





Neonatal central diabetes insipidus (CDI) with or without adipsia is a very rare complication of various complex hypothalamic disorders. It is associated with greater morbidity and a high risk of developing both hypernatremia and hyponatremia, due to the condition itself or secondary to treatment with vasopressin analogs or fluid administration. Its outcomes have yet to be evaluated.
To investigate the clinical outcomes of patients with neonatal-onset CDI or adipsic CDI with hypernatremia.
All patients diagnosed with neonatal CDI in a university hospital-based observational study and followed between 2005 and 2015 were included and analyzed retrospectively.
The various causes of CDI were grouped. Clinical outcome and comorbidities were analyzed.
Ten of the 12 patients had an underlying condition with brain malformations: optic nerve hypoplasia (n = 3), septo-optic dysplasia (n = 2), semilobar holoprosencephaly (n = 1), ectopic neurohypophysis (n = 3), and unilateral absence of the internal carotid artery (n = 1). The other two were idiopathic cases. During the median follow-up period of 7.8 (4.9–16.8) years, all but one patient displayed anterior pituitary deficiency. Transient CDI was found in three (25%) patients for whom a posterior pituitary hyperintense signal was observed with (n = 2) and without (n = 1) structural hypothalamic pituitary abnormalities, and with no other underlying cerebral malformations. Patients with permanent CDI with persistent adipsia (n = 4) and without adipsia (n = 5) required adequate fluid intake and various doses of desamino-D-arginine-8-vasopressin. Those with adipsia were more likely to develop hypernatremia (45 vs 33%), hyponatremia (16 vs 4%) (P < .0001), and severe neurodevelopmental delay (P < .05) than those without adipsia. Comorbidities were common. The underlying cause remains unknown at the age of 23 years for one patient with CDI and normal thirst.
Neonatal CDI may be transient or permanent. These vulnerable patients have high rates of comorbidity and require careful monitoring.
Central diabetes insipidus (CDI) may have two main causes. First, it may be due to a lack of arginine vasopressin (AVP) secretion, with hypotonic polyuria and thirst secondary to an increase in plasma osmolality. Alternatively, it may be due to dysregulation of the osmotically stimulated release of AVP, mostly due to inadequate AVP synthesis, with weak or absent thirst sensations after an increase in plasma osmolality, generally referred to as adipsic diabetes insipidus (DI) or essential or neurogenic hypernatremia or hypothalamic adipsic hypernatremia syndrome (1). In adipsic DI with hypernatremia, the site of the lesion is the osmoreceptor cells in the anterior hypothalamus rather than the supraoptic and paraventricular nuclei, which synthesize AVP, or the posterior pituitary (PP), from which AVP is released into the bloodstream (2). The detection of PP hyperintensity on cerebral magnetic resonance imaging (MRI) is consistent with a diagnosis of adipsic DI, indicating a persistent pituitary store of AVP, although DI is always associated with a loss of PP signal due to the lack of AVP storage in the PP (3).
CDI is rare in neonates and, with the exception of one known family (4), it is not usually related to an identified genetic defect in neurophysin production because it takes time for the toxic effects on AVP-producing neurons to result in a decrease in AVP production (5–7). Possible causes include intraventricular hemorrhage, hypoxemia, tumors, infections, and congenital cerebrovascular abnormalities, as reported in exceptional cases (8–12). Adipsic DI in infants may be due to complex and rare congenital malformations with cerebral midline defects, such as septo-optic dysplasia (SOD) or holoprosencephaly. It may be associated with structural hypothalamic-pituitary abnormalities, such as anterior pituitary hypoplasia, ectopic PP (EPP), and/or the absence of a visible pituitary stalk (13–19). Isolated EPP without an additional midline defect has not been reported to cause adipsic DI with thirst and electrolyte disturbances. Patients with CDI can replace urinary water loss through the oral intake of fluid. They should therefore be able to maintain a normal plasma sodium concentration. However, neonates, in particular, are at risk of hypernatremia, particularly if their access to fluids is limited due to a lack of recognition of the condition by parents or caregivers. Patients with adipsic DI feel no thirst and therefore have no urge to replace fluids. They are therefore susceptible to hypernatremia. After a certain degree of dehydration has been reached, the volume receptors respond, leading to a release of AVP and the maintenance of clinical hypernatremia with concentrated urine. When sufficient water is administered to correct the hypovolemia, an inability to respond to high osmolality is demonstrated by excessive urine output, which may be corrected by treatment with AVP analogs (20–23). Only one case series has been reported for neonatal patients, due to the rarity of these complex conditions, but the long-term outcome of these patients has yet to be studied (19).
We therefore aimed to characterize the initial clinical characteristics of our cohort and to describe the outcome of children with neonatal-onset CDI or adipsic hypernatremia DI.
Patients and Methods
This observational cohort study included all patients (n = 12) identified in the database of the Pediatric Endocrinology Department of Robert Debré Hospital in Paris between 2005 and 2015 and followed for CDI or adipsic hypernatremia DI for more than 1 year after disease onset during infancy.
The diagnosis of CDI or adipsic hypernatremia was based on abnormal thirst (polydipsic/adipsic) with hypernatremia (≥146 mmol/L) associated with an inappropriately low urine osmolality (<300 mosm/kg) in cases of CDI and/or a context of brain malformation constituting a significant risk factor for CDI or adipsic hypernatremia, with an appropriate desamino-D-arginine-8-vasopressin (DDAVP) response for urine osmolality (normal, >750 mosm/kg). Plasma creatinine and urea concentrations were normal in all patients. All patients were regularly managed by our team of pediatric endocrinologists, by individual adjustment of fluid intake, as a function of the thirst abnormality, and with nasal or, preferably, oral (once it became available) DDAVP, initially administered once, twice, or three times daily.
Study protocol
Clinical data for the patients were obtained from their medical records. Demographic characteristics, perinatal events, such as gestational age, birth weight, mode of delivery, and neonatal distress (Apgar score < 7 or neonatal resuscitation), age, symptoms and biological data at diagnosis, cerebral MRI findings, and anterior pituitary function at diagnosis and during follow-up were recorded. We then recorded the management of DDAVP treatment during follow-up, total DDAVP treatment duration, the use of enteral nutritional and fluid support, medication for seizures and/or sleep disorders (central sleep apnea, insufficient duration and quality of sleep), clinical and laboratory test results, and outcome. The pediatricians did not follow a predefined treatment protocol, and patients were managed on an individual basis.
The study protocol was approved by the Ethics Review Committee for Biomedical Research Projects Paris Nord (no. 12–029). Informed consent was obtained from the parents.
Methods
Polyuria was defined as a urine volume ≥ 150 mL/kg/d during the first month of life, ≥ 120 mL/kg/d until the age of 2 years, and ≥ 50 mL/kg/d thereafter. Adipsia was defined as difficulty or refusal to feed despite hypernatremia.
Plasma sodium concentration data were recorded for our patients during follow-up. Serum electrolyte concentrations were checked at each visit, at 3- to 6-month intervals, with the precise timing determined by the patient's underlying condition, and with interim monitoring by the primary care physician in cases of abnormal findings (acute excess weight gain, behavior change, seizures). Plasma sodium concentrations were used to define mild (131–134 mmol/L) or severe (≤ 130 mmol/L) hyponatremia, normonatremia (135–145 mmol/L), and mild (146–149 mmol/L) or severe (≥150 mmol/L) hypernatremia (24).
