic variants
To predict the functional consequences of the identified LHX4 variants, we first checked if they were described in 1000_Genomes and Exome Aggregation Consortium databases. The p.Ala65Val, Arg221Gln, and p.Arg235Gln variants are described with an allele frequency lower than 1/5000 (Table 1). Allele frequencies in control populations indicate that these rare missense variations could be involved in the patients’ disease. Using MaxEntScan (developed by Gene Yeo and Chris Burg from the Massachusetts Institute of Technology, http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html), a splice site recognition tool, we found no splicing alteration resulting from the identified LHX4 variations, except for the splice mutation (c.606+1G>T), for which the donor site score decreased from 9.22 to 0.72.
Most importantly, interspecies alignment of the amino acid sequences of LHX4 showed that all residues targeted by the missense variations identified in this study (i.e., Ala65, Thr163, Arg221, and Arg235) are invariant in vertebrates. The Thr163 and Arg221 residues are conserved throughout evolution, even in drosophila [Fig. 2(C)]. Such a high degree of conservation of these residues therefore strongly suggests that amino acid substitutions at these positions could be detrimental to protein function.
The p.Ala65Val variation lies within the LIM1 domain, a cysteine-rich structure that consists of 2 consecutive zinc-finger motifs. Residue Ala65 is located away from the covalent bounds formed between cysteines and zinc atoms, suggesting that although it is not involved in zinc stability, this residue may interact with protein partners [Fig. 2(B)].
Functional consequences of the identified variants on subcellular localization, transcriptional activity of LHX4, and on the interaction of LHX4 with protein partners
In addition to the expression vectors encoding each of the 7 identified LHX4 variants, we also generated expression vectors encoding 3 other LHX4 variants: a LIM1-domain–deleted variant (p.ΔLIM1), a LIM2-domain–deleted variant (p.ΔLIM2), and a third variant in which alanine residues substituted for cysteines 57 and 60 (p.Cys57-60Ala). When expressed in heterologous HEK293T cells and revealed by western blotting, each LHX4 variant was detected at the expected size [Fig. 4(A)].
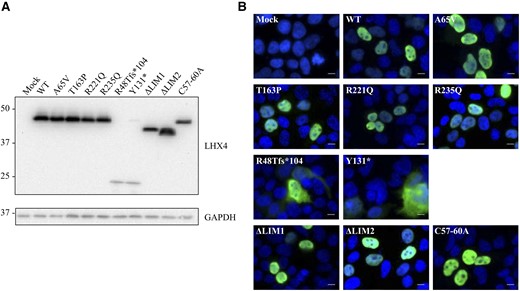
Expression analysis of HA-tagged wild-type (WT) and mutant LHX4 proteins expressed in HEK293T. (A) Protein expression level was assessed by western blotting using a monoclonal anti-HA antibody. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control. Molecular weights are indicated in kDa. (B) Subcellular localization of LHX4 WT and missense variants, as visualized by immunocytofluorescence. Putative truncated proteins corresponding to p.(Arg48Thrfs*104) and p.(Tyr131*) display an abnormal cytoplasmic localization. Other variants are expressed in the nucleus. HA-tagged LHX4 (green); DAPI (blue). White bar is 15 µm.
The subcellular localization of each LHX4 variant was then analyzed by immunocytofluorescence after their transient expression in HEK293T. As expected, the wild-type LHX4 protein was found to be localized in the nucleus. Except for the nonsense and the frameshift mutants, which were found to localize to the cytoplasm, all of the other LHX4 variants displayed a similar nuclear localization [Fig. 4(B)].
