





Glucocorticoids are powerful steroid hormones that regulate development, metabolism, and immune response. However, glucocorticoid unresponsiveness or resistance is observed in the treatment of inflammatory, autoimmune, and lymphoproliferative diseases and significantly limits their efficacy.
In Cushing's disease, although some glucocorticoid-mediated suppression of pituitary-derived ACTH is seen, corticotroph tumors exhibit relative resistance to glucocorticoid action. We previously demonstrated that testicular orphan receptor 4 (TR4) binds to the pro-opiomelanocortin (POMC) promoter to induce corticotroph tumor POMC expression and ACTH secretion, and we hypothesized that TR4 may interact with glucocorticoid signaling to modulate POMC expression and action.
Here we demonstrate that TR4 abrogates glucocorticoid receptor (GR)- or dexamethasone-mediated POMC and activator protein-1 transrepression in both murine and human pituitary corticotroph tumor cells. Co-immunoprecipitation studies indicate that TR4 and GR interact directly with each other, resulting in TR4-mediated disruption of GR binding to the POMC promoter.
These results demonstrate that TR4 binds GR to play an important role in glucocorticoid-directed corticotroph tumor POMC regulation in addition to modulating glucocorticoid actions on other GR targets. Characterization of this pathway may offer important insights into glucocorticoid resistance and may identify a novel approach for the treatment of Cushing's disease and the glucocorticoid-resistant states.
We demonstrate a direct physical interaction between TR4 and GR to disrupt GR binding the promoter of POMC, underlying the derangement of the negative feedback observed in human corticotroph tumors.
Glucocorticoids (GCs), a group of lipophilic steroid hormones produced by the adrenal cortex, play a central role in development, metabolism, immunosuppression, and the stress response (1). The powerful actions of GCs are mainly mediated by the GC receptor (GR), which is ubiquitously expressed (2). Upon ligand binding, the GR disassociates from its cytoplasmic oligomeric complex and translocates to the nucleus where the homodimers bind to palindromic promoter GC response elements (GREs) to modulate target gene expression (3). Maintenance of GC homeostasis depends on complex feedback interactions within the hypothalamo-pituitary-adrenal (HPA) axis to coordinate the regulation of immune, endocrine, and neurological responses (2). Under normal physiological conditions, GCs inhibit their own synthesis by transrepression of hypothalamic CRH and pituitary ACTH via the GR (2). Pro-opiomelanocortin (POMC), the precursor peptide to ACTH, is a well-defined GR responsive gene that is transrepressed by liganded GR via a negative GRE (nGRE, −69 to −61) located in the POMC promoter (3).
Cushing's disease (CD) is characterized by excess pituitary tumor-derived ACTH secretion (4). The consequent adrenal-derived excess cortisol secretion leads to disabling symptoms, including diabetes, hypertension, osteoporosis, and obesity, resulting in an approximately 4-fold increased mortality (5). Although the recent findings of ubiquitin-specific protease 8 mutations (6, 7) and increased expression of heat shock protein 90 (8) in pituitary corticotroph tumors provide new insight into this disorder, the molecular mechanisms underlying CD in many patients remain largely unsolved. Furthermore, although novel therapies such as the next-generation somatostatin receptor ligand pasireotide and the GR blocker mifepristone are now approved treatment (9, 10), the management of many patients with CD remains challenging.
Our recent findings show that testicular receptor 4 (TR4, also known as NR2C2), an orphan nuclear receptor, is strongly expressed in the nucleus of corticotroph pituitary tumors and that TR4 potently induces POMC transcription as well as murine corticotroph tumor cell proliferation and invasion (11). Given the essential roles of the GR in transrepression of POMC and the importance of GC resistance in CD, leukemia, and inflammatory and autoimmune disorders, we sought to better understand the potential interactions between TR4 and GR in POMC regulation.
Materials and Methods
Cell culture and reagents
AtT20/D16V-F2 murine corticotroph tumor cells secreting ACTH were obtained from American Type Culture Collection and cultured as a monolayer in DMEM (Life Technologies, Inc) containing 10% fetal bovine serum (Sigma-Aldrich) and penicillin/streptomycin (Life Technologies, Inc.) at 37°C, 5% CO2 (11). Dexamethasone (Dex), BSA, collagenase, and hyaluronidase were purchased from Sigma-Aldrich.
Primary cultures of human corticotroph tumor
Surgically resected human ACTH-producing pituitary tumor tissues (n = 5) were washed, minced, and digested with DMEM containing 0.5% BSA, 0.35% collagenase, and 0.1% hyaluronidase at 37°C for 20 minutes. After centrifugation, cell pellets were resuspended in culture medium and cultured in an incubator for 24 hours. Then cells were transfected with the pCMX-TR4 plasmid encoding human TR4 (11) or the empty vector pCMX in combination with V5-GR plasmid and/or activator protein-1 (AP-1)-Luciferase reporter (Stratagene). Twenty-four to 48 hours later, cells were harvested to quantitate POMC mRNA expression and AP-1 luciferase activity. Endogenous TR4-GR association was evaluated by immunoprecipitation using an anti-GR antibody (Cell Signaling Technology) in primary cultures of human pituitary corticotroph tumors.
