





Abstract
Multiple endocrine neoplasia type 2 (MEN2) is usually caused by missense mutations in the proto-oncogene, RET.
This study aimed to determine the mutation underlying MEN2A in a female patient diagnosed with bilateral pheochromocytoma at age 31 years and with medullary thyroid carcinoma (MTC) 6 years later.
Leukocyte DNA was used for exome and Sanger sequencing. Wild-type (WT) RET and mutants were expressed in HEK293 cells. Activation of MAPK/ERK and PI3K/AKT was analyzed by Western blotting and luciferase assay. The effect of RET mutants on cell proliferation was tested in a colony forming assay.
Exome sequencing revealed a 6-nucleotide/2-amino acid in-frame deletion in exon 7 of RET (c.1512_1517delGGAGGG, p.505_506del). In vitro expression showed that phosphorylation of the crucial tyrosine 905 was much stronger in the p.505_506del RET mutant compared with WT RET, indicating ligand-independent autophosphorylation. Furthermore, the p.505_506del RET mutant induced a strong activation of the MAPK/ERK pathway and the PI3K/AKT pathway. Consequently, the p.505_506del RET mutant cells increased HEK293 colony formation 4-fold compared with WT RET.
The finding of bilateral pheochromocytoma and MTC in our patient was highly suspicious of a RET mutation. Exome sequencing revealed a 6-base-pair deletion in exon 7 of RET, an exon not yet associated with MEN2. Increased ligand-independent phosphorylation of the p.505_506del RET mutant, increased activation of downstream pathways, and stimulation of cell proliferation demonstrated the pathogenic nature of the mutation. We therefore recommend screening the whole sequence of RET in MTC and pheochromocytoma patients with red flags for a genetic cause.
Multiple endocrine neoplasia type 2 (MEN2) is an autosomal-dominant genetic syndrome with two variants, MEN2A and MEN2B, depending on the underlying activating RET (rearranged during transfection) proto-oncogene mutation. The most common features of MEN2 are medullary thyroid cancer (MTC), which occurs in 70–100% of MEN2 patients, and pheochromocytoma with a frequency of 40–50% (1, 2).
RET is encoded by 21 exons, but RET germline mutations are mostly localized in eight exons (exons 5, 8, 10, 11, and 13–16). Gain-of-function mutations of RET lead to autophosphorylation within the tyrosine kinase domain (2). RET activates different signaling pathways including the RAS/RAF pathway, which results in activation of ERK1/2 (3, 4), and the PI3K/AKT pathway (3, 5, 6). Furthermore, PLC-γ (7), p38MAPK (8), ERK5 (9), and Jun NH2-terminal protein kinase (JNK) (10, 11) are activated by RET. Constitutive activation of RET and subsequently these pathways increases cell proliferation and survival and ultimately results in cell transformation. Screening for RET germline mutations in first-degree relatives of MEN2 patients is recommended to identify affected individuals and allows for prophylactic thyroidectomy prior to onset of MTC (12, 13). Here we report a novel six-nucleotide in-frame deletion in exon 7 of RET, identified in a patient with clinical diagnosis of MEN2A, in whom routine genetic testing for a RET germline mutation had been negative.
Patients and Methods
Patient
A 31-year-old Caucasian woman presented with severe headache, hypertension (170/100 mm Hg), tachycardia (100 bpm), and deteriorating general condition. Clinically, pheochromocytoma was suspected. The diagnosis was biochemically confirmed by high plasma and urinary metanephrines (plasma: normetanephrine, 432 ng/mL [normal < 200]; metanephrine, 927 ng/mL [normal < 90]; urine: normetanephrine, 2523 μg/d [normal < 600]; metanephrine, 4110 μg/d [normal < 350]). Magnetic resonance imaging revealed bilateral adrenal masses with increased tracer accumulation on I-131 meta-iodobezylguanidine positron emission tomography–computed tomography (Figure 1A). Histopathology after bilateral adrenalectomy confirmed the diagnosis of bilateral pheochromocytoma (Supplemental Figure 1A). In search of a germline mutation as an explanation for the bilateral occurrence of pheochromocytoma, we tested VHL (exons 1–3), SDHB (exons 1–8), SDHD (exons 1–4), or RET (exons 10, 11, and 13) with negative results. Still, given the bilateral disease, we suspected a genetic cause and the patient was regularly seen in our outpatient clinic with special attention paid to potential manifestations of syndromic disease. Six years later, at age 37 years, thyroid ultrasound showed a thyroid nodule with a diameter of 5 mm (Figure 1B). Basal calcitonin, normal in all visits before, was now mildly elevated with 7.9 pg/mL (normal < 5.0). After stimulation with pentagastrin, calcitonin increased to 492 pg/mL within 2 minutes and MTC was strongly suspected. The patient underwent thyroidectomy and central lymphadenectomy. Histopathology confirmed a 4-mm MTC with stromal desmoplasia (Supplemental Figure 1B), which was positive for calcitonin and chromogranin A in immunohistochemical staining. Lymph nodes were free of tumor cells and the final TNM classification was pT1a, N0, L0, V0, R0. The patient had plasma metanephrines within normal range (< 88 pg/mL) and nondetectable serum calcitonin (<2.0 pg/mL), and is considered to be in complete remission. PTH and serum calcium were always within the normal range. The patient has no mucosal neuromas of the tongue or lips, no skeletal deformations or joint laxity, and no Marfanoid habitus. Therefore, the clinical diagnosis in this patient is MEN2A. After histological diagnosis of MTC, sequencing of RET was repeated and included exons 8–11 and 13–16, again with negative results.