A complete evaluation of anterior pituitary functions was carried out for all patients at diagnosis and was repeated during follow-up if deemed necessary on the basis of clinical examination and serum free T4 and morning cortisol determinations. GH deficiency (GHD) was diagnosed on the basis of the presence of an ectopic neurohypophysis on cerebral MRI findings and/or symptoms or signs of endocrine dysfunction (hypoglycemia and/or micropenis and/or a decrease in growth velocity during follow-up), low serum IGF-1 concentration, and a serum GH peak of < 10 μg/L or 20 mIU/L in a pharmacological stimulation test or during spontaneous hypoglycemia. TSH deficiency was diagnosed on the basis of a serum free T4 concentration below 10 pmol/L. ACTH deficiency was diagnosed on the basis of morning basal serum cortisol concentrations below 180 nmol/L and/or below 450 nmol/L during hypoglycemia or on the basis of low-dose short tetracosactide (Synacthen) tests. If morning cortisol concentrations exceeded 275 nmol/L, the corticotrophin reserve was not routinely evaluated. Because most of the patients were prepubertal, gonadotropin function was not evaluated in most cases. Such evaluations were reserved for those with symptoms (micropenis, cryptorchidism, agenesis of the olfactory bulbs, and/or sulcus agenesis) during the first 3 months of life. Gonadotropin deficiency was suspected in patients displaying no pubertal development at ages at which puberty would normally occur. It was assessed by measuring serum sex steroid concentrations and FSH and LH levels after GnRH stimulation or after induced puberty. Hyperprolactinemia was defined as basal serum prolactin concentrations above 25 ng/mL (to convert values to mIU/L, this value must be multiplied by 21.2) in children over the age of 3 months.
Body mass index (BMI; weight in kilograms divided by height in meters squared) is expressed as a standard deviation score for sex and chronological age (25). Developmental outcome was defined as normal (no impairment), moderate (intellectual disability), or severe (encephalopathy and psychomotor retardation, abnormal movements, unable to speak or to walk) global delay.
All MRI images (1.5 Tesla Magnet Philips Intera; Philips Medical Systems) were reviewed by the same investigator (M.E.), blind to the endocrinological data. Sagittal and coronal thin (1.5 mm) slices of the pituitary area were acquired with a gradient echo T1-weighted sequence, and coronal slices of the brain were acquired with a T2-weighted sequence. If the pituitary stalk was not visible, gadolinium injection was used. The height and appearance of the anterior pituitary were recorded and judged to be normal, hypoplastic (< −2 SD for age) according to normative data for children (26) or absent (not seen). The presence or absence of the pituitary stalk and the location of the PP hyperintense signal (PPHS) were recorded. Associated brain abnormalities were also described.
Statistical analysis
The results are expressed as numerical values (percentages) for categorical variables and as median (25–75th percentiles) for continuous variables. Comparisons were based on χ2 tests for categorical variables. All statistical analyses were carried out with SAS software, version 9.12 (SAS Institute Inc).
Results
Initial characteristics and underlying congenital disorders
The initial characteristics of the 12 patients are presented in Table 1. Hypernatremia was diagnosed during the first month of life in eight of the 12 patients and between the ages of 1.5 and 8 months in four patients. Patients were diagnosed on the basis of eye and/or endocrine symptoms or signs (including thirst abnormalities, hypoglycemia, cryptorchidism, and micropenis) and/or midline abnormalities (n = 6), or isolated failure to thrive (n = 5), with one patient diagnosed incidentally (n = 1). All but one of the patients presented fetal and/or neonatal distress or were born by cesarean section, and seven (58%) patients had recurrent episodes of fever. Nine patients (75%) had multiple pituitary hormone deficiency (MPHD).
Initial Clinical Characteristics of the 12 Patients With Neurogenic Hypernatremia or Central Neonatal DI
| Patient No. | Sex | Gestational Age, wk | Birth Weight, kg | Fetal Neonatal Distress | Delivery | Age at Diagnosis, d | Circumstances Diagnosis | Plasma Na+, mmol/L | Maximum Urine Osmolality, mOsmol/kg | Thirst | Polyuria | Hypothalamic Dysfunction | Other Abnormalities | Anterior Pituitary Deficiency |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 42 | 3.5 | No | Cesarean | 18 | Failure to thrive | 158 | 109 | Adipsic | No | Thermal dysregulation, central apnea | Cryptorchidism, hypoglycemia | GH, TSH, ACTH |
| 2 | F | 41 | 3.4 | Yes | Cesarean | 240 | SOD | 147 | 595 | Polydipsia | Yes | No | Microcephaly, hypoglycemia | GH, TSH |
| 3 | F | 41 | 3.5 | Yes | Vaginal | 3 | Failure to thrive | 159 | 207 | Adipsic | Yes | Thermal dysregulation | None | TSH, ACTH |
| 4 | F | 39 | 3.5 | No | Cesarean | 5 | Failure to thrive, thermal dysregulation | 166 | 163 | Adipsic | No | Thermal dysregulation | Palpebral ptosis, hypoglycemia | GH, TSH |
| 5 | F | 40 | 3 | Yes | Cesarean | 180 | SOD | 152 | 317 | Polydipsia | Yes | No | Angioma, plagiocephaly, CHD hypoglycemia | no |
| 6 | F | 40 | 3.1 | No | Vaginal | 90 | Before intervention for cleft lip palate | 160 | 170 | Adipsic | Yes | No | Microcephaly, cleft lip-palate hypercholesterolemia | no |
| 7 | F | 38 | 3.2 | Yes | Vaginal (forceps) | 30 | Failure to thrive | 158 | 455 | Adipsicc | No | Thermal dysregulation | CNPAS, SMMCI, coloboma hypoglycemia | GH, TSH, ACTH |
| 8 | M | 39 | 3.4 | Yes | Vaginal | 11 | Micropenis, hypoglycemia | 146 | 173 | Adipsic | Yes | No | Cryptorchidism, micropenis, hypoglycemia | GH, TSH, ACTH |
| 9 | F | 41 | 3 | Yes | Cesarean | 10 | Hypoglycemia | 150 | 123 | Adipsic | No | Thermal dysregulation | Cleft lip-palate, renal hypoplasia, hypoglycemia, chromosomal abnormalityd | GH, TSH, ACTH |
| 10 | M | 39a | 2.4 | Yes | Cesarean | 50 | Failure to thrive | 170 | 169 | Polydipsia | Yes | Thermal dysregulation | Cryptorchidism, micropenis, | GH, TSH |
| 11 | M | 30b | 1.5 | Yes | Vaginal | 10 | Polyuro-polydipsia | 157 | 125 | Polydipsia | Yes | Thermal dysregulation | GER, inguinal hernia | no |
| 12 | M | 39 | 2.4 | Yes | Vaginal (forceps) | 5 | Failure to thrive cryptorchidism, micropenis | 154 | 390 | Adipsic | No | No | Cryptorchidism, micropenis, dextrocardia, GER, butterfly vertebra, renal cyst, hypoglycemia | GH, TSH |
| Patient No. | Sex | Gestational Age, wk | Birth Weight, kg | Fetal Neonatal Distress | Delivery | Age at Diagnosis, d | Circumstances Diagnosis | Plasma Na+, mmol/L | Maximum Urine Osmolality, mOsmol/kg | Thirst | Polyuria | Hypothalamic Dysfunction | Other Abnormalities | Anterior Pituitary Deficiency |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 42 | 3.5 | No | Cesarean | 18 | Failure to thrive | 158 | 109 | Adipsic | No | Thermal dysregulation, central apnea | Cryptorchidism, hypoglycemia | GH, TSH, ACTH |
| 2 | F | 41 | 3.4 | Yes | Cesarean | 240 | SOD | 147 | 595 | Polydipsia | Yes | No | Microcephaly, hypoglycemia | GH, TSH |
| 3 | F | 41 | 3.5 | Yes | Vaginal | 3 | Failure to thrive | 159 | 207 | Adipsic | Yes | Thermal dysregulation | None | TSH, ACTH |
| 4 | F | 39 | 3.5 | No | Cesarean | 5 | Failure to thrive, thermal dysregulation | 166 | 163 | Adipsic | No | Thermal dysregulation | Palpebral ptosis, hypoglycemia | GH, TSH |
| 5 | F | 40 | 3 | Yes | Cesarean | 180 | SOD | 152 | 317 | Polydipsia | Yes | No | Angioma, plagiocephaly, CHD hypoglycemia | no |
| 6 | F | 40 | 3.1 | No | Vaginal | 90 | Before intervention for cleft lip palate | 160 | 170 | Adipsic | Yes | No | Microcephaly, cleft lip-palate hypercholesterolemia | no |
| 7 | F | 38 | 3.2 | Yes | Vaginal (forceps) | 30 | Failure to thrive | 158 | 455 | Adipsicc | No | Thermal dysregulation | CNPAS, SMMCI, coloboma hypoglycemia | GH, TSH, ACTH |
| 8 | M | 39 | 3.4 | Yes | Vaginal | 11 | Micropenis, hypoglycemia | 146 | 173 | Adipsic | Yes | No | Cryptorchidism, micropenis, hypoglycemia | GH, TSH, ACTH |
| 9 | F | 41 | 3 | Yes | Cesarean | 10 | Hypoglycemia | 150 | 123 | Adipsic | No | Thermal dysregulation | Cleft lip-palate, renal hypoplasia, hypoglycemia, chromosomal abnormalityd | GH, TSH, ACTH |
| 10 | M | 39a | 2.4 | Yes | Cesarean | 50 | Failure to thrive | 170 | 169 | Polydipsia | Yes | Thermal dysregulation | Cryptorchidism, micropenis, | GH, TSH |
| 11 | M | 30b | 1.5 | Yes | Vaginal | 10 | Polyuro-polydipsia | 157 | 125 | Polydipsia | Yes | Thermal dysregulation | GER, inguinal hernia | no |
| 12 | M | 39 | 2.4 | Yes | Vaginal (forceps) | 5 | Failure to thrive cryptorchidism, micropenis | 154 | 390 | Adipsic | No | No | Cryptorchidism, micropenis, dextrocardia, GER, butterfly vertebra, renal cyst, hypoglycemia | GH, TSH |
Abbreviations: M, male; F, female; GER, gastroesophageal reflux; CHD, congenital hip dislocation.
