





Abstract
Primary adrenal insufficiency (PAI) is a life-threatening condition that is often due to monogenic causes in children. Although congenital adrenal hyperplasia occurs commonly, several other important molecular causes have been reported, often with overlapping clinical and biochemical features. The relative prevalence of these conditions is not known, but making a specific diagnosis can have important implications for management.
The objective of the study was to investigate the clinical and molecular genetic characteristics of a nationwide cohort of children with PAI of unknown etiology.
A structured questionnaire was used to evaluate clinical, biochemical, and imaging data. Genetic analysis was performed using Haloplex capture and next-generation sequencing. Patients with congenital adrenal hyperplasia, adrenoleukodystrophy, autoimmune adrenal insufficiency, or obvious syndromic PAI were excluded.
The study was conducted in 19 tertiary pediatric endocrinology clinics.
Ninety-five children (48 females, aged 0–18 y, eight familial) with PAI of unknown etiology participated in the study.
A genetic diagnosis was obtained in 77 patients (81%). The range of etiologies was as follows: MC2R (n = 25), NR0B1 (n = 12), STAR (n = 11), CYP11A1 (n = 9), MRAP (n = 9), NNT (n = 7), ABCD1 (n = 2), NR5A1 (n = 1), and AAAS (n = 1). Recurrent mutations occurred in several genes, such as c.560delT in MC2R, p.R451W in CYP11A1, and c.IVS3ds+1delG in MRAP. Several important clinical and molecular insights emerged.
This is the largest nationwide study of the molecular genetics of childhood PAI undertaken. Achieving a molecular diagnosis in more than 80% of children has important translational impact for counseling families, presymptomatic diagnosis, personalized treatment (eg, mineralocorticoid replacement), predicting comorbidities (eg, neurological, puberty/fertility), and targeting clinical genetic testing in the future.
Primary adrenal insufficiency (PAI) is a potentially life-threatening condition that requires accurate diagnosis and urgent treatment with glucocorticoid and often mineralocorticoid replacement. Because the symptoms and signs of adrenal insufficiency are often nonspecific, the diagnosis may be easily overlooked (1).
In contrast to the predominance in autoimmune etiologies in adults, most causes of PAI in childhood have an inherited, monogenic origin (1–3). Genetic causes of pediatric PAI can be classified into four major groups according to the underlying pathogenesis; 1) impaired steroidogenesis, 2) adrenal hypoplasia, 3) familial glucocorticoid deficiency (FGD) and FGD-like disorders, and 4) adrenal destruction.
Congenital adrenal hyperplasia (CYP21A2, CYP11B1, HSD3B2, CYP17A1, POR deficiencies) constitutes the largest subgroup of impaired steroidogenesis and is the most common cause of PAI in children (1, 2, 4). In contrast, there are other individually rare causes of PAI. Several genetic causes of adrenal hypoplasia (NR0B1/dosage-sensitive sex reversal, adrenal hypoplasia congenita critical region, on the X chromosome, gene 1 [DAX-1], NR5A1/SF-1, CDKN1C gene defects), congenital lipoid adrenal hyperplasia (CYP11A1, STAR gene defects), familial glucocorticoid deficiency (FGD) and FGD-like conditions (MC2R [FGD1], MRAP [FGD2], STAR, MCM4, NNT, TXNRD2 gene defects) and adrenal destruction (AIRE, ABCD1, PEX1, LIPA gene defects) are now well established (5–16). However, it is also emerging that there is considerable overlap in the clinical and biochemical presentation of these conditions. For example, FGD/FGD-like conditions (MC2R, MRAP, NNT gene defects) can present with salt loss suggestive of adrenal hypoplasia, and alterations in STAR and CYP11A1 resulting in partial loss of protein function may have a predominant FGD-like phenotype (17–20).
Establishing a specific genetic diagnosis of PAI is extremely valuable for identifying presymptomatic children who could benefit from treatment before the onset of potentially life-threatening symptoms and for counseling family members appropriately about the risk of passing the condition on to their children (1, 3, 20, 21). Knowing the genetic etiology can also help to modify treatments, such as the need for long-term mineralocorticoid replacement, and can predict potential comorbidities, such as impaired puberty or fertility and neurological dysfunction.
Next-generation sequencing (NGS) approaches are revolutionizing our ability to sequence large numbers of genes quickly and cost effectively. In this study, a custom panel-based NGS approach has been used to sequence all known PAI-associated genes in a national cohort of 95 children with PAI of unknown etiology.
Patients and Methods
Patients
A pediatric cohort study was performed with PAI patients recruited from 19 pediatric endocrinology clinics in Turkey. Inclusion criteria of a PAI phenotype was defined as the presence of signs and symptoms of adrenal insufficiency together with high plasma ACTH and low serum cortisol and intermediary glucocorticoid metabolites at initial presentation. Exclusion criteria were as follows: 1) congenital adrenal hyperplasia (21α-hydroxylase, 11β-hydroxylase, 3β-hydroxysteroid dehydrogenase 2, 17α-hydroxylase, or cytochrome P450 reductase deficiencies) diagnosed by a distinctive serum steroid hormone profiles; 2) X-linked adrenoleukodystrophy in boys with neurological findings and elevated very long-chain fatty acids, or a family history of affected males with adrenoleukodystrophy; 3) clinical and biochemical evidence of autoimmune adrenal failure; and 4) known syndromic causes of PAI (specifically, classic Triple A syndrome or Xp deletion syndrome involving NR0B1/DAX-1 with Duchenne muscular dystrophy) (Figure 1).