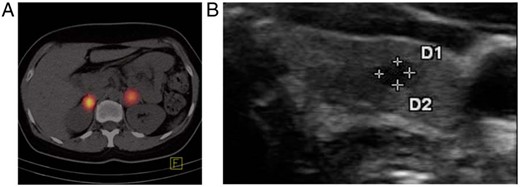
A, I-131 meta-iodobezylguanidine positron emission tomography–computed tomography revealed bilateral adrenal masses with increased tracer accumulation suggesting bilateral pheochromocytoma. B, Thyroid ultrasound showed a 4-mm hypoechogenic nodule in the left thyroid lobe. D, diameter.
Sequencing
Informed consent was obtained from all individuals before sequencing. DNA was extracted from leukocytes (QIAamp DNA Blood Maxi Kit, QIAGEN) and quantified with NanoDrop 2000c Spectrophotometer (Thermo Scientific). Genomic DNA (500 ng) was fragmented by Covaris shearing following the manufacturer's instructions. Genomic libraries were prepared using NEB Next Ultra kit and appropriate barcodes following the manufacturer's instructions (New England Biolabs) with eight postligation PCR cycles. Exome enrichment was performed on a pool consisting of three adapter-ligated DNA samples (333 ng each) using the Roche Nimblegen EZSeqCap EZ Human Exome Library v2.0 protocol and 18 postcapture PCR amplification cycles. Sequencing of enriched DNA fragments was performed on an Illumina HiSeq2500 platform using the paired-end sequencing protocol with a read length of 101 base pairs (bp) each. The p.505_506del mutation in exon 7 of RET was confirmed on gDNA by PCR using 5′taggggtggtcacctca 3′ as forward primer and 5′cagcatgttcctaaagggca 3′ as reverse primer. The PCR product was isolated (QIAquick Gel Extraction Kit, QIAGEN) and sequenced (Seqlab).
Mutagenesis of RET9 plasmids
An expression vector containing the wild-type (WT) short isoform of the human protoRET gene (pBabe-RET9 vector, kindly provided by Massimo Santoro, Pisa, Italy) was used for transient and stable transfections. The p.505_506del RET mutation found in the patient with MEN2A and a C634R mutation were introduced by site-directed mutagenesis (Quick Change Site Directed Mutagenesis KIT, Agilent Technologies) with the following primers: p.505_506del forward 5′-cagctgcttgtaacagtgtcatatgtggccgagg-3′, reverse 5′-cctcggccacatatgacactgttacaagcagctg-3′ and C634R forward 5′-gacgagctgcgccgcacgg-3′, reverse 5′-ccgtgcggcgcagctcgtc-3. After transformation into XL1-blue competent cells, DNA was isolated from cultured single clones and sequencing confirmed successful mutagenesis (Seqlab).
Cell culture, transfection, and Western blotting
Twenty-four hours before transfection, 0.5 × 106 HEK293 cells (obtained from ATCC) were seeded in a six-well plate and cultured in DMEM (Gibco) supplemented with 10% FCS (Gibco) and 1% ZellShield (Minerva Biolabs). HEK293 cells were transiently transfected with RET9 WT, C634, and p.505_506del plasmids using Lipofectamine 2000 (Life Technologies). Fourty-eight hours after transfection, cells were lysed and PhosSTOP and protease inhibitor (Roche) were added. Protein (10 μg) were separated on a 6 or 10% SDS-polyacrylamide gel, transferred onto polyvinylidene fluoride (PVDF) filters, and blocked with 5% milk powder. Proteins of interest were detected with the following primary antibodies: RET (Cell Signaling, No. 3223 Lot:6), phospho-RET (Tyr905) (Cell Signaling, No. 3221 Lot:3), pAKT (S473) (Cell Signaling, No. 4060 Lot:16), or pERK1/2 (T202,Y204) (Cell Signaling, No. 4370 Lot:12). Stable transfection of HEK293 cells was achieved with Lipofectamine 2000 and subsequent culture in medium supplemented with 500 ng/mL puromycin (Gibco). Nonreducing conditions were achieved by omitting β-mercaptoethanol from Laemmli's buffer.