Small for gestational age.
Prematurity.
Became polydipsic during follow-up.
Microdeletion 18p11.32.
Initial Clinical Characteristics of the 12 Patients With Neurogenic Hypernatremia or Central Neonatal DI
| Patient No. | Sex | Gestational Age, wk | Birth Weight, kg | Fetal Neonatal Distress | Delivery | Age at Diagnosis, d | Circumstances Diagnosis | Plasma Na+, mmol/L | Maximum Urine Osmolality, mOsmol/kg | Thirst | Polyuria | Hypothalamic Dysfunction | Other Abnormalities | Anterior Pituitary Deficiency |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 42 | 3.5 | No | Cesarean | 18 | Failure to thrive | 158 | 109 | Adipsic | No | Thermal dysregulation, central apnea | Cryptorchidism, hypoglycemia | GH, TSH, ACTH |
| 2 | F | 41 | 3.4 | Yes | Cesarean | 240 | SOD | 147 | 595 | Polydipsia | Yes | No | Microcephaly, hypoglycemia | GH, TSH |
| 3 | F | 41 | 3.5 | Yes | Vaginal | 3 | Failure to thrive | 159 | 207 | Adipsic | Yes | Thermal dysregulation | None | TSH, ACTH |
| 4 | F | 39 | 3.5 | No | Cesarean | 5 | Failure to thrive, thermal dysregulation | 166 | 163 | Adipsic | No | Thermal dysregulation | Palpebral ptosis, hypoglycemia | GH, TSH |
| 5 | F | 40 | 3 | Yes | Cesarean | 180 | SOD | 152 | 317 | Polydipsia | Yes | No | Angioma, plagiocephaly, CHD hypoglycemia | no |
| 6 | F | 40 | 3.1 | No | Vaginal | 90 | Before intervention for cleft lip palate | 160 | 170 | Adipsic | Yes | No | Microcephaly, cleft lip-palate hypercholesterolemia | no |
| 7 | F | 38 | 3.2 | Yes | Vaginal (forceps) | 30 | Failure to thrive | 158 | 455 | Adipsicc | No | Thermal dysregulation | CNPAS, SMMCI, coloboma hypoglycemia | GH, TSH, ACTH |
| 8 | M | 39 | 3.4 | Yes | Vaginal | 11 | Micropenis, hypoglycemia | 146 | 173 | Adipsic | Yes | No | Cryptorchidism, micropenis, hypoglycemia | GH, TSH, ACTH |
| 9 | F | 41 | 3 | Yes | Cesarean | 10 | Hypoglycemia | 150 | 123 | Adipsic | No | Thermal dysregulation | Cleft lip-palate, renal hypoplasia, hypoglycemia, chromosomal abnormalityd | GH, TSH, ACTH |
| 10 | M | 39a | 2.4 | Yes | Cesarean | 50 | Failure to thrive | 170 | 169 | Polydipsia | Yes | Thermal dysregulation | Cryptorchidism, micropenis, | GH, TSH |
| 11 | M | 30b | 1.5 | Yes | Vaginal | 10 | Polyuro-polydipsia | 157 | 125 | Polydipsia | Yes | Thermal dysregulation | GER, inguinal hernia | no |
| 12 | M | 39 | 2.4 | Yes | Vaginal (forceps) | 5 | Failure to thrive cryptorchidism, micropenis | 154 | 390 | Adipsic | No | No | Cryptorchidism, micropenis, dextrocardia, GER, butterfly vertebra, renal cyst, hypoglycemia | GH, TSH |
| Patient No. | Sex | Gestational Age, wk | Birth Weight, kg | Fetal Neonatal Distress | Delivery | Age at Diagnosis, d | Circumstances Diagnosis | Plasma Na+, mmol/L | Maximum Urine Osmolality, mOsmol/kg | Thirst | Polyuria | Hypothalamic Dysfunction | Other Abnormalities | Anterior Pituitary Deficiency |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 42 | 3.5 | No | Cesarean | 18 | Failure to thrive | 158 | 109 | Adipsic | No | Thermal dysregulation, central apnea | Cryptorchidism, hypoglycemia | GH, TSH, ACTH |
| 2 | F | 41 | 3.4 | Yes | Cesarean | 240 | SOD | 147 | 595 | Polydipsia | Yes | No | Microcephaly, hypoglycemia | GH, TSH |
| 3 | F | 41 | 3.5 | Yes | Vaginal | 3 | Failure to thrive | 159 | 207 | Adipsic | Yes | Thermal dysregulation | None | TSH, ACTH |
| 4 | F | 39 | 3.5 | No | Cesarean | 5 | Failure to thrive, thermal dysregulation | 166 | 163 | Adipsic | No | Thermal dysregulation | Palpebral ptosis, hypoglycemia | GH, TSH |
| 5 | F | 40 | 3 | Yes | Cesarean | 180 | SOD | 152 | 317 | Polydipsia | Yes | No | Angioma, plagiocephaly, CHD hypoglycemia | no |
| 6 | F | 40 | 3.1 | No | Vaginal | 90 | Before intervention for cleft lip palate | 160 | 170 | Adipsic | Yes | No | Microcephaly, cleft lip-palate hypercholesterolemia | no |
| 7 | F | 38 | 3.2 | Yes | Vaginal (forceps) | 30 | Failure to thrive | 158 | 455 | Adipsicc | No | Thermal dysregulation | CNPAS, SMMCI, coloboma hypoglycemia | GH, TSH, ACTH |
| 8 | M | 39 | 3.4 | Yes | Vaginal | 11 | Micropenis, hypoglycemia | 146 | 173 | Adipsic | Yes | No | Cryptorchidism, micropenis, hypoglycemia | GH, TSH, ACTH |
| 9 | F | 41 | 3 | Yes | Cesarean | 10 | Hypoglycemia | 150 | 123 | Adipsic | No | Thermal dysregulation | Cleft lip-palate, renal hypoplasia, hypoglycemia, chromosomal abnormalityd | GH, TSH, ACTH |
| 10 | M | 39a | 2.4 | Yes | Cesarean | 50 | Failure to thrive | 170 | 169 | Polydipsia | Yes | Thermal dysregulation | Cryptorchidism, micropenis, | GH, TSH |
| 11 | M | 30b | 1.5 | Yes | Vaginal | 10 | Polyuro-polydipsia | 157 | 125 | Polydipsia | Yes | Thermal dysregulation | GER, inguinal hernia | no |
| 12 | M | 39 | 2.4 | Yes | Vaginal (forceps) | 5 | Failure to thrive cryptorchidism, micropenis | 154 | 390 | Adipsic | No | No | Cryptorchidism, micropenis, dextrocardia, GER, butterfly vertebra, renal cyst, hypoglycemia | GH, TSH |
Abbreviations: M, male; F, female; GER, gastroesophageal reflux; CHD, congenital hip dislocation.