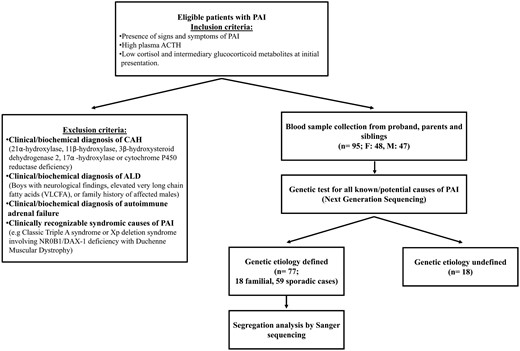
Overview of the study design, recruitment, and outcome of genetic analysis.
ALD, adrenoleukodystrophy.
All patients were assessed by a pediatric endocrinologist. A structured questionnaire was used to systematically evaluate all clinical, biochemical, and imaging data related to the diagnosis and treatment of PAI and all other relevant medical and family history. Studies were performed with the approval of the Ethics Committee of the Marmara University Faculty of Medicine (Istanbul, Turkey; B.30.2.MAR.0.01.02/AEK/108). Patients and/or parents provided written informed consent, and all studies were conducted in accordance with the principles of the Declaration of Helsinki.
A total of 95 PAI patients (48 females and 47 males) from 85 families and their unaffected siblings and parents were included (Figure 1). The most common presenting features were hyperpigmentation (94%), salt-wasting crisis/electrolyte imbalance (51%), hypoglycemia with/without convulsions (47%), vomiting/abdominal pain (26%), prolonged jaundice (24%), fatigue (18%), neonatal respiratory distress (17%), frequent infections (11%), and failure to thrive or weight loss (7%). Seven patients had 46,XY disorders of sex development (DSD). Detailed clinical findings are provided in Supplemental Table 1.
Parents of patients were consanguineous in 63 (74%) families, whereas 22 families did not report consanguinity. A total of 51 patients were on hydrocortisone treatment alone, whereas 44 were also commenced on mineralocorticoid replacement due to salt wasting, high plasma renin activity, or low aldosterone. Eight families had multiple affected siblings (six pairs, two trios, n = 18).
Molecular analyses
DNA samples
Genomic DNA was extracted from whole blood of patients, parents, and available unaffected siblings using a QIAamp DNA blood maxikit (QIAGEN Inc).
Design of targeted gene panel
A custom HaloPlex DNA target enrichment panel (Agilent Technologies Inc) was designed (SureDesign) to capture 160 known and candidate genes involved in adrenal development and function. All coding exons and 100 base pairs of intronic flanking sequence were included. The panel covered known genes potentially causing PAI, congenital adrenal hyperplasia-related genes, potential syndrome-related genes and candidate genes based on data from biochemical/biological pathways, mouse models of adrenal dysfunction, and gene expression (Supplemental Methods).
Sequence capture and NGS
Sequence capture was performed according to the HaloPlex Target Enrichment Protocol version D.5 (Agilent Technologies Inc) for Illumina sequencing (Supplemental Methods). Patient genomic DNA aliquots (225 ng) were processed in batches of 24 samples at a time with an enrichment control DNA sample as a positive control. Sequencing was performed on a MiSeq next-generation sequencer (Illumina Inc).
Variant analysis
Sequence alignment and variant calling were performed using SureCall (version 2.0) software (Agilent Technologies Inc). All potential disease causing variants were confirmed by PCR and Sanger sequencing. Variants in known disease genes were considered highly likely to be pathogenic if they segregated with the phenotype with an appropriate inheritance pattern within families, were determined damaging or likely damaging by several bioinformatic prediction models (Ensembl Variant Effector Predictor; SIFT; PolyPhen2; and Mutation Taster) and if they had been reported previously. In addition, novel missense changes were absent in at least 200 Turkish control samples and had a minor allele frequency less than 1:100 000 in the Exome Aggregation Consortium (ExAC) browser (ExAC; Cambridge, MA, http://exac.broadinstitute.org; accessed July 2015).
More detailed description of methods, workflows, coverage, and quality control are provided in Supplemental Methods.
Results
A molecular genetic diagnosis was obtained in 81% children with PAI (77 of 95) using this targeted NGS approach. A total of 43 different deleterious nonsynonymous variations were detected in nine different genes (Table 1). These changes were all confirmed by Sanger sequencing and included missense mutations (n = 24), nonsense mutations (n = 7), frameshift mutations (n = 5), in-frame single codon deletions (n = 3), splice site disruptions (n = 2), and whole gene/exon deletions (n = 2). Of these variations, 22 (51%) had not been reported previously but were considered to be causative because they segregated with the phenotype in the family, they were classed as damaging or probably damaging by several bioinformatic predictions, and they were not found in the control samples or databases (Supplemental Table 2). The remaining 21 mutations have been reported (Supplemental Table 1). A molecular diagnosis was reached in all eight families with multiple affected siblings, including novel changes in five families (Table 1).