Dual-luciferase reporter assay
MAPK/ERK pathway activation was measured with a luciferase reporter gene assay (Promega). HEK293 cells (4 × 104) were seeded in a 24-well plate as triplicates and cultured to approximately 75% confluence. Cells were then transiently cotransfected with the RET9 WT, C634, or p.505_506del plasmids (17 ng/well), a reporter construct encoding the firefly luciferase gene (pGL4.33[luc2P/SRE/Hygro] Vector, Promega, 280 ng/well,) as well as renilla luciferase expression vector under the control of the serum response factor (also known as serum response element [SRE]) promoter (pRL Renilla Luciferase Reporter Vector, Promega, 33 ng/well). SRE regulates the transcription of luciferase reporter gene after activation of MAPK/ERK pathway. For transfection, the FuGENE (Promega) reagent was used in a 1:3 (DNA:FuGENE) ratio. Cells were collected 48 hours after transfection and the activities of firefly and renilla luciferases were determined with the Dual-Glo Luciferase Assay System (Promega) in a Sirius luminometer (Berthold Detection Systems). Firefly luciferase luminescence values were divided by renilla luciferase luminescence values from the same transfection sample to control for differences in transfection efficiency.
Colony formation assay
HEK293 cells (1 × 105) were seeded in a six-well plate in triplicate and the RET9 WT, C634, and p.505_506del plasmids were transfected with polyethylenimine (Polysciences Inc.) in a 1:4 (DNA:polyethylenimine) ratio. Medium was changed to 5% FCS in DMEM and 1 μg/mL puromycin 24 hours after transfection and then every 2–3 days. After 14 days, colonies were fixed and stained (0.5% crystal violet [Roth], 1% formaldehyde, and 1% methanol in PBS). This experiment was repeated three times.
Statistical analysis
Statistical analysis was performed using one-way ANOVA (GraphPad Prism 6, GraphPad). Differences were considered significant if *, P < .05; **, P < .01; ***, P < .001; ****, P < .0001.
Results
A novel RET mutation was identified in this patient with MEN2A
Exome sequencing from leukocyte DNA revealed a heterozygous in-frame deletion of six nucleotides in exon 7 of RET (c.1512_1517delGGAGGG), leading to loss of two amino acids, glutamic acid, and glycine (p. 505_506del) (Figure 2A). Sanger sequencing confirmed the mutation (Figure 2B).
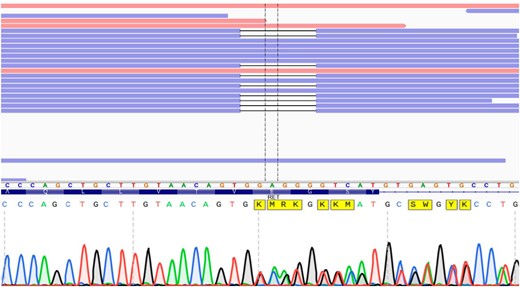
A, Visualization of RET sequencing exon 7 (amino acids on blue background) and right flanking intron with the Integrative Genomics Viewer. Reads that match the reference sequence are displayed in blue and red. Deletions are indicated with a black dash (—). Exome sequencing revealed a heterozygous in-frame deletion of 6-bp (c.1512_1517delGGAGGG)/2 amino acids (glutamic acid and glycine). B, Sanger sequencing confirmed the mutation.
The RET p.505_506 deletion leads to increased MAPK pathway activation
To study the consequences of the p.505_506 deletion in RET, we constructed expression vectors for the WT RET9, the most frequent activating RET mutation C634R (14, 15), located in the cysteine-rich region and previously found in patients with MEN2A, and the novel p505_506 deletion. After expression in HEK293 cells, the p.505_506del RET9 mutant led to significantly higher MAPK/ERK activation than WT RET9 (Figure 3). MAPK/ERK activation by the positive control C643R was significantly higher than both RET9 WT and RET9 p.505_506del.