Transfection and reporter assays
Murine corticotroph tumor AtT20 cells were transfected with POMC- or AP-1-Luciferase reporters, V5-TR4, V5-GR, and control vector pcDNA3.1/V5-HisB plasmids using Lipofectamine 2000 (Life Technologies, Inc). Because TR4 has been reported to alter viral promoter activity (10), protein concentration was used as an internal control to normalize firefly luciferase results.
Real-time PCR
Total RNA was extracted with RNeasy (QIAGEN). RNA quantification and integrity were assessed by measurement of absorbance at 260 and 280 nm. Total RNA was reverse-transcribed into first-strand cDNA using a cDNA synthesis kit (Life Technologies). Quantitative PCRs were carried out using a CFX Real-time PCR Detection System (Bio-Rad Laboratories Inc). Primer sequences (Life Technologies) were as follows: mouse actin forward primer, 5′-GGC TGT ATT CCC CTC CAT CG-3′; mouse actin reverse primer, 5′-CCA GTT GGT AAC AAT GCC ATG T-3′; mouse pomc forward primer, 5′-CCA TAG ATG TGT GGA GCT GGT G-3′; and mouse pomc reverse primer, 5′-CAT CTC CGT TGC CAG GAA ACA C-3′.
ACTH ELISA assay
Murine and human ACTH ELISA kits were purchased from Calbiotech. The ELISA assays were performed in triplicate.
Plasmid constructs
The plasmid containing 1-kb human POMC promoter fused with luciferase reporter was a gift from Dr. Jacques Drouin (Institute de Recherches Cliniques de Montreal, Montréal, Québec, Canada). The AP-1 luciferase reporter was from Stratagene. The plasmids of pCMX-TR4 encoding human TR4 and the empty vector pCMX were gifts from Dr. Chawnshang Chang (University of Rochester Medical Center, Rochester, NY). The plasmids of V5-TR4 and V5-GR containing mouse TR4 or GRα fused to the 5′ of V5 tag respectively were generously provided by Dr. Michael R. Downes and Dr. Ronald M. Evans (The Salk Institute for Biological Studies, San Diego, CA). The plasmids encoding the different regions of GR including 1–278aa, 1–432aa, 427–792aa, 501–792aa, and 518–792aa were generated by the QuikChange II XL site-directed mutagenesis kit (Stratagene) using the above-mentioned V5-GR plasmid as template. The pairs of primers for construction of these GR truncation variants were listed as follows: V5-GR 1–278aa forward primer, 5′-CCT AAA ATT CAG GAT ACT GGA GAA AAG GGC AAT TCT GCA GAT-3′; and reverse primer, 5′-ATC TGC AGA ATT GCC CTT TTC TCC AGT ATC CTG AAT TTT AGG-3′; V5-GR 1–432aa forward primer, 5′-CAC AGC AAC GGG ACC AGA AAA GGG CAA TTC TG-3′; and reverse primer, 5′-CAG AAT TGC CCT TTT CTG GTC CCG TTG CTG TG-3′; V5-GR 427–792aa forward primer, 5′-CCT TCC ACC ATG GAC TCC ACA GCA ACG GGA-3′; and reverse primer, 5′-TCC CGT TGC TGT GGA GTC CAT GGT GGA AGG-3′; V5-GR 501–792aa forward primer, 5′-CCC TTC CAC CAT GGA CGG AAT GAA CCT GGA AG-3′; and reverse primer, 5′-CTT CCA GGT TCA TTC CGT CCA TGG TGG AAG GG-3′; V5-GR 518–792aa forward primer, 5′-CCT TCC ACC ATG GAC CAA GCC ACT GCA GGA-3′; and reverse primer, 5′-TCC TGC AGT GGC TTG GTC CAT GGT GGA AGG-3′. The plasmids containing various regions of TR4 were generated by insertion of the respective PCR products into the KpnI/XhoI site of the pcDNA 3.1/V5-HisB vector. The pairs of primers for construction of these TR4 truncation variants were listed as follows: V5-TR4 1–106aa forward primer, 5′-CGG GGT ACC GAG CTC GGA TCC ACT A-3′; and reverse primer, 5′-CCG CTC GAG TTT GCC TTC CCC AGC AAA CGC TCCA-3′; V5-TR4 1–329aa forward primer, 5′-CGG GGT ACC GAG CTC GGA TCC ACT A-3′; and reverse primer, 5′-CCG CTC GAG TAT GGA GGT GAA GCA CTA TCT GTG GTA-3′; V5-TR4 205–596aa forward primer, 5′-CGG GGT ACC GCT GCT TCC ACT GAG AAG ATC-3′; and reverse primer, 5′-CCG CTC GAG TAT AGA CTG GCT CCA GTG ATC TG-3′. All sequences were confirmed by automatic DNA sequencing.