Sequence Variations Detected in Our Cohort of 95 Children With Primary Adrenal Insufficiency
| Gene (Chromosome) | Familial, n | Sporadic, n | Total, n | Variants | n | Consanguinity | Mineralocorticoid Treatment |
|---|---|---|---|---|---|---|---|
| MC2R (18p11.21) | 4 | 21 | 25 | 22/25 (88%) | 2/25 (8%) | ||
| p.D103N | 1 | ||||||
| p.G116V | 2 | ||||||
| p.R137W | 1 | ||||||
| p.V142L | 1 | ||||||
| p.T143S | 1 | ||||||
| p.L225R | 1 | ||||||
| p.G226R | 1 | ||||||
| p.A233P | 2 | ||||||
| p.C251W | 2 | ||||||
| c.560delT (p.V187Afs*29) | 10 | ||||||
| Deletion | 3 | ||||||
| NR0B1 (Xp21.2)a | 6b | 6 | 12 | 3/12 (25%) | 12/12 (100%) | ||
| p.W235* | 3 | ||||||
| p.W236* | 1 | ||||||
| p.E256* | 3 | ||||||
| p.W291C | 1 | ||||||
| p.L299R | 1 | ||||||
| p.Y378* | 1 | ||||||
| p.C396* | 1 | ||||||
| p.V269del | 1 | ||||||
| STAR (8p11.23) | 2 | 9 | 11 | 8/11 (72%) | 11/11 (100%) | ||
| p.S13P | 3 | ||||||
| p.W96C | 2 | ||||||
| p.L157P | 1 | ||||||
| p.E169K | 1 | ||||||
| p.R182H | 1 | ||||||
| p.W250*/p.I166M | 1 | ||||||
| p.S12Afs*9 | 1 | ||||||
| p.K159del | 1 | ||||||
| CYP11A1 (15q24.1) | 2 | 7 | 9 | 8/9 (89%) | 6/9 (67%) | ||
| p.R451W | 9 | ||||||
| MRAP (21q22.11) | 2 | 7 | 9 | 5/9 (56%) | 2/9 (22%) | ||
| p.L53P | 1 | ||||||
| c.IVS3ds + 1insT | 1 | ||||||
| c.IVS3ds + 1delG | 5 | ||||||
| p.K30del | 2 | ||||||
| NNT (5p12) | 2 | 5 | 7 | 7/7 (100%) | 2/7 (29%) | ||
| p.D178G | 1 | ||||||
| p.H370R | 1 | ||||||
| c.1769dupA (p.D590Efs*29) | 1 | ||||||
| c.2396delC (p.P799Qfs*22) | 1 | ||||||
| c.127_128delTG (p.W43Vfs*2) | 2 | ||||||
| Deletion (exon 2–3) | 1 | ||||||
| ABCD1 (Xq28)a | 0 | 2 | 2 | 0/2 | 1/2 | ||
| p.G512S | 1 | ||||||
| p.Y547C | 1 | ||||||
| NR5A1 (9q33.1) | 0 | 1 | 1 | 0/1 | 1/1 | ||
| p.R92Q | 1 | ||||||
| AAAS (12q13.13) | 0 | 1 | 1 | 1/1 | 0/1 | ||
| p.R445* | 1 | ||||||
| Total | 18 | 59 | 77 | 54/77 (70%) | 37/77 (48%) |
| Gene (Chromosome) | Familial, n | Sporadic, n | Total, n | Variants | n | Consanguinity | Mineralocorticoid Treatment |
|---|---|---|---|---|---|---|---|
| MC2R (18p11.21) | 4 | 21 | 25 | 22/25 (88%) | 2/25 (8%) | ||
| p.D103N | 1 | ||||||
| p.G116V | 2 | ||||||
| p.R137W | 1 | ||||||
| p.V142L | 1 | ||||||
| p.T143S | 1 | ||||||
| p.L225R | 1 | ||||||
| p.G226R | 1 | ||||||
| p.A233P | 2 | ||||||
| p.C251W | 2 | ||||||
| c.560delT (p.V187Afs*29) | 10 | ||||||
| Deletion | 3 | ||||||
| NR0B1 (Xp21.2)a | 6b | 6 | 12 | 3/12 (25%) | 12/12 (100%) | ||
| p.W235* | 3 | ||||||
| p.W236* | 1 | ||||||
| p.E256* | 3 | ||||||
| p.W291C | 1 | ||||||
| p.L299R | 1 | ||||||
| p.Y378* | 1 | ||||||
| p.C396* | 1 | ||||||
| p.V269del | 1 | ||||||
| STAR (8p11.23) | 2 | 9 | 11 | 8/11 (72%) | 11/11 (100%) | ||
| p.S13P | 3 | ||||||
| p.W96C | 2 | ||||||
| p.L157P | 1 | ||||||
| p.E169K | 1 | ||||||
| p.R182H | 1 | ||||||
| p.W250*/p.I166M | 1 | ||||||
| p.S12Afs*9 | 1 | ||||||
| p.K159del | 1 | ||||||
| CYP11A1 (15q24.1) | 2 | 7 | 9 | 8/9 (89%) | 6/9 (67%) | ||
| p.R451W | 9 | ||||||
| MRAP (21q22.11) | 2 | 7 | 9 | 5/9 (56%) | 2/9 (22%) | ||
| p.L53P | 1 | ||||||
| c.IVS3ds + 1insT | 1 | ||||||
| c.IVS3ds + 1delG | 5 | ||||||
| p.K30del | 2 | ||||||
| NNT (5p12) | 2 | 5 | 7 | 7/7 (100%) | 2/7 (29%) | ||
| p.D178G | 1 | ||||||
| p.H370R | 1 | ||||||
| c.1769dupA (p.D590Efs*29) | 1 | ||||||
| c.2396delC (p.P799Qfs*22) | 1 | ||||||
| c.127_128delTG (p.W43Vfs*2) | 2 | ||||||
| Deletion (exon 2–3) | 1 | ||||||
| ABCD1 (Xq28)a | 0 | 2 | 2 | 0/2 | 1/2 | ||
| p.G512S | 1 | ||||||
| p.Y547C | 1 | ||||||
| NR5A1 (9q33.1) | 0 | 1 | 1 | 0/1 | 1/1 | ||
| p.R92Q | 1 | ||||||
| AAAS (12q13.13) | 0 | 1 | 1 | 1/1 | 0/1 | ||
| p.R445* | 1 | ||||||
| Total | 18 | 59 | 77 | 54/77 (70%) | 37/77 (48%) |
Novel variants are marked in bold. All mutations are homozygous except for hemizygous mutations in X-linked genes and p.W250*/p.I166M in STAR which was compound heterozygous.