We also tested the ability of each LHX4 variant to activate the proximal promoters of the POU1F1, GH, and PRL genes. Consistent with previous studies (12, 21), wild-type LHX4 successfully activated those promoters in a dose-dependent manner. No activation was observed after cotransfection of the POU1F1 reporter plasmid with a plasmid expressing the p.(Arg48Thrfs*104), p.(Tyr131*), or p.Thr163Pro variant [Fig. 5(A)]. Similar results were obtained with the GH and PRL reporter plasmids [Fig. 5(B) and 5(C)]. The other LHX4 variants, p.Ala65Val and p.Arg221Gln, displayed a slightly reduced transactivation capacity, depending on the promoter tested. For all tested promoters, the deletion or disruption of the LHX4 LIM1 domain abolished luciferase reporter activation, whereas the deletion of LIM2 domain only caused a partial loss of function. To investigate the mechanism underlying the dominant expression of the identified LHX4 variants, we assayed the POU1F1 reporter gene transactivation in the presence of both wild-type and mutant LHX4 proteins. Cotransfection of equal amounts of each LHX4 variants with wild-type LHX4 plasmid showed no significant difference compared with a half dose of the wild-type LHX4 plasmid, excluding a dominant negative effect of these variants over the wild-type LHX4 protein [Fig. 5(D)].
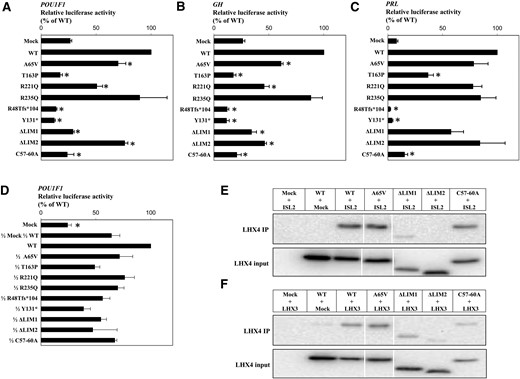
Functional assessment of the identified LHX4 variants. (A–D) Transactivation of the POU1F1, GH, and PRL proximal promoters by LHX4 variants. CHO cells were transiently cotransfected with 30 ng of cDNA expression vectors for wild-type (WT) or mutated LHX4 and 100 ng of a luciferase reporter gene under the control of the POU1F1, GH, or PRL promoter. Promoter activity was assessed by measuring luciferase activity 24 hours after transfection. For negative and half-dose controls, the total amount of plasmid was adjusted to 30 ng with the empty expression vector. Results are expressed in relative light units normalized by the total amount of protein (in µg) and adjusted to WT activity. The data are the mean ± SD of 3 independent experiments, each performed in triplicate. (A–C) P < 0.01 compared with WT (*). (D) P < 0.01 compared with 1/2 mock, 1/2 WT (*). (E) Coimmunoprecipitation of HA-LHX4 and FLAG-ISL2. HEK293T whole-cell lysates were immunoprecipitated with anti-FLAG antibody (ISL2). LHX4 was revealed using anti-HA antibody. (F) Coimmunoprecipitation of HA-LHX4 and FLAG-LHX3. HEK293T whole-cell lysates were immunoprecipitated with anti-FLAG antibody (LHX3). LHX4 was revealed using anti-LHX4 antibody (Santa Cruz Biotechnology). Input represents 10% of total lysates.
To further investigate the effect of the p.Ala65Val variant found in the LIM1 domain and to assess the importance of this domain, we studied its interaction with 2 protein partners (ISL2 and LHX3). Coimmunoprecipitation experiments in HEK293T cells showed an interaction between wild-type LHX4 and ISL2 and also, to our knowledge, a previously unknown interaction between LHX4 and LHX3. Interaction studies between those partners and the p.Ala65Val LHX4 variant did not reveal any loss of binding ability [Fig. 5(E) and 5(F)]. The artificial constructs p.ΔLIM1 and p.Cys57-60Ala, which impair the first LIM domain of LHX4, showed a partial alteration of LHX4 binding capacity; p.ΔLIM2 almost abolished interactions between LHX4 and other proteins.