Co-immunoprecipitation and Western blotting
After cotransfection of V5-tagged mouse TR4 and/or GR plasmids into AtT20 cells for 48 hours, cells were harvested and lysed in cell lysis buffer (Cell Signaling Technology) containing complete protease inhibitor (Roche) and 100 mm phenylmethylsulfonylfluoride (Cell Signaling Technology). The majority (85%) of the cell lysates were then incubated with Protein A/G agarose beads (Santa Cruz Biotechnology, Inc) and anti-V5 antibody (Life Technologies) or mouse IgG at 4°C overnight with agitation. The resin was then washed three times with PBS. The pulled-down protein complexes were run on SDS-PAGE gels and immunoblotted using anti-V5 antibody and anti-TR4 antibodies. The remaining cell lysates (input, 15%) were also analyzed on SDS-PAGE gels and immunoblotted in similar fashion. Anti-TR4 antibody (ab109301; Abcam) and normal rabbit IgG (Santa Cruz Biotechnology, Inc) were used for endogenous immunoprecipitation, and individual membranes were immunoblotted with anti-GR (M-20), anti-mineralocorticoid receptor (anti-MCR; H-300), and anti-progesterone receptor (anti-PR; C-19) antibodies (Santa Cruz Biotechnology, Inc). The densitometric analyses of the protein bands vs the individual loading controls were performed using the ImageQuant 5.2 software (GE Healthcare). The results shown were representative of three independent experiments.
Chromatin immunoprecipitation assay
To characterize the binding region of TR4 on the POMC promoter, AtT20 cells were transiently transfected with V5-TR4 and empty vector pcDNA 3.1-His B. Twenty-four hours later, a chromatin immunoprecipitation (ChIP) assay was carried out using EZ-ChIPChromatin Immunoprecipitation Kit (EMD Millipore) and anti-V5 antibody or mouse IgG control (Life Technologies). Co-immunoprecipitated DNA was analyzed by real time PCR using paired POMC-promoter-specific primers, including primer pair 1 (forward, 5′-GCC AGG TGA CAC ACT CC-3′; and reverse, 5′-GTT TTG TTT GGG GGA TTC C-3′), primer pair 2 (forward, 5′-AGA TTA GGC AGG CAC CCC GAC TG-3′; and reverse, 5′- GAA TGG TCT GGG TGG GGA TTG TCT G-3′), primer pair 3 (forward, 5′-TCT TGC AGA TCG GAG TGG AAT TAC C-3′; and reverse, 5′-TGG CGC TGG TGG TTA GGA AGA ACT T-3′), and primer pair 4 (forward, 5′-TTT CCA GGC AGA TGT GCC TTG CGC T-3′; and reverse, 5′-CAG GGT TGG GTG GGT GAG CCT TGG A-3′). To examine interactions of TR4 on GR binding to the POMC promoter, AtT20 cells were transiently transfected with V5-TR4 and empty vector pcDNA 3.1-His B. Twenty-four hours later, ChIP assay was carried out using EZ-ChIP Chromatin Immunoprecipitation Kit (EMD Millipore) and anti-GR antibody or rabbit IgG control (Cell Signaling Technology). Coimmunoprecipitated DNA was analyzed by real-time PCR using primer pair 4. The results shown were representative of three independent experiments.
Statistics
At least three independent experiments were performed for all studies. Results were expressed as mean ± SEM. Differences were assessed using Student's t test. P values < .05 were considered significant.
Results
TR4 attenuates Dex- and GR-mediated POMC suppression and AP-1 transrepression
Our previous studies demonstrated that TR4 induces POMC expression by binding to the POMC promoter (11). To investigate potential interactions of TR4 on GC-mediated POMC repression, murine corticotroph tumor AtT20 cells were cotransfected with POMC-promoter luciferase reporter and TR4-expressing (pCMX-TR4) or control (pCMX) plasmids. Eighteen hours later, AtT20 transfectants were treated with Dex (10 and 100 nm) or vehicle for an additional 6 hours. As previously reported by us (11) and depicted in Figure 1A, TR4 overexpression resulted in an approximately 2-fold increase in POMC promoter reporter activity compared to control vector transfected cells. As expected, the addition of Dex suppressed POMC promoter activity by approximately 30% in control vector transfected cells (Figure 1A). In contrast, Dex treatment of corticotroph tumor cells transfected with TR4 did not result in POMC promoter suppression (Figure 1A). To further validate the observed actions of TR4 on POMC transcription, we compared actions of a specific short hairpin RNA (shRNA) to TR4 on POMC transcription (11) to a nonsense control in the murine corticotroph tumor cells. Consistently, Dex treatment inhibited POMC promoter reporter activity by approximately 20% in control cells. In contrast, TR4 knockdown induced an approximately 80% reduction in POMC promoter reporter activity, which was more pronounced than Dex treatment (Figure 1B), suggesting that abrogation of corticotroph tumor TR4 expression effectively represses POMC expression. We also tested actions of TR4 