





Abstract
A majority of patients with resistance to thyroid hormone (RTH) are asymptomatic, whereas some patients show signs of hyperthyroidism, or hypothyroidism, or both. Thyrotoxic periodic paralysis is the most common form of acquired periodic paralysis. However, it has not been reported in a patient with RTH up to now.
We evaluated a 36-year-old male patient from China with elevated serum free T4 and free T3 and inappropriately high TSH who presented with periodic paralysis.
Clinical, biochemical, and radiological assessments, as well as DNA sequencing, were performed.
The patient's laboratory tests revealed the following: TSH, 6.14 mIU/L (0.27–4.2 mIU/L); free T3, 12.85 pmol/L (2.8–7.1 pmol/L); free T4, 33.62 pmol/L (9.05–25.5 pmol/L); and serum SHBG, 19.4 nmol/L (18.3–54.1 nmol/L). No significant suppression of TSH was observed in the rapid TSH suppression test with somatostatin analogs. Compound muscle action potential after exercise of the patient was reduced by 58%. Sequencing of thyroid hormone receptor genes confirmed a C446S mutation in the THRβ gene.
This is the first report of periodic paralysis as a new phenotype of RTH syndrome.
Caused by mutation of the thyroid hormone (TH) receptors (THRs) comprised of α- and β-subunits, resistance to TH (RTH) is fairly rare, with a prevalence of approximately one in 40 000 or 50 000 individuals (1, 2). Since it was first reported by Refetoff et al (1) in 1967, approximately 3000 cases from 1000 families worldwide have been reported. The clinical manifestations of RTH are variable. Most affected individuals are completely asymptomatic, although some may show signs of elevated TH, such as tachycardia and hyperactivity, and others present with symptoms of reduced TH deprivation, such as growth retardation and learning disabilities. Symptoms of both TH deficiency and TH elevation have also been observed in some patients (3).
Thyrotoxic periodic paralysis (TPP) is characterized by recurring episodes of muscle weakness in the presence of thyrotoxicosis, the most common form of acquired periodic paralysis, with a higher prevalence in Asian males (4). Possible etiologies of TPP include Graves' disease, toxic adenoma or multinodular goiter, TSH-producing pituitary tumor, and lymphocytic thyroiditis. Periodic paralysis in patients with RTH has not yet been reported. Here, we present the first report of such a patient with RTH who presented with hypokalemic periodic paralysis as the onset and chief manifestation.
Case Report
A 36-year-old male patient was admitted to our hospital presenting with recurrent paralysis over the past 8 years. In March of 2007, he experienced his first episode of paralysis after a transfusion of fluid containing glucose when he had a fever caused by respiratory infection. He could only move his fingers at that time. Fortunately, his breathing muscles were not affected. Blood tests performed shortly after his attack showed hypokalemia, elevated free T3 (FT3) and free T4 (FT4), and abnormally high TSH levels. He recovered with an iv supplement of potassium. He was given an antithyroid drug for approximately 1 year, and there was no effect on thyroid function or clinical symptoms. Paralysis of the whole body or weakness of the extremities occurred repeatedly, usually after glucose transfusion or vigorous physical activity, including long-time exercise. Oral or iv potassium supplements were effective. Dietary carbohydrate intake or alcohol did not trigger any attack in this patient. Since 2010, he had intermittent palpitations, tremors, and irritability. From 2013 on, paralysis or weakness of recently exercised muscles has occurred more frequently. After recovering from the last severe attack of general paralysis in late June, he was admitted to our hospital for further diagnosis and treatment.
He was the only child of his parents, and his mother had died. After he was diagnosed with an abnormal thyroid function, his father and two daughters were also evaluated. Elevated FT3 and FT4 and unsuppressed TSH were found in his father (TSH, 1.54 mIU/L [0.27–4.2 mIU/L]; FT3, 9.23 pmol/L [2.8–7.1 pmol/L]; and FT4, 32.14 pmol/L [9.05–25.5 pmol/L]) and his elder daughter (TSH, 4.45 mIU/L [0.27–4.2 mIU/L]; FT3, 10.72 pmol/L [2.8–7.1 pmol/L]; and FT4, 40.6 pmol/L [9.05–25.5 pmol/L]). The results of his younger daughter were lost. Although abnormal thyroid function was found, his father did not present with any thyroid-associated symptoms. His elder daughter, who was 10 years old, had learning disabilities and lagged in response. No growth or developmental abnormalities were found in his younger daughter, who was 3 years old.
No abnormalities were found in the patient, except for a slight goiter. Antibodies against thyroperoxidase, thyroglobulin, and TSH receptors were negative, and SHBG was normal. Thyroid ultrasonography showed diffuse lesions with multiple cysts. Electrocardiograms and echocardiographs were normal. Nuclear magnetic resonance imaging of the pituitary was not abnormal. The thyroid alone was clearly demonstrated in the somatostatin receptor scintigraphy. Compound muscle action potential (CMAP) after exercise was reduced by 58%. A rapid TSH suppression test with somatostatin analogs was performed by subcutaneously injecting 100 mg Sandostatin three times every 8 hours. Serum TSH levels were measured at 0, 2, 4, 6, 8, and 24 hours after the first injection. No significant suppression of TSH was observed.