Hemizygous mutations in X-linked genes.
Familial cases included sibling pairs except for NR0B1 in which two sibling trios were identified. Nucleotide position of variants is shown in Supplemental Table 1.
Sequence Variations Detected in Our Cohort of 95 Children With Primary Adrenal Insufficiency
| Gene (Chromosome) | Familial, n | Sporadic, n | Total, n | Variants | n | Consanguinity | Mineralocorticoid Treatment |
|---|---|---|---|---|---|---|---|
| MC2R (18p11.21) | 4 | 21 | 25 | 22/25 (88%) | 2/25 (8%) | ||
| p.D103N | 1 | ||||||
| p.G116V | 2 | ||||||
| p.R137W | 1 | ||||||
| p.V142L | 1 | ||||||
| p.T143S | 1 | ||||||
| p.L225R | 1 | ||||||
| p.G226R | 1 | ||||||
| p.A233P | 2 | ||||||
| p.C251W | 2 | ||||||
| c.560delT (p.V187Afs*29) | 10 | ||||||
| Deletion | 3 | ||||||
| NR0B1 (Xp21.2)a | 6b | 6 | 12 | 3/12 (25%) | 12/12 (100%) | ||
| p.W235* | 3 | ||||||
| p.W236* | 1 | ||||||
| p.E256* | 3 | ||||||
| p.W291C | 1 | ||||||
| p.L299R | 1 | ||||||
| p.Y378* | 1 | ||||||
| p.C396* | 1 | ||||||
| p.V269del | 1 | ||||||
| STAR (8p11.23) | 2 | 9 | 11 | 8/11 (72%) | 11/11 (100%) | ||
| p.S13P | 3 | ||||||
| p.W96C | 2 | ||||||
| p.L157P | 1 | ||||||
| p.E169K | 1 | ||||||
| p.R182H | 1 | ||||||
| p.W250*/p.I166M | 1 | ||||||
| p.S12Afs*9 | 1 | ||||||
| p.K159del | 1 | ||||||
| CYP11A1 (15q24.1) | 2 | 7 | 9 | 8/9 (89%) | 6/9 (67%) | ||
| p.R451W | 9 | ||||||
| MRAP (21q22.11) | 2 | 7 | 9 | 5/9 (56%) | 2/9 (22%) | ||
| p.L53P | 1 | ||||||
| c.IVS3ds + 1insT | 1 | ||||||
| c.IVS3ds + 1delG | 5 | ||||||
| p.K30del | 2 | ||||||
| NNT (5p12) | 2 | 5 | 7 | 7/7 (100%) | 2/7 (29%) | ||
| p.D178G | 1 | ||||||
| p.H370R | 1 | ||||||
| c.1769dupA (p.D590Efs*29) | 1 | ||||||
| c.2396delC (p.P799Qfs*22) | 1 | ||||||
| c.127_128delTG (p.W43Vfs*2) | 2 | ||||||
| Deletion (exon 2–3) | 1 | ||||||
| ABCD1 (Xq28)a | 0 | 2 | 2 | 0/2 | 1/2 | ||
| p.G512S | 1 | ||||||
| p.Y547C | 1 | ||||||
| NR5A1 (9q33.1) | 0 | 1 | 1 | 0/1 | 1/1 | ||
| p.R92Q | 1 | ||||||
| AAAS (12q13.13) | 0 | 1 | 1 | 1/1 | 0/1 | ||
| p.R445* | 1 | ||||||
| Total | 18 | 59 | 77 | 54/77 (70%) | 37/77 (48%) |
| Gene (Chromosome) | Familial, n | Sporadic, n | Total, n | Variants | n | Consanguinity | Mineralocorticoid Treatment |
|---|---|---|---|---|---|---|---|
| MC2R (18p11.21) | 4 | 21 | 25 | 22/25 (88%) | 2/25 (8%) | ||
| p.D103N | 1 | ||||||
| p.G116V | 2 | ||||||
| p.R137W | 1 | ||||||
| p.V142L | 1 | ||||||
| p.T143S | 1 | ||||||
| p.L225R | 1 | ||||||
| p.G226R | 1 | ||||||
| p.A233P | 2 | ||||||
| p.C251W | 2 | ||||||
| c.560delT (p.V187Afs*29) | 10 | ||||||
| Deletion | 3 | ||||||
| NR0B1 (Xp21.2)a | 6b | 6 | 12 | 3/12 (25%) | 12/12 (100%) | ||
| p.W235* | 3 | ||||||
| p.W236* | 1 | ||||||
| p.E256* | 3 | ||||||
| p.W291C | 1 | ||||||
| p.L299R | 1 | ||||||