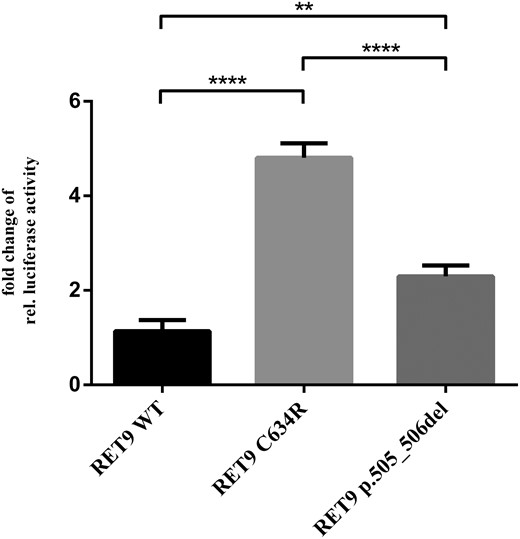
For RET9 p.505_506del, MAPK pathway activation is significantly stronger compared with RET9 WT.
HEK293 cells were transfected with the indicated plasmid together with luciferase reporters induced by MAPK activation. Relative luciferase activity is expressed as fold increase of RET9 WT normalized for renilla luciferase. (One-way ANOVA, **, P < .01; ****, P < .0001; mean ± SEM; n = 3).
The RET9 p.505_506 deletion leads to increased RET, AKT, and ERK phosphorylation
Western blot analysis confirmed that HEK293 cells do not express endogenous RET (Figure 4A). After transfection of the WT, C634R, and p.505_506del mutant plasmids, we detected double bands representing the glycosylated and partially glycosylated RET proteins: the partially glycosylated protein is predominant in WT RET9, the p.505_506del mutant, and RET9 C634R mutant used as a positive control (Figure 4A). Without addition of a ligand, phosphorylation of the crucial amino acid tyrosine 905 (16–18) occurred in RET9 C634R and p.505_506del but not in WT RET9, which suggests ligand-independent autophosphorylation (Figure 4B). Next, the effect on the MAPK/ERK and PI3K/AKT signaling pathways was determined. We found a stronger phosphorylation of ERK1/2 (Figure 4C) and also AKT (Figure 4E) after transfection of RET9 p.505_506del and RET9 C634R compared with WT RET9. Therefore, the p.505_506del mutant activates the MAPK/ERK and PI3/AKT pathways but to a lesser degree than the C634R mutant. Nonreducing conditions revealed dimerization for RET9 C634R but not for the p.505_506 deletion mutant (data not shown).

(Auto)phosphorylation and activation of downstream pathways by RET9 p.505_506del after transient transfection of HEK293.
10-μg protein lysate from HEK293-transfected cells with RET9 WT (negative control), C634R (positive control), p505_506del, or nontransfected HEK293 cells were separated on SDS-PAGE and transferred on a PVDF membrane (n = 4). PVDF membranes were stained with antibodies anti-RET (A), anti-pRET (B), anti pERK1/2 (C), anti-α-Tubulin (control for A–C) (D), anti pAKT (E), or anti β-Actin (control for C and D) (F).
The RET9 p.505_506 deletion leads to increased cell proliferation
The influence of RET9 p.505_506del on cell growth was analyzed in a colony-formation assay. HEK293 cells were transfected with the RET plasmids and cultured for 14 days in selection medium. HEK293 cells formed significantly more colonies after transfection with RET9 p.505_506del and RET9 C634R compared with RET9 WT (Figure 5).

RET9 p.505_506del forms significantly more colonies than RET9 WT.
A, HEK293 cells were stably transfected with plasmid encoding for the respective plasmid as indicated. Cells were cultured in a humidified incubator at 37°C and 5% carbon dioxide for 14 days under 5% FCS in DMEM supplemented with 1 μg/mL puromycin. (mean ± SEM; n.s., not significant; ****, P < .0001; one-way ANOVA; n = 3). B, Example of one colony formation assay.
Family screening reveals that RET p.505_506del is a de novo mutation
After establishing the pathogenic nature of the 505_506del RET mutation, we contacted the family and tested the proposita's parents (mother, age 66 y; father, age 71 y) and brother (age 43 y). No one of these relatives had a history of MTC, pheochromocytoma, or primary hyperparathyroidism, and no one harbored the proposita's RET mutation, which is therefore a de novo mutation (Figure 6).

Sanger sequencing of the parents and the brother of the proposita revealed that the mutation occurred de novo.
