





Abstract
Pituitary stalk interruption syndrome (PSIS) and holoprosencephaly (HPE) are congenital midline defects. Rare mutations in the sonic hedgehog (SHH) signaling gene CDON have recently been reported in patients with HPE.
To report a unique case of PSIS with a maternally inherited nonsense mutation in the SHH signaling protein CDON.
We performed exome sequencing on a case of PSIS. Control databases (1000 Genomes, dbSNP, Exome Variant Server, ExAC Browser) and an ancestry-matched control panel were screened upon identification of CDON mutation.
We identified a novel heterozygous nonsense mutation (c.2764T>C, Glu922Ter) in a case of PSIS without HPE who presented with neonatal hypoglycemia and cholestasis associated with GH, TSH, and ACTH deficiencies. This mutation was absent in all control databases and from 400 healthy ancestry-matched control subjects. The mutation was inherited from the patient's mother, who was operated on in childhood for strabismus. The absence of this variant in control samples suggests that it is likely to be responsible for the phenotype.
We report for the first time a mutation in the CDON gene associated with PSIS.
The pituitary gland regulates growth, reproduction, stress response, and metabolism by secreting hormones in response to signals from the hypothalamus (1). The posterior lobe is derived from the pituitary stalk by an evagination of the neuroectoderm lining the floor of the ventral diencephalon. A number of genes including HESX1, LHX3, LHX4, POU1F1, PROP1, SIX6, OTX2, PTX2, GLI2, SOX2, and SOX3 are involved in anterior pituitary development, and mutations in these genes are associated with a range of pituitary phenotypes including combined pituitary hormone deficiencies (CPHDs) and isolated GH deficiency (1, 2). However, little is known about the genes controlling the development of the pituitary stalk and posterior pituitary.
Pituitary stalk interruption syndrome (PSIS) is a rare disorder characterized by the combination of specific findings on magnetic resonance imaging (MRI): interrupted pituitary stalk, absent or ectopic posterior pituitary, and anterior pituitary hypoplasia. PSIS can be associated with other midline and ophthalmic abnormalities and variable endocrine disorders including hypoglycemia, micropenis, growth failure, or CPHD (3). The etiology of PSIS is unknown in 95% of cases, although genetic causes are suspected. Mice lacking HesX1, Lhx2, Nkx2.1, Rax, and Tbx3, as well as Hex1/Hes5 double knockouts fail to form a pituitary stalk (1, 4). In the human, mutations or sequence variants in LHX4, OTX2, HESX1, SOX3, PROKR2, and GPR161 have been postulated to be involved in PSIS (4, 5). Recently, mutations in holoprosencephaly (HPE)-related genes have been reported in two patients with PSIS or isolated pituitary hypoplasia, presenting with CPHD and without HPE (6).
CDON (cell adhesion associated, oncogene regulated) is a cell surface sonic hedgehog (SHH)-binding protein that promotes SHH signaling activity by acting as a coreceptor with PTCH1 (7). Cdon−/− mice display HPE with strain-dependent severity (8). Recently, heterozygous, loss-of-function CDON mutations have been reported in six HPE patients with a range of phenotypic severity (9). One patient had GH deficiency, another had biliary atresia, and another had cryptorchidism, absent of pituitary and adrenal atrophy with hepatic cholestasis (9). Here, we describe a novel heterozygous nonsense mutation in CDON associated with PSIS.
The study was approved by the Comité de Protection des Personnes Ile-de-France (no. IRB00003835). The control panel consisted of 400 unrelated 46,XY males of Moroccan ancestry (normospermic or fathered at least two children). All samples used for this study were collected with informed consent.