Genome DNA was obtained and purified from peripheral leukocytes of the patient and his elder daughter, according to standard techniques. Sequencing of exons of THRA and THRB as well as possible pathogenic genes for periodic paralysis, including CACNA1S, KCNE3, and SCN4A, were performed. Only one point mutation was found in the THRB gene (Figure 1). The patient was heterozygous for the transversion T1336A within exon 11 of the THRB gene (Figure 1). This nucleotide change led to the substitution of a cysteine with a serine at the 446 residue (p.Cys446Ser). The patient's daughter had a mutation at the same point of the THRB gene.
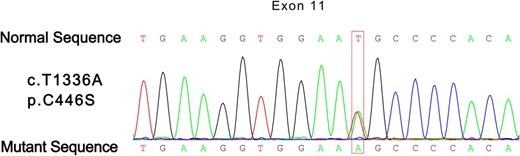
Partial electropherogram showing the heterozygosis for c.1336T>A transition in exon 11, codon 446.
Discussion
Hypokalemic periodic paralysis is characterized by recurrent episodes of muscle weakness and low serum potassium levels. Because CMAP amplitude declines during muscle weakness and muscle weakness can be triggered by exercise during periodic paralysis, electrophysiological exercise testing was recognized as a useful diagnostic tool for periodic paralysis with a relatively high specificity. After 2–5 minutes of intermittent strong muscle contraction, abnormally greater reduction (33% was usually taken as a cutoff) in CMAP amplitude was found during asymptomatic periods in patients with periodic paralysis. Profound impairment during electrophysiological exercise tests was found in this patient, which confirmed the existence of periodic paralysis.
Periodic paralysis could either be hereditary, due to the mutations in the sodium, calcium, or potassium channels, or secondary, due to some pathophysiological disorders like thyrotoxicosis. Compared to familial hypokalemic periodic paralysis, patients with TPP have an older age of onset, a higher male-to-female ratio, a shorter duration of the attacks, lower potassium levels during the attack, relatively more benign clinical course, and lower positivity of confirmed gene mutation (5). KCNJ18 gene mutation was formerly reported to cause susceptibility to TPP. However, approximately 1.8% of Chinese patients with thyrotoxicosis experience paralysis (6), whereas only 3.1% of the patients with TPP were found to have KCNJ18 gene mutation (7). In a recently published study, Kuhn et al (2015) concluded that KCNJ18 alterations are seldom pathogenic, and genetic diagnosis for a patient with TPP could not be established on the basis of KCNJ18 gene mutation alone (8).
Three ion channel genes, CACNA1S, KCNE3, and SCN4A, frequently involved in familial periodic paralysis, were sequenced in this patient. All results were negative, which indicated that this patient was less likely to have familial periodic paralysis.
Further DNA sequencing revealed a heterozygous mutation in codon 446 (C446S), which was oriented in the T3-binding domain of THRβ. Weiss et al (9) previously reported that a kindred with RTH had a mutation at the same point of the THRβ subunit as this patient, but the mutant nucleotide was different. Unlike the T to A transversion in this patient, a single nucleotide substitution of C for T was found in the formerly mentioned kindred, leading to replacement of the normal cysteine-446 with an arginine (C446R). The mutant THRB (C446R) could neither bind T3 nor mediate relevant transactivation. In addition, it inhibited T3-mediated transactivation of the wild-type THRβ (8). Clinical characteristics of the family members with C446R mutations were similar to previous reports—namely, goiter, tachycardia, and learning disabilities. The nucleotide substitution resulting in C446S mutation has been previously reported in a South African kindred with RTH, manifesting typical symptoms of hyperthyroidism in the index cases (10). Although this patient had a mutation at the same point of the THRB gene, this patient showed an apparently different clinical profile, namely, periodic paralysis, which further proved that the phenotype of RTH with the same genotype is variable.
It is known that TH action is mediated by three highly homologous nuclear receptor isoforms (THRα, THRβ1, and THRβ2) encoded by two human genes (THR-α and THR-β). These THRs have different tissue distributions; for example, the pituitary and hypothalamus mainly express THRβ2. RTH mutations involving this isoform usually lead to defective feedback of the hypothalamus-pituitary-thyroid axis and unsuppressed TSH at the presence of increased THs. THRα is predominantly expressed in skeletal muscle; thus, skeletal muscle retains sensitivity to TH in patents with THRB mutation alone. Because skeletal muscle is the major determinant of energy expenditure in humans and raised energy expenditure is recognized as a feature of hyperthyroidism, we speculate that periodic paralysis might be a manifestation of hyperthyroidism in skeletal muscle in patients with mutated THRB genes and normal THRA genes, as in this patient. This finding is in line with a study by Mitchell et al (11), which showed that resting energy expenditure is substantially increased in patients with the THRB mutation.
This was the first case reported with C446S mutation in the THRB gene within the Chinese population. More importantly, this was the first patient found to present with periodic paralysis as the main manifestation of RTH.
Acknowledgments
We extend our deepest gratitude to Ms. Vivian Shi for her kind help in improving the language of this paper.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- CMAP
compound muscle action potential
- FT3
free T3
- FT4
free T4
- RTH
resistance to TH
- TH
thyroid hormone
- THR
TH receptor
- TPP
thyrotoxic periodic paralysis.

{kind=link}