on GR-mediated POMC transcription by measuring POMC-promoter luciferase reporter activities after overexpression of the GR either alone or in combination with TR4. As shown in Figure 1C, POMC-promoter luciferase activity was suppressed either by GR (Figure 1C, lane 3) alone or by the combination of Dex and GR (Figure 1C, lane 4). In contrast, TR4 overexpression in murine corticotroph tumor cells blocked GR (Figure 1C, lane 7), and combination of Dex and GR (Figure 1C, lane 8) induced POMC luciferase suppression. RT-PCR quantification of POMC mRNA in TR4-overexpressing or control vector-transfected cells confirmed the reporter assay experiments, whereby Dex treatment inhibited the POMC mRNA level by approximately 50% of control cells, and TR4 overexpression induced a 2.5-fold increase in POMC mRNA levels compared to control (Figure 1D). In contrast, Dex treatment did not suppress POMC mRNA expression in TR4-overexpressing corticotroph tumor cells (Figure 1D). In support of these findings, TR4 knockdown not only inhibited the basal POMC mRNA level, but also led to an enhancement of Dex-induced POMC mRNA suppression (Figure 1E). We further tested murine corticotroph tumor POMC mRNA changes after Dex treatment with or without co-overexpression of GR either in native cells or in TR4-overexpressing murine corticotroph tumor transfectants. As shown in Figure 1F, the POMC mRNA level was suppressed by either Dex (Figure 1F, lane 2) or GR overexpression (Figure 1F, lane 3) alone or by the combination of Dex and GR overexpression (Figure 1F, lane 4). However, both Dex- and GR-mediated POMC suppression was blocked by coexpression of TR4 (Figure 1F, lanes 6–8). In full support of our findings of TR4 actions to abrogate both Dex- and GC-mediated actions on POMC promoter reporter and POMC mRNA, we also demonstrated that corticotroph TR4 overexpression abrogated Dex- and GR-mediated actions on murine corticotroph tumor ACTH secretion. As shown in Figure 1G, ACTH secretion was suppressed by Dex treatment and GR overexpression alone or in combination (Figure 1G, lanes 2–4 vs lane 1), but Dex- and GR-mediated suppression of ACTH secretion was blocked by coexpression of TR4 (Figure 1G, lanes 6–8 vs lane 5). In primary cultures of freshly resected human pituitary corticotroph tumor cells (from five individual patients), we observed a similar action of TR4 to abrogate either GR- (Figure 1H) or Dex- (Figure 1I) mediated actions on POMC mRNA levels or human corticotroph tumor ACTH secretion (Figure 1J). These results indicated that TR4 acted to blunt both GR- and Dex-mediated POMC transrepression in both murine and human corticotroph tumors in vitro.
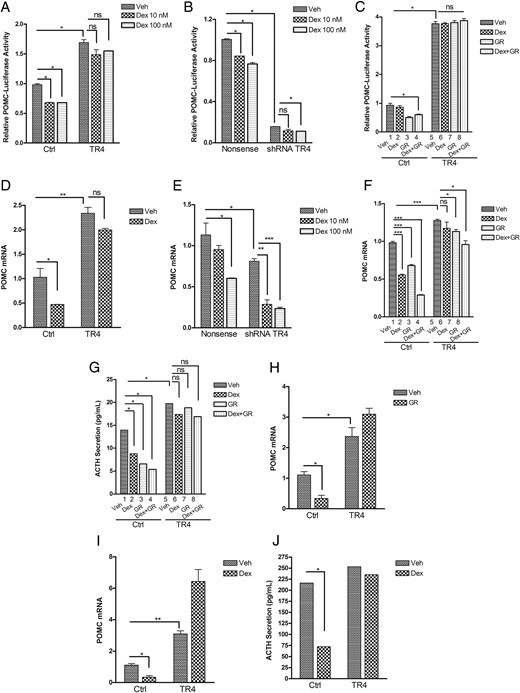
TR4 attenuates the Dex- or GR-mediated POMC suppression.
A, Murine pituitary corticotroph tumor AtT20 cells were cotransfected with POMC-promoter luciferase reporter and TR4-expressing or control plasmids for 18 hours, followed by treatment with Dex (10 or 100 nm) or vehicle for another 6 hours. The results are expressed as POMC promoter induction relative to vector control. B, shRNA TR4 stable transfectants or nonsense control-transfected AtT20 cells were cotransfected with POMC-promoter luciferase reporter. Twenty-four hours later, cells were treated with vehicle or Dex (10 or 100 nm) for another 4 hours, after which POMC luciferase activities were measured and expressed as POMC promoter induction relative to vector control. C, POMC luciferase activities were measured in AtT20 cells transfected with either TR4 or GR alone or with TR4 in combination with GR in the presence or absence of Dex treatment for 4 hours. D and E, POMC mRNA expression was measured by RT-PCR in AtT20 cells overexpressing TR4 (D) or knockdown TR4 (E) after treatment with vehicle or Dex for 6 hours. F, AtT20 cells were cotransfected with TR4 or GR plasmids alone or in combination. Twenty-four hours later, cells were treated with vehicle or Dex for 6 hours, and mRNA was harvested for real-time PCR analysis of POMC mRNA expression. G, Cell supernatants from latter cells were collected for measurement of ACTH secretion by ELISA assay. H–J, Primary cultures of freshly resected human corticotroph tumor cells (representative of five individual tumors studied) were transfected with TR4 or GR alone or with TR4 in combination with GR for 18 hours with or without Dex treatment. POMC mRNA expression was measured by RT-PCR (H and I). ACTH secretion was detected using an ACTH ELISA kit (J). Data shown were representative of at least three independently repeated experiments. Bars indicate the mean ± standard error of the mean of triplicate tests. *, P < .05; **, P < .01; ***, P < .005. ns, Not significant.