| p.Y378* | 1 | ||||||
| p.C396* | 1 | ||||||
| p.V269del | 1 | ||||||
| STAR (8p11.23) | 2 | 9 | 11 | 8/11 (72%) | 11/11 (100%) | ||
| p.S13P | 3 | ||||||
| p.W96C | 2 | ||||||
| p.L157P | 1 | ||||||
| p.E169K | 1 | ||||||
| p.R182H | 1 | ||||||
| p.W250*/p.I166M | 1 | ||||||
| p.S12Afs*9 | 1 | ||||||
| p.K159del | 1 | ||||||
| CYP11A1 (15q24.1) | 2 | 7 | 9 | 8/9 (89%) | 6/9 (67%) | ||
| p.R451W | 9 | ||||||
| MRAP (21q22.11) | 2 | 7 | 9 | 5/9 (56%) | 2/9 (22%) | ||
| p.L53P | 1 | ||||||
| c.IVS3ds + 1insT | 1 | ||||||
| c.IVS3ds + 1delG | 5 | ||||||
| p.K30del | 2 | ||||||
| NNT (5p12) | 2 | 5 | 7 | 7/7 (100%) | 2/7 (29%) | ||
| p.D178G | 1 | ||||||
| p.H370R | 1 | ||||||
| c.1769dupA (p.D590Efs*29) | 1 | ||||||
| c.2396delC (p.P799Qfs*22) | 1 | ||||||
| c.127_128delTG (p.W43Vfs*2) | 2 | ||||||
| Deletion (exon 2–3) | 1 | ||||||
| ABCD1 (Xq28)a | 0 | 2 | 2 | 0/2 | 1/2 | ||
| p.G512S | 1 | ||||||
| p.Y547C | 1 | ||||||
| NR5A1 (9q33.1) | 0 | 1 | 1 | 0/1 | 1/1 | ||
| p.R92Q | 1 | ||||||
| AAAS (12q13.13) | 0 | 1 | 1 | 1/1 | 0/1 | ||
| p.R445* | 1 | ||||||
| Total | 18 | 59 | 77 | 54/77 (70%) | 37/77 (48%) |
Novel variants are marked in bold. All mutations are homozygous except for hemizygous mutations in X-linked genes and p.W250*/p.I166M in STAR which was compound heterozygous.
Hemizygous mutations in X-linked genes.
Familial cases included sibling pairs except for NR0B1 in which two sibling trios were identified. Nucleotide position of variants is shown in Supplemental Table 1.
The range of genetic etiologies found in this cohort were as follows: MC2R (n = 25), NR0B1 (n = 12), STAR (n = 11), CYP11A1 (n = 9), MRAP (n = 9), NNT (n = 7), ABCD1 (n = 2), NR5A1 (n = 1), and AAAS (n = 1) (Figure 2). Most patients were homozygous for recessive changes (62 of 77, 80%), one patient carried compound heterozygous changes (1 of 77, 1.3%), and 14 patients had hemizygous mutations in X-linked genes (NR0B1, ABCD1) (14 of 77, 18%). As expected, consanguinity rates were much higher in families of patients harboring mutations in recessive genes (51 of 63, 81%) compared with X-linked genes (3 of 14, 21%; P < .0001) (Table 1). Recurrent mutations were detected in several genes, such as c.560delT in MC2R (10 patients from nine unrelated families), p.R451W in CYP11A1 (nine patients from eight unrelated families), c.IVS3ds+1delG in MRAP (five patients from five unrelated families), and p.S13P in STAR (three patients from two unrelated families). Geographical hot spots were found for the p.R451W CYP11A1 mutation in eastern Turkey and for the c.IVS3ds+1delG MRAP mutation in the west (Figure 3, A and B).
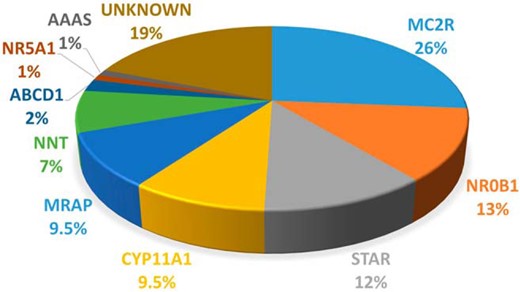
Pie chart showing the percentage of mutations in each gene in this cohort of children with PAI.