Discussion
In this study of a large cohort of patients affected by a rare endocrine disorder that is defined by the association of hypopituitarism with an ectopic posterior pituitary and/or a sella turcica defect, we identified an LHX4 variant in 7 of the 417 unrelated patients. To date, to our knowledge, this is the largest cohort of patients presenting with this phenotype who have been screened for LHX4 mutations. One of the 7 identified variants is an unambiguous splice defect (c.606+1G>T); it destroys the invariant GT sequence of the splice donor site of intron 4. On the basis of our functional assays, 5 other variations are also nonambiguous disease-causing mutations: p.(Tyr131*), p.(Arg48Thrfs*104), p.Ala65Val, p.Thr163Pro, and p.Arg221Gln. The p.(Tyr131*), p.(Arg48Thrfs*104), and p.Thr163Pro mutations result in a loss of function of the recombinant proteins which lose their ability to transactivate at least 3 target promoters (POU1F1, GH, and PRL). For p.(Tyr131*) and p.(Arg48Thrfs*104), nonsense-mediated mRNA decay (NMD) is, however, the most probable mechanism underlying the loss of function of LHX4. The study of LHX4 expression and NMD could not be performed ex vivo because LHX4 is not expressed in accessible cells such as blood cells. Our experiments reflect the loss of function of the variants if the corresponding transcripts escape NMD. As for the p.Ala45Val and p.Arg221Gln missense mutations, our functional studies demonstrated a marked decrease in their ability to transactivate the POU1F1 and GH promoters, in keeping with the in silico analyses, that also argue for their pathogenicity. The p.Arg235Gln variation, which did not result in a loss of function in those experiments, is likely to be a rare polymorphism; however, it cannot be ruled out that this variant could have a deleterious effect in cellular systems closer to the human pituitary tissue than human embryonic kidney or CHO cell lines. However, so far, there is no human pituitary cell line available that would provide a more physiological environment with effectors or protein partners of pituitary-specific transcription factors such as LHX4. Overall, our data show that the prevalence of LHX4 mutations in patients with pituitary deficits is 1.4% (6 mutation carriers among 417 unrelated patients). This proportion is comparable to the proportion of pituitary deficits linked to HESX1 or SOX3 mutations. Cohort studies retrieved HESX1 mutations in about 0.2% (1 of 472 patients) (28) to 1.5% (1 of 64 patients) (29) of patients with hypopituitarism. SOX3 molecular defects have been reported to be responsible for hypopituitarism in 2.6% of male patients (2 of 76 patients) (30).
This study also unveils a wide range of phenotypic signs that can be associated with an LHX4 defect. The previously documented Type I Chiari cerebellar defect associated with LHX4 mutations (7, 15) was present in only 1 of our mutation-positive cases. Most importantly, EPP and sella turcica anomalies are not always present in the event of LHX4 mutation; the patient carrying the p.Ala65Val mutation (individual II2 from family 1) had a normal sella turcica, and the patient with the p.Tyr131* mutation (individual II2 from family 6) presented neither of these defects. Interestingly, patients who carry mutations with a total loss of function (p.Arg48Thrfs*104, p.Tyr131*, c.606+1G>T, and p.Thr163Pro) show a combined pituitary deficit including a corticotrophin deficit. This was not the case for the patients who carried the variants with a partial loss of function (p.Ala65Val and p.Arg221Gln), who only presented with a GH deficit associated or not associated with a follicle-stimulating hormone/luteinizing hormone deficit. Corticotrophin deficiency has been observed in at least half of the reported patients with LHX4 mutations, which may be underestimated, given that it can occur later in life around puberty (17). In the current study, 4 of the 6 patients carrying a nonambiguous LHX4 mutation presented with early-onset corticotrophin deficit; however, none of them demonstrated neonatal respiratory distress syndrome, a feature previously described in 2 patients with an LHX4 defect (11, 18) as well as in the Lhx4 knockout murine model (9). Our patients also presented additional features such as aortic coarctation, pyelocaliceal duplicity, epilepsy, and rounded forehead, which were absent from previous reports. However, with the current epidemiological data, it is difficult to state conclusively that these later anomalies result from the identified LHX4 defects and to draw definitive phenotype:genotype correlations.