We next sought to determine whether the TR4-mediated actions we had demonstrated to block Dex and GR suppression of POMC were specific to the POMC gene or whether TR4 might have a broader role in modifying GC actions. AP-1 interacts with the GR and is negatively regulated by GCs to modulate both inflammatory responses and cell proliferation (12). As demonstrated in Figure 2A, TR4 overexpression markedly increased murine corticotroph tumor cell AP-1 transactivation in a dose-dependent manner. Consistent with the latter finding, TR4 knockdown inhibited AP-1 activation in a time-dependent manner with maximal 90% inhibition by 72 hours (Figure 2B). Dex treatment alone had a weak effect on the AP-1 luciferase reporter activity (Figure 2C, lane 2), whereas GR overexpression alone (Figure 2C, lane 3) or in combination with Dex treatment (Figure 2C, lane 4) suppressed the AP-1 activity by 45 and 55%, respectively. In contrast, TR4 overexpression not only induced AP-1 transactivation, but also abrogated both the GR- and Dex-mediated suppressive effects on AP-1 luciferase (Figure 2C, lanes 6–8). Similar effects were observed in primary cultures of human pituitary corticotroph tumor cells, where TR4 increased AP-1 activity at the 24- and 48-hour time points (Figure 2D). AP-1 transrepression was observed in human corticotroph primary cultures after Dex treatment alone, GR overexpression alone, or the combination of Dex and GR (Figure 2E, lanes 2–4 vs lane 1), but this AP-1 repression was largely blocked by concomitant TR4 overexpression (Figure 2E, lanes 6–8 vs lane 5), supporting our findings in the murine AtT20 cells. In summary, these results indicate that TR4 acts to modulate GC- and GR-directed actions on a variety of GR transcriptional targets, including POMC and AP-1.
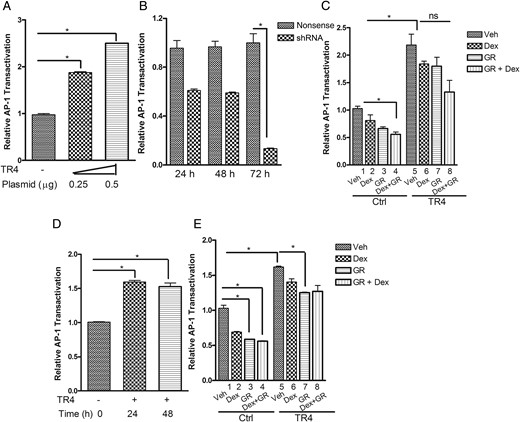
TR4 blocks the Dex- or GR-regulated AP-1 transrepression.
A, V5-TR4 expression plasmid (250 and 500 ng) was transiently transfected into murine corticotroph tumor AtT20 cells stably expressing AP-1 luciferase reporter, and AP-1 luciferase activities were determined 72 hours after transfection. B, An AP-1 luciferase reporter and shRNA TR4 or nonsense control plasmids were transiently transfected into AtT20 cells and harvested at 24, 48, and 72 hours after transfection to quantitate AP-1 luciferase activity. C, Mouse corticotroph tumor AtT20 cells were cotransfected with AP-1 luciferase reporter and/or GR- and TR4-expressing or control plasmids for 48 hours and then treated with Dex (100 nm) or vehicle for an additional 6 hours. Results are expressed as AP-1 luciferase induction relative to vector control. D, An AP-1 luciferase reporter and TR4 expression plasmids were transiently transfected into primary cultures of freshly resected human pituitary corticotroph tumor cells (n = 5) and harvested at 24 and 48 hours after transfection to quantitate AP-1 luciferase activity. E, Primary cultures of human corticotroph tumor cells (n = 5) were cotransfected with AP-1 luciferase reporter and/or GR- and TR4-expressing or control plasmids and then treated with Dex (100 nm) or vehicle for an additional 48 hours to detect AP-1 luciferase activities. Data shown are representative of at least three independently conducted experiments. Bars indicate the mean ± standard error of the mean of triplicate tests. *, P < .05. ns, Not significant.
TR4 interacts with GR
To further analyze the mechanism of TR4 effects on GR function, we tested whether TR4 physically interacted with GR. AtT20 cell lysates were immunoprecipitated using either an anti-TR4 antibody or normal rabbit IgG. As illustrated in Figure 3A, immunoblot of the TR4 precipitation complex demonstrated that GR was present in the protein complex. In contrast, TR4 did not immunoprecipitate the PR and the MCR, indicating specificity of the TR4-GR interaction. In support of the findings in the murine corticotroph tumors, immunoblot of a TR4 precipitation complex from primary cultures of human corticotroph tumor cells confirmed the TR4-GR interaction (Figure 3B). Cotransfection of AtT20 cells with a pCMX-TR4 plasmid and a V5-tagged GR plasmid followed by immunoprecipitation with an anti-V5 antibody or mouse IgG control demonstrated that TR4 was present in the V5-GR precipitated complex (Figure 3C). Reciprocal immunoprecipitation studies using an anti-TR4 antibody demonstrated that GR was present in the TR4-precipitated complex, confirming that TR4 and GR were present together (Figure 3D). Collectively, these studies provide evidence for a physical interaction between the TR4 and GR proteins.