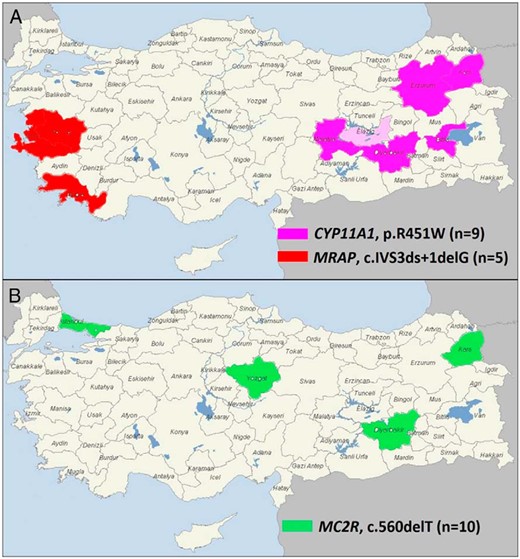
Geographical distribution of recurrent mutations identified in this study.
A, The MRAP c.IVS3ds+1delG mutation was identified mainly in patients from west Turkey, whereas the CYP11A1 p.R451W mutation was found in patients who originated from east Turkey. The first report of the CYP11A1 p.R451W mutation was in a family from Germany who were originally from Elazig (shown in light pink) (20). B, The MC2R c.560delT mutation showed a wider distribution most likely reflecting migration from the east to west of Turkey and has been described previously in a family from northern Iran (17).
Although there was considerable overlap in the clinical and biochemical features of children within this cohort (Supplemental Table 1), several notable findings have emerged. For example, most patients with MC2R, MRAP, and STAR mutations presented in the first weeks or months of life, whereas children with the p.R451W mutation in CYP11A1 presented in early childhood (1–6 y) (Figure 4). Children with nicotinamide nucleotide transhydrogenase (NNT) changes presented at different ages in the first 2 years, whereas boys with NR0B1 (DAX-1) mutations had a bimodal pattern, presenting either in the first month of life or else after 18 months.
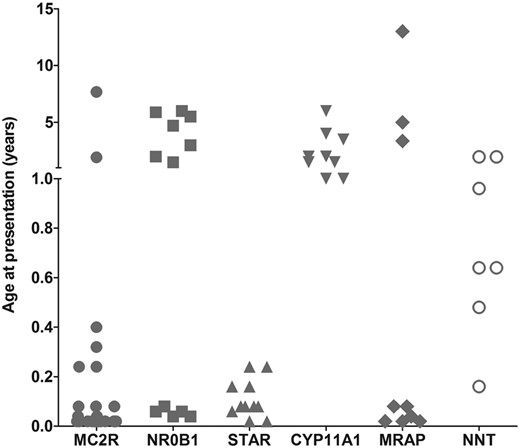
Age at presentation of the patients with PAI.
Most children had extremely high ACTH levels at diagnosis and almost all children, even babies, were clinically hyperpigmented. Hypoglycemia was a frequent finding, and hypoglycemic convulsions at presentation were more common in children with MC2R (22 of 25, 88%) and MRAP (five of nine, 56%) mutations than in children with an alternative diagnosis (5 of 43, 12%) (P < .0001). Salt-wasting states requiring mineralocorticoid replacement occurred in all children with NR0B1 (DAX-1) and STAR mutations and in most children with the CYP11A1 p.R451W change (six of nine, 66%) (Table 1). Fewer children with NNT (two of seven, 28%), MRAP (two of nine, 22%), and MC2R (2 of 25, 8%) mutations required mineralocorticoid replacement, although four additional children with MC2R defects had transient hyponatremia (sodium 117–133 mmol/L) that resolved without fludrocortisone treatment (Supplemental Table 1). Adrenal imaging was generally uninformative, showing normal sized or hypoplastic glands for most of these diagnoses, including many children with STAR mutations (congenital lipoid adrenal hyperplasia) in which enlarged adrenals are reported (22).
Additional clinical features were seen in many children in this cohort, such as altered growth, neuromotor delay, learning difficulties, and cardiac defects (Supplemental Table 1). Abnormalities in thyroid function such as subclinical hypothyroidism were common (n = 20, 26%), whereas thyroglossal cysts (n = 2), primary hypothyroidism (n = 2), and transient hypothyroidism (n = 1) were also found. DSD occurred in all six 46,XY infants with STAR mutations. Five of these children were phenotypic females who presented with a salt-losing crisis, and in two cases a karyotype was not available prior to this genetic analysis. The one boy with severe hypospadias due to STAR deficiency developed hypergonadotropic hypogonadism in puberty and needed T replacement. One of the four boys with the p.R451W mutation in CYP11A1 had a small penis and cryptorchidism. One boy with a NR0B1 mutation and X-linked adrenal hypoplasia congenita (AHC) had macrophallia and another one had cryptorchidism. Coincidental hypospadias and unilateral cryptorchidism was found in one boy with disruption of MC2R.
Stature was variable in the 25 children with MC2R mutations with seven children (28%) having tall stature (>2 SD score or two percentile lines above parental target height), four having short stature (<−2 SD score) and the rest being within the normal range. There was no history of preterm birth associated with CYP11A1 mutations, but there were reports of multiple stillbirths in three families in which a child had severe disruption of MC2R (deletion or c.560delT frameshift). Despite the range of neurological and other features seen, the two boys with mutations in ABCD1 (causing X-linked adrenoleukodystrophy) and one boy with disruption of AAAS (causing Triple A syndrome) appeared to have adrenal only phenotypes and would not have been diagnosed without genetic testing.