Small for gestational age.
Prematurity.
Became polydipsic during follow-up.
Microdeletion 18p11.32.
Cerebral MRI findings identified an underlying congenital malformation in 10 patients: optic nerve hypoplasia (n = 3), SOD (n = 2), semilobar holoprosencephaly (n = 1), structural hypothalamic pituitary abnormalities with an ectopic PPHS at the median eminence (n = 3), including a mild form of holoprosencephaly with congenital nasal pyriform aperture stenosis (CNPAS), and solitary median maxillary central incisor (SMMCI) in one case, and an absence of the left internal carotid artery (n = 1). Two patients, one with and the other without a visible hyperintense signal for the neurohypophysis, had no identifiable underlying cause of CDI on MRI. A high frequency of olfactory bulb and/or sulcus agenesis (n = 5) and brain abnormalities, such as interdigitating frontal gyri (n = 4) or abnormalities of gyration (n = 1) were demonstrated in patients with optic nerve hypoplasia, SOD, and holoprosencephaly (Table 2 and Figure 1).
MRI Findings for the 12 Patients
| Patient No. | Underlying Diagnosis | Anterior Pituitary | Pituitary Stalk | PPHS | Chiasma Optic Nerve | Septum Pellucidum | Olfactory Bulbs and/or Sulcus | Corpus Callosum | Ventricular Distension | Midline Abnormalities | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Optic nerve hypoplasia | Normal | Present | Absent | Hypoplasia | Present | Agenesis | Normal | No | Interdigitating frontal gyri, peduncle cyst, aqueduct stenosis | Arachnoid cyst |
| 2 | Optic nerve hypoplasia | Normal | Present | Absent | Hypoplasia | Present | Agenesis | Thin | No | Interdigitating frontal gyri | No |
| 3 | Optic nerve hypoplasia | Normal | Present | Absent | Hypoplasia | Present | Agenesis | Normal | Yes | Tentorium cerebelli dehiscence | Abnormalities of gyration, ischemic lesion, hypoplastic right oculomotor nerve III |
| 4 | SOD | Hypoplasia | Present | Absent | Hypoplasia | Absent | Present | Normal | Yes | Interdigitating frontal gyri | Hypoplasia of left lenticular and caudate nucleus, hypoplastic left oculomotor nerve III |
| 5 | SOD | Hypoplasia | Present | Absent | Hypoplasia | Absent | Agenesis | Normal | Yes | Frontal brain asymmetry, interdigitating frontal gyri | Frontal calcification, plagiocephaly, arachnoid cyst |
| 6 | Semi lobar holoprosencephaly | Normal | Present | Present | Normal | Present | Agenesis | Partial agenesis | No | Frontal lobe fusion | no |
| 7 | Structural hypothalamic pituitary abnormalities | Aplasia | Absent | Present (ectopic) | Normal | Present | Present | Normal | No | CNPAS, SMMCI | no |
| 8 | Structural hypothalamic pituitary abnormalities | Hypoplasia | Absent | Present (ectopic) | Normal | Present | Present | Normal | No | no | no |
| 9 | Structural hypothalamic pituitary abnormalities | Hypoplasia | Absent | Present (ectopic) | Normal | Present | Present | Normal | No | no | no |
| 10 | Agenesis of the left internal carotid artery | Hypoplasia | Present | Absent | Normal | Present | Present | Normal | No | Basilar impression, cerebral peduncle pathology | Agenesis of the left internal carotid artery, vertebral cervical pathology |
| 11 | No cerebral malformation | Normal | Present | Absent | Normal | Present | Present | Normal | No | no | no |
| 12 | No cerebral malformation | Normal | Present | Present | Normal | Present | Present | Thin | Yes | no | no |
| Patient No. | Underlying Diagnosis | Anterior Pituitary | Pituitary Stalk | PPHS | Chiasma Optic Nerve | Septum Pellucidum | Olfactory Bulbs and/or Sulcus | Corpus Callosum | Ventricular Distension | Midline Abnormalities | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Optic nerve hypoplasia | Normal | Present | Absent | Hypoplasia | Present | Agenesis | Normal | No | Interdigitating frontal gyri, peduncle cyst, aqueduct stenosis | Arachnoid cyst |
| 2 | Optic nerve hypoplasia | Normal | Present | Absent | Hypoplasia | Present | Agenesis | Thin | No | Interdigitating frontal gyri | No |
| 3 | Optic nerve hypoplasia | Normal | Present | Absent | Hypoplasia | Present | Agenesis | Normal | Yes | Tentorium cerebelli dehiscence | Abnormalities of gyration, ischemic lesion, hypoplastic right oculomotor nerve III |
| 4 | SOD | Hypoplasia | Present | Absent | Hypoplasia | Absent | Present | Normal | Yes | Interdigitating frontal gyri | Hypoplasia of left lenticular and caudate nucleus, hypoplastic left oculomotor nerve III |
| 5 | SOD | Hypoplasia | Present | Absent | Hypoplasia | Absent | Agenesis | Normal | Yes | Frontal brain asymmetry, interdigitating frontal gyri | Frontal calcification, plagiocephaly, arachnoid cyst |
| 6 | Semi lobar holoprosencephaly | Normal | Present | Present | Normal | Present | Agenesis | Partial agenesis | No | Frontal lobe fusion | no |
| 7 | Structural hypothalamic pituitary abnormalities | Aplasia | Absent | Present (ectopic) | Normal | Present | Present | Normal | No | CNPAS, SMMCI | no |
| 8 | Structural hypothalamic pituitary abnormalities | Hypoplasia | Absent | Present (ectopic) | Normal | Present | Present | Normal | No | no | no |
| 9 | Structural hypothalamic pituitary abnormalities | Hypoplasia | Absent | Present (ectopic) | Normal | Present | Present | Normal | No | no | no |
| 10 | Agenesis of the left internal carotid artery | Hypoplasia | Present | Absent | Normal | Present | Present | Normal | No | Basilar impression, cerebral peduncle pathology | Agenesis of the left internal carotid artery, vertebral cervical pathology |
| 11 | No cerebral malformation | Normal | Present | Absent | Normal | Present | Present | Normal | No | no | no |
| 12 | No cerebral malformation | Normal | Present | Present | Normal | Present | Present | Thin | Yes | no | no |
Aplasia, ≤1 mm; hypoplasia, <3 mm; normal, ≥3 mm.