The patient was born to nonconsanguineous parents of Moroccan origin. The mother was operated for congenital convergent strabismus at age 7 years. The patient was born at 39 weeks, presented breech, then delivered by cesarean in apparent death with Apgar at 1–5-9; weight, 3240 g; height, 51 cm; and cephalic perimeter, 35.5 cm. At birth, she presented with hypoglycemia (0.23 g/L). She also presented with axial hypotonia, vomiting, and jaundice with increased conjugated bilirubin. Hepatic disease was suspected because of jaundice, hepatomegaly, pale stools, and neonatal cholestasis. Hepatic enzyme serum levels were: aspartate aminotransferase, 98 U/L (normal, <50); alanine aminotransferase, 57 U/L (normal, <45); γ-glutamyl transpeptidase activity, 84 U/L (normal, <45); total bilirubin, 84 μmol/L (normal, < 17); and conjugated bilirubin, 77 μmol/L. The liver ultrasound was normal. Endocrine evaluation was performed at age 1 month and 15 days. GH in hypoglycemia was 5.5 mU/L (N > 20), IGF-1 < 3 ng/mL (N > 40 ng/mL), T4L 8.1 pmol/L (N > 15), TSH 2.25 mU/L (N), cortisol 8 ng/mL (N > 50), ACTH 11 pg/mL. Gonadotropin secretion was not evaluated. MRI evaluation indicated the absence of the pituitary stalk, ectopic posterior pituitary, and small anterior pituitary (2 mm; normal size, 5 mm; Figure 1). There was no evidence of HPE or strabismus. The patient was given substitutive therapy with: T4 (25 μg by mouth), hydrocortisone (2 mg three times daily by mouth), GH (1 U/kg body weight/d delivered sc), and vitamin D substitution. The hepatic parameters were normal 1 month later. She presented seven generalized seizures during the first year associated with hypoglycemia, concomitant to fever. Information on the use of hydrocortisone doses was not correctly applied by the parents, and this partly explains the occurrence of hypoglycemia. The seizures stopped on administration of Depakine 200 mg given orally twice daily. At age 4.5 years, the patient has normal height, growth, and development.
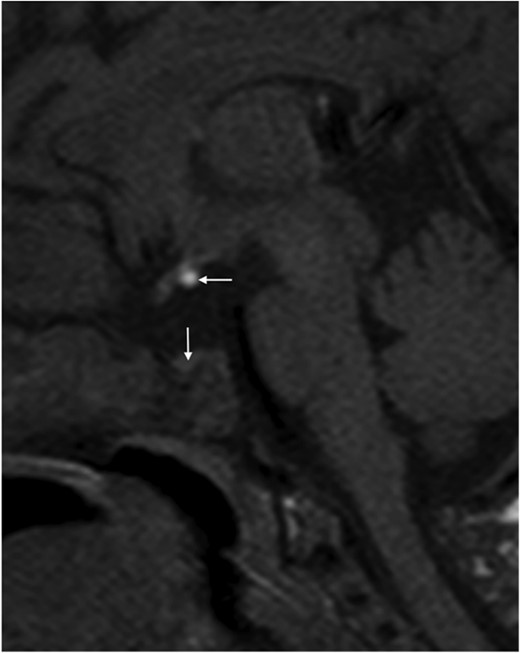
MRI of the patient showing the small anterior pituitary of 2 mm (vertical arrow) and the ectopic posterior lobe (horizontal arrow).
The pituitary stalk is absent.
DNA from the patient was used for exome sequencing. Insufficient DNA was available from the parents for exome sequencing. Exon enrichment was performed using Agilent SureSelect Human All Exon V4. Paired-end sequencing was performed on the Illumina HiSeq2000 platform using TruSeq v3 chemistry. Read files were generated via the manufacturer's proprietary software. Reads were mapped using the Burrows-Wheeler Aligner14 and the GATK version 1.615. Duplicate reads were marked using Picard version 1.62 (http://broadinstitute.github.io/picard/). SNP and indel variants were called using the GATK Unified Genotyper for each sample. SNP novelty was determined against dbSNP138. Variants with a minor allelic frequency of > 0.01 were excluded from subsequent analysis. Rare and novel variants were analyzed by a range of web-based bioinformatics tools using the EnsEMBL SNP Effect Predictor (http://www.ensembl.org/Tools/VEP). All variants were screened against the Human Gene Mutation Database Professional Biobase (http://www.hgmd.cf.ac.uk/ac/index.php). In silico analysis was performed to determine the potential pathogenicity of the variants. Potentially pathogenic mutations were verified using Sanger sequencing. The number of paired-end reads was 20 464 677 (mean coverage of 67.44). The percentage of target bases with > 10 times coverage was 96.57%. The number of variants with predicted serious (involving an essential splice site, a stop codon gained or lost, a complex indel, a frameshift mutation, or a nonsynonymous change) consequences for the child was 11 680. Analyses of the datasets revealed serious mutations in 58 genes, of which 52 were predicted by PolyPhen2 and/or SIFT to be deleterious substitutions for protein function (Supplemental Table 1).