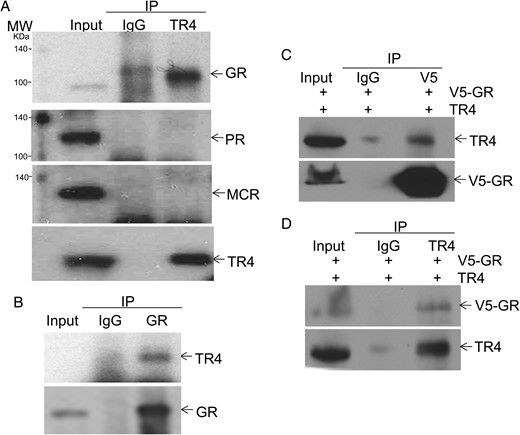
TR4 binds GR.
A and B, Murine (AtT20, A) or human pituitary corticotroph tumor cells (B) were lysed and subjected to immunoprecipitation using specific anti-TR4 or anti-GR antibodies, after which immunocomplexes were analyzed by immunoblot using anti-GR-, PR-, MCR-, and TR4-specific antibodies. C and D, AtT20 cells transfected with plasmids encoding TR4 and V5-tagged GR were harvested and immunoprecipitated with mouse IgG, anti-V5 (GR) antibody (C), or anti-TR4 antibody (D). The immunocomplexes were then analyzed by immunoblotting using specific antibodies to TR4 and V5 (GR). Data shown were representative of at least three independently repeated experiments.
Dex treatment regulates TR4 and GR interaction dynamically
Our previous immunocytochemical studies in corticotroph pituitary tumors demonstrated that TR4 is predominantly located in the nucleus (11). In contrast, prior studies demonstrated that the GR protein dynamically shuttles between the cytosol and nucleus upon ligand binding (13). We next tested whether TR4 alters GR translocation in response to GC binding and whether the TR4-GR interaction could be modulated by GC binding to the GR. To examine this, we extracted cytosolic and nuclear protein lysates after Dex treatment for 2 and 16 hours in both TR4-overexpressing and vector control AtT20 cell transfectants. A swift translocation of GR from the cytosol to the nucleus was observed after Dex treatment beginning at 2 hours and reducing by 16 hours (Figure 4A, control: lanes 1–6; quantified in Figure 4B); the latter was consistent with Dex-directed regulation of GR protein degradation (13). TR4 overexpression (V5-TR4) did not alter the observed Dex-directed GR translocation or its protein stability (Figure 4A, V5-TR4: lanes 7–12; quantified in Figure 4B). Next, we examined whether Dex treatment would alter GR content in the TR4-immunoprecipitated complex. As depicted in Figure 4C and quantified in Figure 4D, in concert with the timing of the Dex-directed GR nucleus translocation, the TR4-GR interaction was also dynamically regulated by Dex treatment with increased GR present in the TR4-immuoprecipitated complex after 2-hour Dex treatment, followed by decreased GR in the TR4-immuoprecipitated complex by 16 hours (Figure 4, C and D). Altogether these studies demonstrated that in unstimulated conditions, the low level nuclear-located GR readily interacts with TR4; and after GC treatment, enhanced availability of nuclear GR results in increased TR4-GR interaction. This provides a mechanistic basis for the regulation of POMC by TR4, with or without GC treatment.
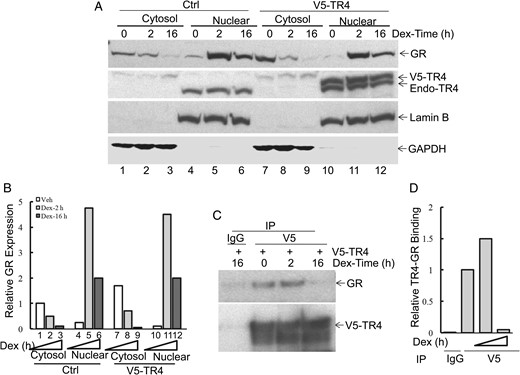
Dex treatment dynamically regulates TR4 and GR interaction.
A and B, AtT20 cells were transiently transfected with V5-TR4 or control plasmid and treated with Dex (100 nm) for 2 and 16 hours. Individual cytosolic and nuclear protein lysates were prepared from the above cells and probed with specific antibodies. A, Lamin B and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were used as nuclear and cytosol markers. B, Protein bands were quantitated by scanning densitometry and compared to controls using the ImageQuant 5.2 software. C and D, Immunoprecipitation was then carried out using an anti-V5 antibody, and the presence of GR in the immunocomplex was detected by immunoblot using specific anti-GR antibody (C). Protein bands were quantitated by scanning densitometry and compared to controls using the ImageQuant 5.2 software (D). The results shown were representative of three independent experiments.