MRI Findings for the 12 Patients
| Patient No. | Underlying Diagnosis | Anterior Pituitary | Pituitary Stalk | PPHS | Chiasma Optic Nerve | Septum Pellucidum | Olfactory Bulbs and/or Sulcus | Corpus Callosum | Ventricular Distension | Midline Abnormalities | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Optic nerve hypoplasia | Normal | Present | Absent | Hypoplasia | Present | Agenesis | Normal | No | Interdigitating frontal gyri, peduncle cyst, aqueduct stenosis | Arachnoid cyst |
| 2 | Optic nerve hypoplasia | Normal | Present | Absent | Hypoplasia | Present | Agenesis | Thin | No | Interdigitating frontal gyri | No |
| 3 | Optic nerve hypoplasia | Normal | Present | Absent | Hypoplasia | Present | Agenesis | Normal | Yes | Tentorium cerebelli dehiscence | Abnormalities of gyration, ischemic lesion, hypoplastic right oculomotor nerve III |
| 4 | SOD | Hypoplasia | Present | Absent | Hypoplasia | Absent | Present | Normal | Yes | Interdigitating frontal gyri | Hypoplasia of left lenticular and caudate nucleus, hypoplastic left oculomotor nerve III |
| 5 | SOD | Hypoplasia | Present | Absent | Hypoplasia | Absent | Agenesis | Normal | Yes | Frontal brain asymmetry, interdigitating frontal gyri | Frontal calcification, plagiocephaly, arachnoid cyst |
| 6 | Semi lobar holoprosencephaly | Normal | Present | Present | Normal | Present | Agenesis | Partial agenesis | No | Frontal lobe fusion | no |
| 7 | Structural hypothalamic pituitary abnormalities | Aplasia | Absent | Present (ectopic) | Normal | Present | Present | Normal | No | CNPAS, SMMCI | no |
| 8 | Structural hypothalamic pituitary abnormalities | Hypoplasia | Absent | Present (ectopic) | Normal | Present | Present | Normal | No | no | no |
| 9 | Structural hypothalamic pituitary abnormalities | Hypoplasia | Absent | Present (ectopic) | Normal | Present | Present | Normal | No | no | no |
| 10 | Agenesis of the left internal carotid artery | Hypoplasia | Present | Absent | Normal | Present | Present | Normal | No | Basilar impression, cerebral peduncle pathology | Agenesis of the left internal carotid artery, vertebral cervical pathology |
| 11 | No cerebral malformation | Normal | Present | Absent | Normal | Present | Present | Normal | No | no | no |
| 12 | No cerebral malformation | Normal | Present | Present | Normal | Present | Present | Thin | Yes | no | no |
| Patient No. | Underlying Diagnosis | Anterior Pituitary | Pituitary Stalk | PPHS | Chiasma Optic Nerve | Septum Pellucidum | Olfactory Bulbs and/or Sulcus | Corpus Callosum | Ventricular Distension | Midline Abnormalities | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Optic nerve hypoplasia | Normal | Present | Absent | Hypoplasia | Present | Agenesis | Normal | No | Interdigitating frontal gyri, peduncle cyst, aqueduct stenosis | Arachnoid cyst |
| 2 | Optic nerve hypoplasia | Normal | Present | Absent | Hypoplasia | Present | Agenesis | Thin | No | Interdigitating frontal gyri | No |
| 3 | Optic nerve hypoplasia | Normal | Present | Absent | Hypoplasia | Present | Agenesis | Normal | Yes | Tentorium cerebelli dehiscence | Abnormalities of gyration, ischemic lesion, hypoplastic right oculomotor nerve III |
| 4 | SOD | Hypoplasia | Present | Absent | Hypoplasia | Absent | Present | Normal | Yes | Interdigitating frontal gyri | Hypoplasia of left lenticular and caudate nucleus, hypoplastic left oculomotor nerve III |
| 5 | SOD | Hypoplasia | Present | Absent | Hypoplasia | Absent | Agenesis | Normal | Yes | Frontal brain asymmetry, interdigitating frontal gyri | Frontal calcification, plagiocephaly, arachnoid cyst |
| 6 | Semi lobar holoprosencephaly | Normal | Present | Present | Normal | Present | Agenesis | Partial agenesis | No | Frontal lobe fusion | no |
| 7 | Structural hypothalamic pituitary abnormalities | Aplasia | Absent | Present (ectopic) | Normal | Present | Present | Normal | No | CNPAS, SMMCI | no |
| 8 | Structural hypothalamic pituitary abnormalities | Hypoplasia | Absent | Present (ectopic) | Normal | Present | Present | Normal | No | no | no |
| 9 | Structural hypothalamic pituitary abnormalities | Hypoplasia | Absent | Present (ectopic) | Normal | Present | Present | Normal | No | no | no |
| 10 | Agenesis of the left internal carotid artery | Hypoplasia | Present | Absent | Normal | Present | Present | Normal | No | Basilar impression, cerebral peduncle pathology | Agenesis of the left internal carotid artery, vertebral cervical pathology |
| 11 | No cerebral malformation | Normal | Present | Absent | Normal | Present | Present | Normal | No | no | no |
| 12 | No cerebral malformation | Normal | Present | Present | Normal | Present | Present | Thin | Yes | no | no |
Aplasia, ≤1 mm; hypoplasia, <3 mm; normal, ≥3 mm.
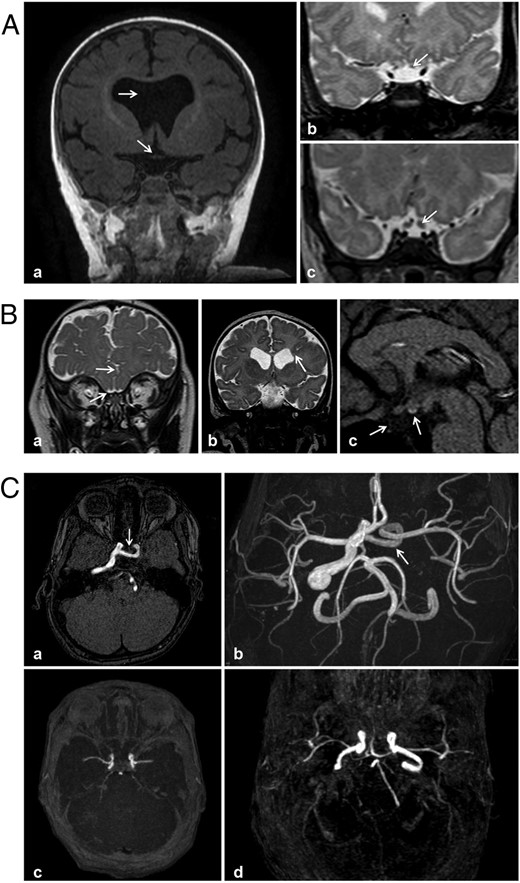
MRI features of patients with neonatal hypothalamic adipsic hypernatremia syndrome (A and B) or CDI (C). Aa, Patient 5; T1-weighted coronal view: septal agenesis and chiasma optic hypoplasia (arrow). Ab and Ac, T2-weighted coronal views: optic nerve hypoplasia (arrow). Ab, patient 1; Ac, normal optic nerve (arrow). Ba, Patient 1; coronal T2-weighted image: interdigitation of the frontal gyri in the midline, agenesis of the olfactory bulbs with existing olfactory gyri, hypoplastic orbital optic nerves. Bb, Patient 3; coronal T2-weighted image through ventricle frontal horns. Square frontal horns with (right) heterotopic subependymal gray matter nodules (arrowheads), and (left) an abnormal deep gyrus (arrow) coming into contact with the lateral wall of the ventricle (without a schizencephalic appearance). Bc, Patient 8; sagittal T1-weighted image: ectopic PP and agenesis of the pituitary stalk and anterior pituitary gland. C, Magnetic resonance time-of-flight angiography. Ca and Cb, Patient 10; agenesis of the cervical and intrapetrous segments of the left internal carotid artery, with an absence of normal flow void in the left ipsilateral cavernous sinus, and with a transsellar anastomotic vessel connecting with the intracavernous internal carotid artery. Cc and Cd, Normal appearance.