Discussion
The past 20 years has seen significant progress in our understanding of the genetic causes of childhood PAI. However, it is unclear how much these genes contribute to pediatric adrenal disease in the clinical setting as most reports have focused on specific categories of adrenal disease such as FGD or adrenal hypoplasia (6, 9).
In this study, an unbiased nationwide cohort of almost 100 children with PAI was recruited from 19 pediatric endocrinology centers across Turkey, and a molecular diagnosis was reached in more than 80% of the children. This represents the largest clinical cohort of children with this rare condition assembled. Because children could die from convulsions, respiratory distress, or salt-losing crises before reaching the hospital or get misdiagnosed with sepsis, the condition may be underdiagnosed. In fact, a history of unexplained death in infancy or childhood in the extended family was common in many of those questioned.
Targeted panel-based capture and high-throughput sequencing proved very effective in reaching a molecular diagnosis in a relatively quick and comprehensive manner. A total of 43 known and novel mutations in nine genes were discovered in 77 patients, with 73 of 77 mutations (95%) occurring in six genes (MC2R, NR0B1, STAR, CYP11A1, MRAP, and NNT). Several recurrent mutations were discovered, which likely represent founder effects. Some of these are localized to certain geographical areas (eg, p.R451W in CYP11A1 in eastern Turkey, c.IVS3ds+1delG in MRAP in western Turkey), which could lead to focused cost-effective clinical genetic testing for patients and families at risk of adrenal insufficiency in these regions (Figure 3A). Other recurrent changes, such as the c.560delT in MC2R showed more diverse geographical distribution across the country, possibly reflecting migration toward the west (Figure 3B). Indeed, this MC2R mutation has been reported previously in the father of two siblings with FGD1 who originated from northern Iran, close to the Turkish border (17). Sanger sequencing for just the three changes in MC2R, CYP11A1, and MRAP would have diagnosed 22 of 85 different families recruited in this nationwide cohort (26%), which could represent a cost-effective approach to first-line clinical genetic analysis. Of note, no mutations were found in MCM4 orTXNRD2, which to date have been described only in Irish Traveler and Kashmiri families, respectively (12, 14, 23)
In addition to defining the population genetics of PAI, this study has provided some useful clinical and novel molecular insight into several of these specific conditions.
Mutations in MC2R (encoding the ACTH receptor) are well established as the cause of FGD1, and approximately 40 different missense changes have been reported (9). Children typically present with hypoglycemia or hyperpigmentation in early infancy or in childhood, but complete loss of function mutations are rare (17, 24). Although diverse missense mutations were common in our cohort, severely disruptive changes in MC2R were found in more than half of the children (13 of 25), all of whom presented in the first 6 months of life with hypoglycemic convulsions, respiratory distress, or both. In addition to the c.560delT mutation discussed above, the first complete deletions of the MC2R locus were found in three patients. Although ACTH plays a facilitative role in supporting mineralocorticoid release, children with FGD1 do not typically have salt loss. However, transient hyponatremia has been reported in several children with severe disruption of the receptor, sometimes leading to a misdiagnosis of adrenal hypoplasia (17, 25). Similar observations were seen in this cohort, with 5 of 25 children having evidence of hyponatremia (sodium 117–133 mmol/L) but only two of them receiving long-term fludrocortisone replacement. On the basis of the genetic diagnosis, it is likely the need for this treatment can be reviewed. The significance of recurrent stillbirths in three families with severe loss of MC2R function is unclear. Although tall stature at diagnosis has been suggested for MC2R mutations, any effects on growth are difficult to interpret because most of our cohort were diagnosed and treated in infancy (26).
Hemizygous mutations in NR0B1 (encoding the nuclear receptor DAX-1) were found in 12 boys, including in two families in which three sons were affected. Mutations in NR0B1 cause X-linked AHC and more than 100 different mutations are reported (5). Most are frameshift or nonsense mutations that disrupt protein function, with a clustering of missense changes in three regions of the ligand-like binding domain (27). The three missense changes identified here (p.V269del, p.W291C, p.L299R) are located in a loop region of helix 3 to helix 5 that interacts with NR5A nuclear receptors (such as steroidogenic factor-1) (28). The novel p.L299 lies adjacent to the frequently mutated p.A300 residue, whereas codons p.V269 and p.W291 have been mutated previously in non-Turkish pedigree (5). All DAX-1-deficient boys presented with salt loss and showed a bimodal distribution pattern of age at presentation as reported previously (Figure 4) (29). Although currently preadolescent, they are likely to develop hypogonadotropic hypogonadism and infertility as part of their condition, so establishing the diagnosis in childhood will help plan endocrine management of puberty, counseling, and potential assisted reproduction (30). Macrophallia in infancy, found in one boy, is emerging as a rare feature of X-linked AHC (31).