Interaction of TR4 with GR requires GR N-terminal domain and TR4 DNA binding domain
GR and TR4 are both highly conserved nuclear receptors and contain several functional protein regions, including an N-terminal domain, a DNA binding domain, a hinge region, a ligand binding domain, and a C-terminal domain (illustrated schematically in Figure 5; A, for GR; C, for TR4). To begin to characterize which regions of the GR protein were involved in its interaction with TR4, protein lysates were harvested from AtT20 cells transfected with V5-tagged murine GR plasmids encoding the GR regions of 427–792aa, 501–791aa, and 518–792aa as well as the full-length GR protein. Proteins were then immunoprecipitated with anti-V5 antibody as before and probed with anti-TR4 antibody (Figure 5B). In contrast to the full-length GR protein, which showed a strong interaction with TR4 (Figure 5B, lane 2), no interaction with TR4 was seen with the other GR fragments (Figure 5B, lanes 3–5). These results demonstrated that TR4 interacts with the GR via its N-terminal region. In parallel, reciprocal immunoprecipitation studies were carried out using various truncation mutants of TR4, including the N-terminal region (1–106aa), DNA binding domain (1–329aa), and its ligand binding domain (205–596aa), as illustrated in Figure 5C. In V5-TR4 immunoprecipitate complexes, we observed that both full-length TR4 (Figure 5C, lane 2) and a 1–329 aa TR4 region (Figure 5C, lane 4) bound to the GR protein. In contrast, the TR4 N-terminal region (1–106aa) and its ligand binding domain (205–596aa) did not bind the GR (Figure 5D, lanes 3 and 5), indicating that the 106–205aa protein sequence (predominantly the DNA binding domain) of TR4 is crucial for the TR4-GR interaction.
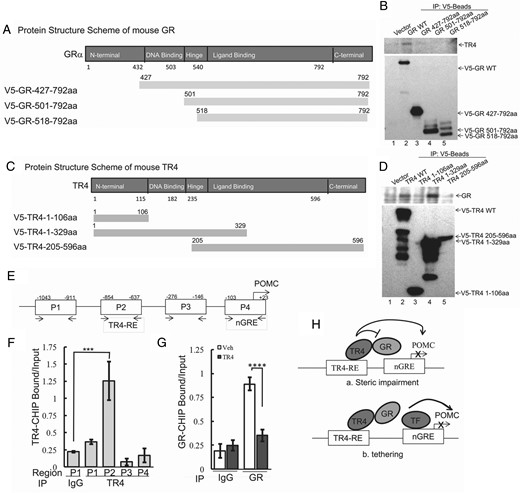
Interaction of TR4 with GR requires the N-terminal GR domain and TR4 DNA binding domain.
A and C, Schematic illustration of the GR protein (A) and TR4 protein (C) functional regions. B, Protein lysates were harvested from AtT20 cells transfected with V5-tagged mutant GR plasmids encoding the C-terminal regions 427–792aa, 501–792aa, and 518–792aa as well as full-length GR protein. Protein lysates were then immunoprecipitated with anti-V5 antibody, and immunocomplexes were analyzed by immunoblot using a specific anti-TR4 antibody. D, AtT20 cells were transfected with either V5-tagged full-length TR4 or 1–106aa, 1–329aa, or 205–596aa deletion mutants, and transfectants were harvested and subjected to immunoprecipitation with anti-V5 antibody. The immunocomplexes were then analyzed by immunoblot using a specific anti-GR antibody. E, Four pairs of primers covering an approximately 1000-bp proximal mouse POMC promoter region (P1–P4) were designed to identify the TR4-binding POMC promoter region. F, ChIP assay was carried out using primers to individual POMC promoter region P1 (−1043 to −911), P2 (−854 to −637), P3 (−276 to −146), and P4 (−103 to +23). G, AtT20 cells were transiently transfected with V5-TR4 and empty vector pcDNA 3.1-His B. Twenty-four hours later, ChIP assay was carried out using an anti-GR antibody or rabbit IgG control to quantify GR binding compared to control. ***, P < .005; ****, P < .01. Data shown are representative of at least three independently repeated experiments. H, Schematic showing the proposed molecular mechanisms underlying TR4 inhibition of negative regulatory GR action on POMC transcription.
Next we sought to examine how our demonstrated TR4-GR interaction modifies POMC transcription and address whether it involves direct TR4 and GR competition for overlapping DNA binding sequences on the POMC promoter, or alternatively a quenching mechanism. To this end, we performed ChIP assays using an anti-TR4 antibody to characterize TR4 binding sites on the POMC promoter. Four pairs of primers covering an approximately 1000-bp proximal mouse POMC promoter region (P1-P4) were tested (illustrated in Figure 5E). As shown in Figure 5F, anti-TR4-immunoprecipitated DNA with the enriched locus was strongly amplified by primer pair 2 (P2), indicating specific TR4 binding to the POMC promoter around this region, which is distinct from the reported GR binding consensus, nGRE presented in the primer pair 4 region (P4) (23). We also used an anti-GR antibody in the ChIP assay to examine TR4 actions on GR binding to the POMC promoter. As shown in Figure 5G, when TR4 was overexpressed, the binding of GR to the nGRE on the POMC promoter was greatly inhibited. Therefore, we concluded that by direct protein-protein interaction, TR4 overrides the inhibitory activity of GR on POMC transcription, leading to up-regulation of POMC expression. Given that TR4 and GR bind different regions of the POMC promoter, we hypothesize that TR4 inhibits the GR binding to POMC by a direct quenching-type action (as depicted in Figure 5H-a) or by facilitating the access of other transactivators, such as Nur77, to the POMC promoter, thereby replacing GR (Figure 5H-b).