Outcome
Table 3 shows the characteristics of the 12 patients at a median age of 8.1 (5.0–16.8) years. The median total duration of follow-up until the last evaluation was 7.8 (4.9–16.8) years.
Characteristics of the 12 Patients at the Last Evaluation
| Patient No. | Age, y | Follow-up Duration, y | BMI, SDS | Anterior Pituitary Dysfunctiona | Neuro- Developmental Delay | Seizures | Visual Impairment | Use of Sleep Medication | Duration of Enteral Nutritional and Fluid Support, y | DDAVP Treatment at the Last Evaluation | DDAVP Oral Melt/Tablets | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose μg/d | Dose μg/kg/d | |||||||||||
| 1 | 5.0 | 4.9 | 0.5 | GH, TSH, ACTH | Severe | Yes | Yes | Yes | — | Yes | 105 | 6.8 |
| 2 | 8.1 | 7.8 | −1.7 | GH, TSH, ACTH | Moderate | No | Yes | No | — | Yes | 30 | 1.6 |
| 3 | 1.5 | 1.3 | 2.5 | GH, TSH, ACTH | Severe | Yes | Yes | Yes | 0.3 | Yes | 15 | 1.2 |
| 4 | 16.4 | 16.0 | −0.2 | GH, TSH | Severe | Yes | Yes | No | — | Yes | 85 | 2.1b |
| 5 | 3.1 | 2.5 | 0.2 | GH | Moderate | No | Yes | No | — | Yes | 150 | 10.3 |
| 6 | 20.0 | 19.8 | 2.3 | No | Moderate | No | No | No | — | Yes | 700 | 12.7b |
| 7 | 18.4 | 18.3 | 0.6 | GH, TSH, ACTH, FSH-LH | Absent | No | No | No | — | Yes | 180 | 3.1 |
| 8 | 16.8 | 16.8 | 1.8 | GH, TSH, ACTH, FSH-LH | Absent | No | No | No | — | Stopped at the age 2.5 y | — | — |
| 9 | 6.2 | 6.0 | 2.6 | GH, TSH, ACTH | Severe | Yes | No | No | 4.5 | Never treated (resolved at 6 mo of age) | — | — |
| 10 | 7.8 | 7.8 | 1.5 | GH, TSH | Moderate | No | No | No | 1.5 | Yes | 600 | 21.3 |
| 11 | 23.0 | 22.1 | 0.7 | GH | Absent | No | No | No | — | Yes | 600 | 8.5b |
| 12 | 8.0 | 7.8 | −0.5 | GH | Severe | Yes | No | Yes | 4.1 | Stopped at the age 3.6 y | No | No |
| Patient No. | Age, y | Follow-up Duration, y | BMI, SDS | Anterior Pituitary Dysfunctiona | Neuro- Developmental Delay | Seizures | Visual Impairment | Use of Sleep Medication | Duration of Enteral Nutritional and Fluid Support, y | DDAVP Treatment at the Last Evaluation | DDAVP Oral Melt/Tablets | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose μg/d | Dose μg/kg/d | |||||||||||
| 1 | 5.0 | 4.9 | 0.5 | GH, TSH, ACTH | Severe | Yes | Yes | Yes | — | Yes | 105 | 6.8 |
| 2 | 8.1 | 7.8 | −1.7 | GH, TSH, ACTH | Moderate | No | Yes | No | — | Yes | 30 | 1.6 |
| 3 | 1.5 | 1.3 | 2.5 | GH, TSH, ACTH | Severe | Yes | Yes | Yes | 0.3 | Yes | 15 | 1.2 |
| 4 | 16.4 | 16.0 | −0.2 | GH, TSH | Severe | Yes | Yes | No | — | Yes | 85 | 2.1b |
| 5 | 3.1 | 2.5 | 0.2 | GH | Moderate | No | Yes | No | — | Yes | 150 | 10.3 |
| 6 | 20.0 | 19.8 | 2.3 | No | Moderate | No | No | No | — | Yes | 700 | 12.7b |
| 7 | 18.4 | 18.3 | 0.6 | GH, TSH, ACTH, FSH-LH | Absent | No | No | No | — | Yes | 180 | 3.1 |
| 8 | 16.8 | 16.8 | 1.8 | GH, TSH, ACTH, FSH-LH | Absent | No | No | No | — | Stopped at the age 2.5 y | — | — |
| 9 | 6.2 | 6.0 | 2.6 | GH, TSH, ACTH | Severe | Yes | No | No | 4.5 | Never treated (resolved at 6 mo of age) | — | — |
| 10 | 7.8 | 7.8 | 1.5 | GH, TSH | Moderate | No | No | No | 1.5 | Yes | 600 | 21.3 |
| 11 | 23.0 | 22.1 | 0.7 | GH | Absent | No | No | No | — | Yes | 600 | 8.5b |
| 12 | 8.0 | 7.8 | −0.5 | GH | Severe | Yes | No | Yes | 4.1 | Stopped at the age 3.6 y | No | No |
None of the patients had hyperprolactinemia.
Tablet.