Discussion
Our patient first developed bilateral pheochromocytoma, which already suggested an underlying genetic defect, but routine sequencing, including part of RET, VHL, SDHB, and SDHD, failed to detect a mutation commonly associated with pheochromocytoma. Considering the possibility of syndromic pheochromocytoma, the patient was followed closely. Six years later, biochemical and ultrasound screening raised the suspicion of a MTC, which was histologically confirmed, establishing the clinical diagnosis of MEN2A. An extended routine genetic screening found no mutations in the eight most commonly affected RET exons (8–11, 13–16) associated with MEN2, constituting an “orphan” status of our patient with clinical evidence of MEN2A, but no mutation. Exome sequencing finally detected a deletion of six nucleotides in exon 7 of RET. The fact that this RET variant has not been described as a polymorphism and was not present in a database of more than 135 healthy probands against which the exome sequencing data were compared, strongly suggests that this deletion is indeed a mutation. This mutation is listed neither in COSMIC nor in the MEN2 database of the department of pathology in Utah (http://arup.utah.edu/database/MEN2/MEN2_display.php) and is considered novel. Mutations in other genes besides RET can predispose for pheochromocytoma, but exome sequencing revealed no mutation in any of these genes in the proposita (NF1, VHL, SDHD, SDHC, SDHB, EGLN1/PHD2, KIF1, SDH5/SDHAF2, IDH1, TMEM127, SDHA, MAX, and HIF2).
The novel p.505_506del RET germline mutation is located in the extracellular part of RET, at the end of the cadherin-like domain and outside the cysteine-rich region. Pathogenic mutations in RET are usually found in the extracellular cysteine-rich domain (exons 10 and 11) or the intracellular tyrosine-kinase domain (16). So far, only one pathogenic mutation is known upstream of the cysteine-rich region. This mutation is located in codon 292 in exon 5 (cadherin like domain 3) and is associated with a mild form of MEN2 (19). For MTC, a genotype-phenotype correlation is well known (20–22). The p.505_506del mutation is a gain-of-function mutation but leads to a weaker activation of MAPK/ERK and PI3/AKT pathway than the most frequent activating C634R mutation, which we used as a positive control for functional characterization of our novel deletion mutation. This may explain the unusual phenotype of our patient with development of bilateral pheochromocytoma 6 years before manifestation of an MTC. The course of disease in our patient and the in vitro characteristics of the 505_506del RET mutant seem to be comparable to the V292M RET mutation in exon 5 (19).
The molecular mechanism underlying the activating effects of the novel 505_506del RET mutant are not fully clear. Usually, mutations in the cysteine-rich region affecting a cysteine lead to an aberrant interchain disulphide bonding, which then mimics ligand-induced RET homodimerization (23). We could not observe homodimerization and assume that the p.505_506del mutation affects the conformation of the tyrosine kinase domain, facilitating ligand-independent downstream signaling.
Although RET has been described as the susceptibility gene for MEN2 25 years ago, neither this 505_506del mutation nor any other activating mutations in exon 7 of RET have been reported. This may be explained by the fact that a tiered approach, starting with analysis of the most commonly affected RET exons, already solved the vast majority of cases. If this first step failed to detect a RET mutation, further search for novel mutations outside those hotspot exons was not mandatory and certainly not clinical routine, possibly leaving rare exon 7 mutations undiscovered (24). In addition, the phenotype of our patient differs from the common MEN2A patient with development of pheochromocytoma many years before an MTC would have become apparent. Limited sequencing of only hotspot exons, especially in pheochromocytoma patients with no other manifestations suggestive of MEN2, may explain why no other mutations in exon 7 of RET have been reported until now. It is conceivable that among patients with pheochromocytoma and no known mutation some actually harbor an uncommon RET mutation and are predisposed for development of MTC, but have not yet been diagnosed as RET mutation carriers and MEN2A patients. This may especially be true for patients with de novo mutations, such as our patient, because the negative family history provides no immediate clues for a germline mutation.
The specific 505_506del RET mutation reported here is certainly not frequent enough to justify whole-exome sequencing and could have been detected by Sanger sequencing. But this novel mutation in exon 7 of RET provides an example that demonstrates the importance of sequencing the entire coding region of RET in patients with MEN2 when no mutation is identified in the commonly affected exons (25).
Acknowledgments
We thank Dr Massimo Santoro for providing the RET9 wil-type plasmid and Sebastian Hoenes for excellent support during the experiments.
This work was supported by Deutsche Forschungsgemeinschaft DFG Grants FU356/8-1 (to D.F.), MO1018/2-1 (to L.C.M), and the IFORES program, Faculty of Medicine, Duisburg-Essen.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- MEN2
multiple endocrine neoplasia type 2
- JNK
Jun NH2-terminal protein kinase
- MTC
medullary thyroid carcinoma
- PVDF
polyvinylidene fluoride
- SRE
serum response element
- WT
wild type.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}