Transactivation studies did not unveil a dominant negative effect of the identified LHX4 mutants over the wild-type protein, an observation that supports a haploinsufficiency mechanism underlying the dominant phenotype associated with LHX4 mutations (13, 18, 21). However, this hypothesis could be challenged by the recent study of Savova et al. (31), who displayed a list of human genes with allele-specific regulation and monoallelic expression. Because this list includes LHX4, it is tempting to speculate that monoallelic expression might be the mechanism behind the supposed haploinsufficiency of LHX4 mutations. Furthermore, random monoallelic expression was also shown in that study and could explain the incomplete penetrance observed in 5 of the 6 families carrying an unambiguous LHX4 defect. On the basis of this hypothesis, the absence of disease in the parents who carry the mutation in the heterozygous state could be a result of the single expression of the normal LHX4 allele. The observation made by Savova et al. (31) could also explain the variability in clinical expression of the disease phenotype in patients with an LHX4 mutation.
LIM domains are known to ensure protein-protein interaction in many fundamental biological processes (23, 32–35). The identification of a missense variation (p.Ala65Val) in the LIM1 domain of LHX4 in 1 of our patients prompted us to study (1) the impact of this variation and of a LIM1 deletion or inactivation, as well as a LIM2 deletion, on the ability of LHX4 to interact with 2 protein partners (ISL2 and LHX3), and (2) the putative role of the LIM1 and LIM2 domains in the biological activity of LHX4. In our experimental conditions, the p.Ala65Val variation did not impair the ability of LHX4 to bind to those protein partners. As for the LIM2 domain, its deletion almost abolished the interaction of LHX4 with either ISL2 or LHX3. Similarly, mutations in the LIM2 domain of the closely related LIM-HD transcription factor, LHX3, have been shown to impair the ability of LHX3 to interact with POU1F1 (36). In a yeast 2-hybrid screen, Howard et al. (24) found that only the LIM2 domain is required for LHX4 interaction with IFT172. Taken together, these data clearly show that the LIM2 domain is crucial for protein-protein interaction. These results strongly support the fact that the LIM2 domain alone is able to bind ISL2 and LHX3. Notably, these interactions remain partially intact in the presence of the p.Ala65Val missense variation or even after deletion or inactivation of LIM1 domain. Our transcriptional activation assay showed that a LIM1 deletion or inactivation has a greater impact on LHX4 transcriptional activity than a LIM2 deletion. Thus, the LIM1 domain seems more important than the LIM2 domain for the interactions that result in DNA binding. This is an example of LHX4 LIM1 and LIM2 domains having potentially different roles for protein function.
In conclusion, this study provides evidence of the scarcity of molecular defects found in LHX4 that result in syndromic congenital hypopituitarism. The wide range of identified phenotypic features extends the phenotypic spectrum associated with LHX4 mutations. This study also provides additional insight into the function of LHX4 through identification of a protein partner and better characterization of its LIM domains.
Abbreviations:
- CHO
Chinese hamster ovary
- CPHD
combined pituitary hormone deficiency
- EPP
ectopic posterior pituitary
- GH
growth hormone
- IGHD
isolated growth hormone deficiency
- LIM-HD
LIM homeodomain
- MRI
magnetic resonance imaging
- NMD
nonsense-mediated mRNA decay
- PCR
polymerase chain reaction
- SD
standard deviation
Acknowledgments
We are grateful to the patients and their families, whose cooperation made this study possible, and all referring physicians.
This work was supported by grants from the Société Française d’Endocrinologie et Diabétologie Pédiatrique and the Fondation pour la Recherche Médicale (Grant No. PLP20131028838).
Disclosure Summary: The authors have nothing to disclose.
References
Author notes
Address all correspondence and requests for reprints to: Marie Legendre, PhD, U.F. de Génétique Moléculaire, Hôpital Trousseau, 26 avenue du Docteur Arnold Netter, 75012 Paris, France. E-mail: [email protected].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}