Characteristics of the 12 Patients at the Last Evaluation
| Patient No. | Age, y | Follow-up Duration, y | BMI, SDS | Anterior Pituitary Dysfunctiona | Neuro- Developmental Delay | Seizures | Visual Impairment | Use of Sleep Medication | Duration of Enteral Nutritional and Fluid Support, y | DDAVP Treatment at the Last Evaluation | DDAVP Oral Melt/Tablets | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose μg/d | Dose μg/kg/d | |||||||||||
| 1 | 5.0 | 4.9 | 0.5 | GH, TSH, ACTH | Severe | Yes | Yes | Yes | — | Yes | 105 | 6.8 |
| 2 | 8.1 | 7.8 | −1.7 | GH, TSH, ACTH | Moderate | No | Yes | No | — | Yes | 30 | 1.6 |
| 3 | 1.5 | 1.3 | 2.5 | GH, TSH, ACTH | Severe | Yes | Yes | Yes | 0.3 | Yes | 15 | 1.2 |
| 4 | 16.4 | 16.0 | −0.2 | GH, TSH | Severe | Yes | Yes | No | — | Yes | 85 | 2.1b |
| 5 | 3.1 | 2.5 | 0.2 | GH | Moderate | No | Yes | No | — | Yes | 150 | 10.3 |
| 6 | 20.0 | 19.8 | 2.3 | No | Moderate | No | No | No | — | Yes | 700 | 12.7b |
| 7 | 18.4 | 18.3 | 0.6 | GH, TSH, ACTH, FSH-LH | Absent | No | No | No | — | Yes | 180 | 3.1 |
| 8 | 16.8 | 16.8 | 1.8 | GH, TSH, ACTH, FSH-LH | Absent | No | No | No | — | Stopped at the age 2.5 y | — | — |
| 9 | 6.2 | 6.0 | 2.6 | GH, TSH, ACTH | Severe | Yes | No | No | 4.5 | Never treated (resolved at 6 mo of age) | — | — |
| 10 | 7.8 | 7.8 | 1.5 | GH, TSH | Moderate | No | No | No | 1.5 | Yes | 600 | 21.3 |
| 11 | 23.0 | 22.1 | 0.7 | GH | Absent | No | No | No | — | Yes | 600 | 8.5b |
| 12 | 8.0 | 7.8 | −0.5 | GH | Severe | Yes | No | Yes | 4.1 | Stopped at the age 3.6 y | No | No |
| Patient No. | Age, y | Follow-up Duration, y | BMI, SDS | Anterior Pituitary Dysfunctiona | Neuro- Developmental Delay | Seizures | Visual Impairment | Use of Sleep Medication | Duration of Enteral Nutritional and Fluid Support, y | DDAVP Treatment at the Last Evaluation | DDAVP Oral Melt/Tablets | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose μg/d | Dose μg/kg/d | |||||||||||
| 1 | 5.0 | 4.9 | 0.5 | GH, TSH, ACTH | Severe | Yes | Yes | Yes | — | Yes | 105 | 6.8 |
| 2 | 8.1 | 7.8 | −1.7 | GH, TSH, ACTH | Moderate | No | Yes | No | — | Yes | 30 | 1.6 |
| 3 | 1.5 | 1.3 | 2.5 | GH, TSH, ACTH | Severe | Yes | Yes | Yes | 0.3 | Yes | 15 | 1.2 |
| 4 | 16.4 | 16.0 | −0.2 | GH, TSH | Severe | Yes | Yes | No | — | Yes | 85 | 2.1b |
| 5 | 3.1 | 2.5 | 0.2 | GH | Moderate | No | Yes | No | — | Yes | 150 | 10.3 |
| 6 | 20.0 | 19.8 | 2.3 | No | Moderate | No | No | No | — | Yes | 700 | 12.7b |
| 7 | 18.4 | 18.3 | 0.6 | GH, TSH, ACTH, FSH-LH | Absent | No | No | No | — | Yes | 180 | 3.1 |
| 8 | 16.8 | 16.8 | 1.8 | GH, TSH, ACTH, FSH-LH | Absent | No | No | No | — | Stopped at the age 2.5 y | — | — |
| 9 | 6.2 | 6.0 | 2.6 | GH, TSH, ACTH | Severe | Yes | No | No | 4.5 | Never treated (resolved at 6 mo of age) | — | — |
| 10 | 7.8 | 7.8 | 1.5 | GH, TSH | Moderate | No | No | No | 1.5 | Yes | 600 | 21.3 |
| 11 | 23.0 | 22.1 | 0.7 | GH | Absent | No | No | No | — | Yes | 600 | 8.5b |
| 12 | 8.0 | 7.8 | −0.5 | GH | Severe | Yes | No | Yes | 4.1 | Stopped at the age 3.6 y | No | No |
None of the patients had hyperprolactinemia.
Tablet.
A transient form of adipsic neurogenic hypernatremia DI was demonstrated in three patients (patients 8, 9, and 12) with either isolated structural hypothalamic pituitary abnormalities (n = 2) or no cerebral malformation (n = 1) on MRI. A PPHS was detected in all three patients at a normal (n = 1) or ectopic (n = 2) location. In one patient who never received DDAVP treatment, hypernatremia resolved at the age of 6 months. Two patients received DDAVP treatment until the ages of 2.5 and 3.6 years. No recurrence of hypernatremia was reported during the subsequent follow-up of these patients. All three patients had an anterior pituitary deficiency, with either isolated GHD (n = 1) or MPHD (n = 2). Two of these patients had severe neurodevelopmental delay.
All the other patients (n = 9) received oral DDAVP treatment twice (n = 3) or three times (n = 6) daily, at doses ranging from 15 μg/d for the youngest patient (age, 1.5 y) to 700 μg/d. Two of the three patients receiving more than 200 μg/d DDAVP had no PPHS, with left internal carotid agenesis in one patient (patient 10) and no cerebral malformation in the other (patient 11; this patient remained an exceptional case). The presence of normal thirst in these patients, with polydipsia if undertreated, was suggestive of probable CDI without hypothalamic adipsic hypernatremia syndrome. Three other patients had normal thirst (patients 2, 5, and 7) with underlying optic nerve hypoplasia, SOD, and ectopic neurohypophysis, respectively. They required various doses of DDAVP (from 30 to 180 μg/d) and displayed moderate (n = 2) or no neurodevelopmental delay (n = 1). Four patients (patients 1, 3, 4, and 6) had persistent adipsic DI, with severe (n = 3) or moderate (n = 1) neurodevelopmental delay.
Overall, five (42%) patients had severe developmental delay and required medication for seizures, and three required medication for sleep disorders (insufficient duration and quality of sleep). Four patients required nocturnal enteral feeding to maintain an adequate intake of nutrients and fluids. Three patients were obese (BMI > 2 SD).
Plasma sodium concentrations during follow-up are indicated for each patient in Figure 2, as a function of the frequency of normonatremia and dysnatremia (percentage). In total, 563 plasma sodium concentrations were analyzed, with a median of 50 (range, 15 to 98) determinations per patient. Plasma sodium concentrations were considered for patients with transient adipsic DI, until this condition resolved. As expected, the most common abnormality was hypernatremia, although seven patients suffered severe episodes of hyponatremia. In these patients, plasma sodium concentration at admission ranged from 117 to 130 mmol/L. When the three patients with a transient form of adipsic hypernatremia DI were excluded and compared with patients with persistent adipsia (patients 1, 3, 4, and 6), those with normal thirst (patients 2, 5, 7, 10, and 11) were found to be more likely to have normal plasma sodium concentrations (63 vs 39%) and less frequent hyper- or hyponatremia (33 vs 45% and 4 vs 16%, respectively) (P < .0001) (Figure 3). They were also found to be more likely to require higher DDAVP doses and were less frequently affected by neurodevelopmental delay (P < .05) than patients with persistent adipsia (Supplemental Table 1).
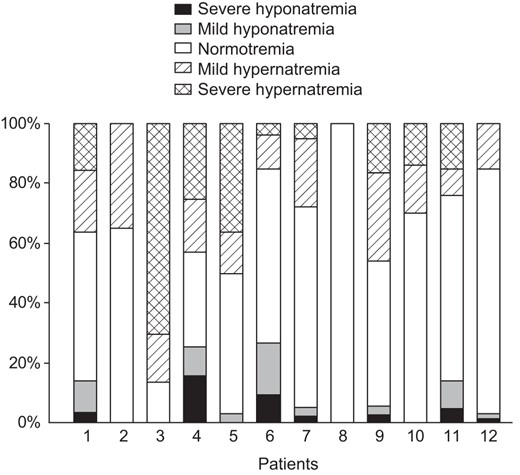
Plasma sodium concentrations during follow-up in 12 patients treated with DDAVP for neonatal CDI or adipsic DI, as a function of the frequency of normal and abnormal results for natremia, for each patient.
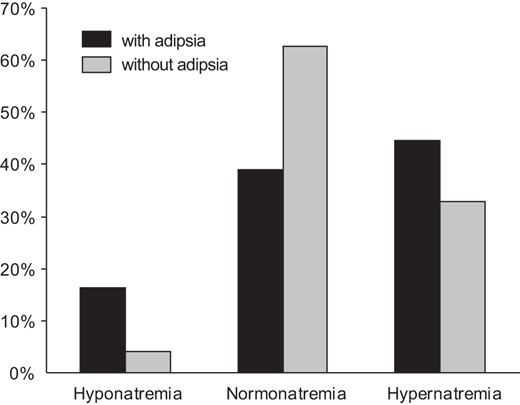
Frequency of normal and abnormal sodium concentrations in patients followed for congenital permanent CDI (n = 9) with and without adipsia.