Discussion
In the normal physiological state, pituitary-derived ACTH is under negative regulation by feedback from adrenal-derived cortisol, resulting in a carefully balanced availability of GCs. This rapid and specific negative regulation of ACTH mediated by GC action in the anterior pituitary is largely attributable to the binding of the liganded GR to the 5′-flanking region of the POMC gene (14). In CD, although some GC-mediated suppression of pituitary-derived ACTH is seen, corticotroph tumors exhibit relative resistance to this GC action (15). We have recently shown that TR4 is strongly expressed in human corticotroph tumors and that it induces murine and human corticotroph tumor POMC transcription and ACTH secretion (11). In the current studies, we demonstrate that TR4 acts to attenuate the effects of both GCs and the GR to inhibit POMC gene transcription via a direct protein-protein interaction. We also demonstrated that TR4 potentially induces AP-1 transactivation, providing further mechanistic insight into the potential mode of action by which TR4 induces proliferation (11). Additionally, we show here that TR4 acts to block GR-mediated AP-1 transrepression, pointing to a broader significance of the TR4-GR interaction we describe.
Orphan nuclear receptors represent a diverse group of the nuclear receptor superfamily, which is highly conserved throughout evolution. They function as ligand-mediated transcription factors to modulate numerous physiological and pathophysiological processes (16). TR4 belongs to subfamily 2 of nuclear receptors and, together with its homology gene TR2, plays key roles in spermatogenesis, lipoprotein regulation, and central nervous system development (17). In addition to direct DNA binding, TR4 can serve as a coactivator or corepressor by interacting with other nuclear receptors (18). Although the mechanism(s) for increased TR4 expression in corticotroph tumors are not fully understood, it is possible that increased phosphorylated active TR4 could result from enhanced activity of growth factor signaling, as has been recently highlighted due to ubiquitin specific protease 8 mutation in human corticotroph tumors (6, 7).
GR-mediated gene repression occurs via several mechanisms, which may be mediated by direct DNA binding or independent of DNA binding (19). GR-mediated POMC repression involves antagonism of Nur responsive element activity by the GR physically interacting with Nur77 protein to disrupt its binding to the POMC promoter (20). Additionally, the GR can also bind the nGRE located upstream of the TATA-box in the POMC promoter to inhibit POMC transcription (21). In our studies, unlike Nur77, TR4-induced activation of POMC transcription cannot be antagonized by GR expression or Dex treatment in corticotroph tumor cells. Based on our co-immunoprecipitation studies, which demonstrate that TR4 binds the GR via its N-terminal domain, we hypothesize that TR4 binding to GR prevents the GR accessing the nGRE to mediate POMC suppression (Figure 5H, schematic a). However, we cannot entirely exclude other mechanisms. For example, it is possible that upon TR4 disrupts GR binding to POMC promoter, other transactivators bind to the same GR binding sequences and abolish GR-mediated POMC suppression (Figure 5H, schematic b). Nonetheless, our demonstration that both V5-tagged TR4 and endogenous TR4 immunoprecipitated the GR protein is more supportive of a direct action of TR4 to disrupt GR action. Our studies also demonstrate a novel feature of the TR4-GR interaction, namely the requirement of the N-terminal region of GR for protein-protein interaction. When GR forms heterodimers with other nuclear receptors, such as chicken ovalbumin upstream transcription factor II (22), MCR (23), and androgen receptor (24), the hinge domain or DNA domain of GR has been implicated for maintaining the protein-protein interaction. Given that the GR N-terminal region is involved with its interactions with cofactors to modulate gene expression (25), we hypothesize that TR4 binding to the N-terminal GR impairs its ability to bind other cofactors. In tandem, given that TR4 itself is a strong coactivator of POMC (18), this combined action may totally abrogate the gene repressive effect of GR, providing a dual molecular mechanism for the negative effect of TR4 on GR actions.
Based on our results, we believe that TR4 modulates GC and GR actions on a variety of GR targets and plays an important role in GC resistance. Further characterization of this pathway may offer important insights into GC resistance in CD and other diseases such as steroid-resistant asthma, inflammatory bowel disease, and GC-resistant leukemia.
Acknowledgments
We thank Dr Jacques Drouin (Institute de Recherches Cliniques de Montreal, Montréal, Québec, Canada) for his generous gift of plasmid of human POMC luciferase reporter; Dr Michael R. Downes and Dr Ronald M. Evans (The Salk Institute for Biological Studies, San Diego, CA) for their gifts of plasmids of V5-TR4 and V5-GR; and Dr Chawnshang Chang (University of Rochester Medical Center, Rochester, NY) for the gift of pCMX-TR4 plasmid.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- AP-1
activator protein-1
- CD
Cushing's disease
- ChIP
chromatin immunoprecipitation
- Dex
dexamethasone
- GC
glucocorticoid
- GR
GC receptor
- GRE
GC response element
- HPA
hypothalamo-pituitary-adrenal
- MCR
mineralocorticoid receptor
- nGRE
negative GRE
- POMC
pro-opiomelanocortin
- PR
progesterone receptor
- shRNA
short hairpin RNA
- TR4
testicular receptor 4.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}