True CYP11A1 deficiency (encoding P450 side chain cleavage [P450scc]) is a relatively recently established endocrine condition because it was thought that disruption of this enzyme, which facilitates the first three steps in conversion of cholesterol to pregnenolone, would be incompatible with fetal survival in humans (4). However, several children with this condition are now reported (32, 33). Severe loss of CYP11A1/P450scc function is usually associated with severe salt-losing adrenal failure in the neonatal period and a female phenotype in 46,XY children (46,XY DSD). Milder changes can present later with adrenal insufficiency and a history of hypospadias in boys (34, 35). The p.R451W variant found in all our patients was described recently in two brothers of Turkish origin residing in Germany who had normal genitalia and childhood-onset PAI (20). Their family originated from the Elazig region of Eastern Anatolia, a province bordering two regions where our patients lived (Figure 3A). The p.R451W mutation disrupts hydrogen bonds with residues F428 and Y449 in the K-L loop of the enzyme, causing partial loss of function consistent with the late presentation and normal genitalia in three of the four boys (20). Nevertheless, all children will need careful follow-up through puberty and into adult life to monitor sex hormone production and fertility. These findings confirm that mild loss of CYP11A1/P450scc function can present with isolated adrenal insufficiency and normal male genitalia, similar to mild loss of function of STAR (18–20).
Defects in STAR (encoding steroidogenic acute regulatory protein) disrupt the transport of cholesterol into mitochondria and classically lead to congenital lipoid adrenal hyperplasia (4, 22). A range of mutations were found, including recurrent p.S13P changes in three patients including two siblings. This is by far the most aminoterminal STAR mutation reported to date and affects the mitochondrial leader that is involved in targeting and localizing STAR to the outer mitochondrial membrane (36). Classic congenital lipoid adrenal hyperplasia is associated with severe salt-losing adrenal failure and 46,XY DSD due to a block in both adrenal and testicular steroidogenesis. The two-hit hypothesis suggests that after an initial reduction in steroid production and increased ACTH drive, accumulation of intracellular lipid has a secondary toxic effect on cells (22). Interestingly, nine of the children presented between 3 weeks and 3 months of age, consistent with the two-hit hypothesis, whereas only two presented in the first week (22). Two girls were found to have 46,XY DSD only after genetic analysis revealed STAR deficiency, highlighting the importance of obtaining a karyotype in phenotypic girls with salt-losing adrenal failure. As with CYP11A1/P450scc deficiency, these children all need endocrine follow-up for life to monitor puberty and ovarian function in 46,XX girls and for puberty induction and sex steroid replacement in 46,XY DSD. Indeed, the one patient with hypospadias raised male is now showing evidence of hypergonadotropic hypogonadism in adolescence.
Mutations in MRAP (encoding melanocortin 2 receptor-associated protein) and NNT (encoding nicotinamide nucleotide transhydrogenase) cause adrenal insufficiency without other features and can only be diagnosed by genetic analysis, as shown here. MRAP is essential for trafficking the ACTH receptor (melanocortin 2 receptor) to the cell membrane, and mutations causing FGD2 were first described in 2005 (9, 10, 24). The intron 3 splice site is especially vulnerable, often resulting in early-onset adrenal insufficiency. Patients with novel aminoterminal point mutations (p.K30del, p.L53P) presented unusually late (3.5–13 y) (Figure 4), consistent with reports of children with p.V26A and p.Y59D changes (37).
NNT mutations affect cellular oxidation and were first decribed in 2012; approximately 20 children have been reported to date (9, 13, 38). The six novel changes found here include familial homozygous deletions of exons 2–3 and homozygous missense mutations in the mitochondrial matrix region (p.D178G, p.H370R). These findings confirm the importance of NNT for adrenal function in an independent cohort. Unlike in other studies, compound heterozygous mutations were not found (13).
Heterozygous mutations in NR5A1 (encoding the nuclear receptor steroidogenic factor-1) usually cause 46,XY DSD or primary ovarian insufficiency (5). Adrenal insufficiency is extremely rare and has been reported only once, in a 46,XX girl (39). The homozygous p.R92Q mutation found here in a 46,XX girl with early-onset PAI is fascinating because the same homozygous change was reported in a 46,XY phenotypic female with adrenal failure from central Turkey in 2002 (40). This finding reinforces the importance of the A-box motif of steroidogenic factor 1 in monomeric binding to DNA and provides conclusive evidence that severe disruption of steroidogenic factor 1 can cause adrenal insufficiency in humans.
Finally, mutations identified in AAAS (typically causing Triple A syndrome: achalasia, alacrima, Addison disease) and ABCD1 (typically causing X-linked adrenoleukodystrophy) in three children without other features shows how genetic screening can identify adrenal-only phenotypes in young people who may be at risk of developing other symptoms in later life (11, 15).
This nationwide cohort study of high-throughput genetic screening of children with rare causes of PAI has provided many novel and supportive clinical and molecular insights and has significant impact on the management of these patients and their families. New genetic technologies are a powerful tool in defining population genetics of rare conditions and will allow more focused clinical genetic screening programs to be established.
Acknowledgments
We thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about.
This work was supported by Turkish Pediatric Endocrinology Research Grant UPE-2014-2. J.C.A. is a Wellcome Trust Senior Research Fellow in Clinical Science (Grant 098513/Z/12/Z), with support from the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London. T.G. is a European Community, Marie-Curie research fellow (Grant PIEF-GA-2012-328959).
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- AHC
adrenal hypoplasia congenita
- DAX-1
dosage-sensitive sex reversal, adrenal hypoplasia congenita critical region, on the X chromosome, gene 1
- DSD
disorders of sex development
- FGD
familial glucocorticoid deficiency
- NGS
next-generation sequencing
- NNT
nicotinamide nucleotide transhydrogenase
- PAI
primary adrenal insufficiency
- P450scc
P450 side chain cleavage.

{kind=link}
{kind=link}
{kind=link}
{kind=link}