Discussion
The results of this study extend our knowledge about the characteristics of CDI in infants with and without hypothalamic adipsic hypernatremia syndrome. There are clear clinical differences in the presentation of CDI in infants and older children. In cases of neonatal DI, adipsic hypernatremia syndrome associated with congenital cerebral malformations was common. Only two patients in our study had isolated idiopathic neonatal CDI, suggesting that this isolated condition is much rarer in infancy than later in life (5, 7). Fetal and/or neonatal distress was frequent in our study, but none of our patients presented any signs of fetal or neonatal intraventricular hemorrhage, congenital infection, tumor, or thickening of the pituitary stalk, as described in older patients with CDI (5, 27). The adipsic DI of older patients generally results from lesions of the osmoreceptor cells, mostly after surgery for anterior communicating artery aneurysm or craniopharyngioma (28).
In our case series, optic nerve hypoplasia, with or without SOD, was the most common underlying diagnosis. However, the frequencies of water homeostasis impairments, ranging from subtle defects to complete forms of CDI and/or adipsic DI, differ considerably between studies (17, 18, 29–31), with such disorders having a frequency as low as 5% in cases of optic nerve hypoplasia (30). Other midline abnormalities, such as holoprosencephaly, EPP with an absence of the pituitary stalk and anterior pituitary hypoplasia, CNPAS with SMMCI, part of the holoprosencephaly spectrum, have been reported less frequently (13–15, 18, 32, 33). Other types of alteration to brain structures were found in our patients, in which the frequency of interdigitating frontal gyri was high. Contrary to our findings, hyperprolactinemia and/or corpus callosum hypoplasia were reported to be more frequently associated with such midline defects in previous studies (29–31). An impairment of hypothalamic neurohypophyseal function is likely, although the underlying pathophysiological mechanism has yet to be clearly determined (2).
Unilateral (right or left) agenesis of the internal carotid artery is a very rare congenital abnormality, and only 11 previous cases have been reported, diagnosed during childhood (n = 7) or in adults (n = 3) or at an unknown age (n = 1) (34, 35). As reported in a recent review (34), in most cases, an enlarged posterior communicating artery or anterior communicating artery (as in our case) supplies the affected hemisphere with blood. All but one of these patients had pituitary hypoplasia, with the visible hyperintense signal of the PP in a normal or even ectopic (n = 4) location. The hyperintense signal of the PP was not visible in only two patients. The only patient in our study with this vascular abnormality also had no visible hyperintense signal for the PP. Moreover, MPHD was demonstrated in all patients with unilateral internal carotid artery agenesis, but CDI was reported in only two patients. Other midline craniofacial abnormalities were identified in four patients. Complex neural crest differentiation and/or migration disorders probably account for these associations, although it is not possible to exclude a role for hemodynamic mechanisms (34).
Given the total absence of follow-up data for cohorts of neonatal patients with DI (19), we also documented the clinical long-term follow-up of these patients. Leaving aside transient CDI caused by a temporary dysfunction of antidiuretic hormone-producing neurons and resolving spontaneously within a few days of transsphenoidal surgery (36), and contrary to what is seen later in life, when the disorder is more likely to be permanent (27, 37), the key finding of this study was that 25% of our patients (three of the 12) between the ages of 0.5 and 3.6 years displayed transient adipsic DI. The remission of congenital DI has been reported for only one case to date, in a patient with SOD and after 8 years of DDAVP treatment (38). Our three patients with transient adipsic DI had a PPHS and either isolated structural hypothalamic pituitary abnormalities (n = 2) without any other cerebral malformation or no detectable malformation (n = 1) on MRI. The pathophysiological mechanism underlying such cases of transient adipsic DI remains to be elucidated.
Finally, the only patient with permanent congenital DI associated with GHD but with no detectable malformation was unique, with no similar case ever having been described, to our knowledge.
Another interesting finding of this study is the documentation, for the first time, of the pattern of dysnatremia observed for this complex condition in patients in an ambulatory care setting. Hypo- and hypernatremia result in similar clinical symptoms, such as irritability, lethargy, weakness, nausea, seizures, and coma, but mortality is higher for hyponatremia (39). Therefore, in clinical practice, particularly for patients with permanent adipsia and cognitive dysfunction, which were more likely to be associated with complex congenital cerebral malformations, hyponatremia should be avoided by careful modulation of the control of hypernatremia by fluid intake, with fluids being administered because these patients fail to drink due to a lack of thirst, resulting in dehydration with hypernatremia. Polyuria control by DDAVP should also be carefully balanced to prevent DDAVP-induced hyponatremia. These patients are at risk of severe hyponatremia if they receive both excessive amounts of fluids and DDAVP treatment. Daily weighing of the patients should make it possible to detect abnormal weight gain, and plasma sodium concentration should be monitored frequently. Delaying the administration of a dose of DDAVP, or not administering a dose, once or twice per week, allowing diuresis to occur, would reduce the risk of hyponatremia (40).
Most of our patients have comorbid conditions rendering CDI management more difficult. These conditions include developmental delay, visual defects, seizures, sleep disorders, thermal regulation problems, feeding problems, and obesity/overweight. The findings for our cohort also indicate that anterior pituitary deficiencies are common and that most patients have more than one deficiency, with ACTH deficiency complicating the management of water and electrolyte balance (41). The patients of our cohort are also probably at high risk of gonadotropin deficiency, as suggested by the high rate of olfactory bulb agenesis, but most were too young for the diagnosis of gonadotropin deficiency.
One of the strengths of this unique follow-up study is that all patients diagnosed with neonatal DI from one clinical center were included. Its main limitation was the observational nature of retrospective data collection. The numbers of plasma sodium determinations and dysnatremia detections were limited during follow-up; no information was available concerning the duration of the dysfunction, and the frequency of dysnatremia between routine tests was unknown. This may have led to cases of dysnatremia being missed. However, this would be unlikely to result in a substantial misclassification bias, given the large number of plasma sodium determinations. Despite the inclusion of all patients with neonatal DI, the number of subjects investigated was small because this complex condition is very rare. Our study was thus unable to provide further insight into the mechanism underlying permanent and transient congenital DI.
In conclusion, based on our findings, we identified three groups of children with neonatal DI. One group had the classical form of CDI, with or without permanent adipsia, and with complex congenital midline malformations, such as optic nerve hypoplasia, SOD, holoprosencephaly, or unilateral agenesis of the internal carotid artery. The patients of this group required adequate hydration and various doses of DDAVP, with lower doses generally required for patients with adipsia than for those with normodipsia. The second group contained patients with transient adipsic DI with or without isolated structural hypothalamic pituitary abnormalities, but with no other complex cerebral malformation, with spontaneous resolution of the DI within a few months. The final group consisted of a single patient with a unique combination of CDI and normal thirst associated with GHD diagnosed during childhood, with no underlying etiology detectable on MRI.
These original findings for patients with neonatal CDI have important clinical implications for their long-term management and highlight the need for appropriate and vigilant clinical and electrolyte monitoring to prevent unnecessary treatment and to decrease the impact of comorbid conditions in these vulnerable patients.
Acknowledgments
The authors thank Emmanuel Ecosse for assistance with the statistical analysis.
This work was supported in part by the French Ministry of Health (Rare Disease Plan). A.D. held a clinical research fellowship from the European Society for Pediatric Endocrinology. Data collection and analysis, data interpretation, and the decision to submit the paper for publication were the responsibility of the authors alone.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- AVP
arginine vasopressin
- BMI
body mass index
- CDI
central DI
- CNPAS
congenital nasal pyriform aperture stenosis
- DDAVP
desamino-D-arginine-8-vasopressin
- DI
diabetes insipidus
- EPP
ectopic PP
- GHD
GH deficiency
- MPHD
multiple pituitary hormone deficiency
- MRI
magnetic resonance imaging
- PP
posterior pituitary
- PPHS
PP hyperintense signal
- SDS
standard deviation score
- SMMCI
solitary median maxillary central incisor
- SOD
septo-optic dysplasia.

{kind=link}
{kind=link}
{kind=link}