The analysis of the 58 genes revealed a candidate mutation for the phenotype. The patient carries a novel nonsense mutation in the CDON gene (c.2764T>C, Glu922Ter; Figure 2). This mutation is also carried by her mother, who had congenital convergent strabismus. Bae et al (9) described a very broad spectrum of HPE phenotypes in association with CDON mutations that included absent pituitary (case 5288). Furthermore, two patients were noted as having biliary atresia (case 5308) or hepatic cholestasis (case 5288). It is possible therefore that the hepatic disease described in the patient here could be due to the CDON mutation. However, in our experience, cholestasis is frequently seen in PSIS patients during the neonatal period, and it is associated with profound cortisol deficiency.
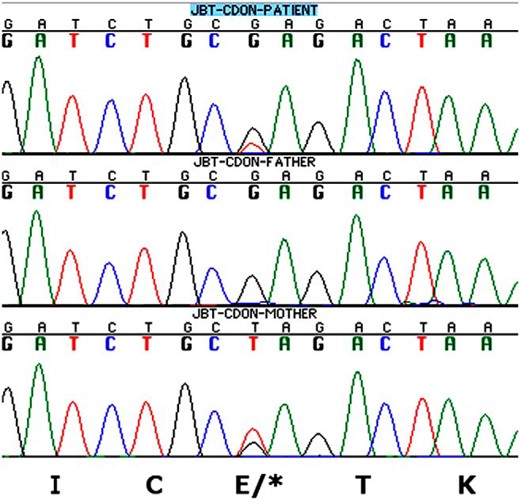
Representative chromatograms of the heterozygous c.2764T>C, Glu922Ter CDON mutation showing patient (top), father (middle), and mother (bottom).
The mutation was absent in the ancestry-matched control cohort.
The patient's mother suffered from strabismus and had a normal height (161 cm), yet also carried the CDON mutation. Bae et al (9) reported hypotelorism in two of six cases, and this may reflect a milder form of the phenotype. In families with pituitary anomalies associated with heterozygous frameshift and missense mutations involving the HESX1 and LHX4 genes, there is a highly variable phenotype together with incomplete penetrance. Here, the phenotypic variability seen in the patient and her mother could be due to the influence of other genetic variants present in the patient (either in the exome in Supplemental Table 1 or in regulatory sequences) or it could be due to gene-environment interactions. Recently, a dramatic synergy between a Cdon mutation in the mouse and a suspected HPE teratogen, ethanol was reported (10). Mice lacking Cdon and in utero ethanol exposure in 129S6 mice give little or no phenotype individually but together produced a broad spectrum of HPE phenotypes. Such environment-gene interactions may explain the spectrum of phenotypes seen in PSIS families.
Acknowledgments
We are grateful for the assistance of Dr Dalila Habes and Dr Bogdan Hermeziu, Service d'hépatologie pédiatrique, and Dr Fernando Sariego, Service de radiologie pédiatrique, Assistance Publique-Hôpitaux de Paris, Hôpital Bicêtre. We also thank Dr Jean Furioli, Service de pédiatrie, Hôpital de Mantes-la-jolie, for assistance in the clinical workup of this family.
This work was supported by the Laboratoire Lilly France and Projet Blanc Institut Pasteur/Assistance Publique-Hopitaux de Paris 2011. A.B and H.R. are funded in part by the program Actions Concertees Inter Pasteuriennes.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- CPHD
combined pituitary hormone deficiency
- HPE
holoprosencephaly
- MRI
magnetic resonance imaging
- PSIS
pituitary stalk interruption syndrome.

{kind=link}
{kind=link}