





The TSH receptor (TSHR) A-subunit shed from the cell surface contributes to the induction and/or affinity maturation of pathogenic TSHR autoantibodies in Graves' disease.
This study aimed to determine whether the quaternary structure (multimerization) of shed A-subunits influences pathogenic TSHR autoantibody generation.
The isolated TSHR A-subunit generated by transfected mammalian cells exists in two forms; one (active) is recognized only by Graves' TSHR autoantibodies, the second (inactive) is recognized only by mouse monoclonal antibody (mAb) 3BD10. Recent evidence suggests that both Graves' TSHR autoantibodies and mAb 3BD10 recognize the A-subunit monomer. Therefore, if the A-subunit monomer is an immunogen, Graves' sera should have antibodies to both active and inactive A-subunits. Conversely, restriction of TSHR autoantibodies to active A-subunits would be evidence of a role for shed A-subunit multimers, not monomers, in the pathogenesis of Graves' disease. Therefore, we tested a panel of Graves' sera for their relative recognition of active and inactive A-subunits.
Of 34 sera from unselected Graves' patients, 28 were unequivocally positive in a clinical TSH binding inhibition assay. None of the latter sera, as well as 8/9 sera from control individuals, recognized inactive A-subunits on ELISA. In contrast to Graves' sera, antibodies induced in mice, not by shedding from the TSHR holoreceptor, but by immunization with adenovirus expressing the free human A-subunit, were directed to both the active and inactive A-subunit forms.
The present study supports the concept that pathogenic TSHR autoantibody affinity maturation in Graves' disease is driven by A-subunit multimers, not monomers.
Hyperthyroidism in Graves' disease is caused by an autoimmune response to the TSH receptor (TSHR) mediated by thyroid stimulating autoantibodies (TSAb). There is substantial evidence that the TSHR structure itself contributes to this response. Unlike the closely related gonadotropin receptors, some TSHRs on the cell surface undergo intramolecular cleavage into disulfide-linked A- and B-subunits, followed by A-subunit shedding. Data from an animal model of Graves' disease suggest that the isolated A-subunit is more effective in inducing TSAb than the identical A-subunit when it remains part of the holoreceptor on the cell surface (1–4).
Very recent data have suggested the importance of the quaternary structure (multimerization) of the shed A-subunit in the induction and/or affinity maturation of pathogenic TSHR autoantibodies, detected in a TSH binding inhibition (TBI) assay or by bioassay (TSAb). This concept arose from the early observation of two distinct forms of recombinant, mammalian A-subunit, each of which can be purified separately (5). One form (termed active) is recognized by pathogenic TSHR Ab and not by mouse monoclonal antibody (mAb) 3BD10. Conversely, the second A-subunit form (termed inactive) is recognized by 3BD10 and not by pathogenic TSHR autoantibodies. Despite this reciprocal recognition, based on their crystal structures, TSAb M22 (6) and 3BD10 (7) can bind simultaneously to the monomeric form of the TSHR A-subunit, as shown in Figure 1. Consequently, because TSAb M22 is known to specifically recognize the active A-subunit form (8, 9), the monomeric A-subunit cannot explain the reciprocally exclusive TSAb and 3BD10 binding that is observed experimentally (5). Instead, computer analysis suggested an alternative explanation, namely that TSAb and 3BD10 recognize A-subunits multimers of different valency (7).
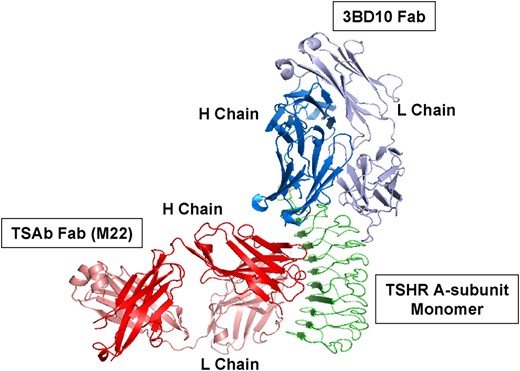
The TSHR A-subunit monomer cannot represent both forms (active and inactive) of purified A-subunits.
The crystal structure of a Fab for human monoclonal TSAb M22 (red) in complex with the major component of TSHR A-subunit (amino acids 22–260; green) has been solved (6) (protein data base 3GO4), as has the crystal structure of a Fab for mouse monoclonal antibody (mAb) 3BD10 (blue/violet) generated to the human A-subunit (7) (protein data base 4QT5). TSAb M22 specifically recognizes the active, not the inactive, A-subunit form (8, 9). Conversely, mAb 3BD10 only recognizes inactive A-subunits (5). Guided by amino acids contributing to its epitope, the 3BD10 Fab can be docked in silico with the M22/TSHR structure (7). Because both M22 and 3BD10 can simultaneously bind to the TSHR A-subunit monomer, the latter cannot explain the reciprocally exclusive TSAb and 3BD10 binding that is observed experimentally (5).
Because of the importance of the TSHR A-subunit structure in the pathogenesis of Graves' disease, in the present study we sought direct experimental evidence to support the concept that an A-subunit multimer, and not a monomer, is the primary immunogen for pathogenic TSHR autoantibody induction and/or affinity maturation. Very simply, if the TSHR A-subunit immunogen is a monomer, sera from Graves' patients should have autoantibodies to both active and inactive forms of the A-subunit, as observed in animals immunized with vectors coding for the isolated A-subunit (10). Conversely, the presence in Graves' sera of autoantibodies solely to the active A-subunit form would be evidence of a role for shed A-subunit multimers, not monomers, in the pathogenesis of Graves' disease.
Materials and Methods
Human and mouse sera
Human sera were from 34 patients with a diagnosis of Graves' disease, sent to us by colleagues from other institutions without identifiable private information and banked at −80°C. Mouse sera were from CXB-recombinant inbred animals immunized with adenovirus coding for the human TSHR A-subunit, as previously reported (11), and selected for the presence of TSHR antibodies as measured in the TSH binding inhibition (TBI) assay (described below).
TBI assay
Sera were tested for TSHR antibodies using a commercial clinical assay kit (Kronus, Boise, ID). In brief, serum aliquots (50 μL for human, 25 μL for mouse) were incubated with detergent-solubilized TSHR; 125I-TSH was added and the TSHR-antibody complexes were precipitated with polyethylene glycol. TBI values were calculated from the formula: [1 − (TSH binding in test serum − nonspecific binding)/(TSH binding in normal serum − nonspecific binding)] × 100. In this assay, TBI values < 10%, 10–15%, and > 15% are regarded as negative, borderline, and unequivocally positive, respectively.
ELISA
IgG class TSHR antibodies (IgG class) were assayed as previously reported (1). Human TSHR A-subunit protein secreted into medium by Chinese Hamster Ovary cells with an amplified transgenome (2) was purified using a mouse monoclonal antibody 3BD10 that is specific for the inactive form of A-subunit (3). Test sera (1:50 dilution for human, 1:100 dilution for mouse; duplicate aliquots) were added to ELISA wells coated with this protein. Antibody binding was detected with horseradish peroxidase–conjugated antihuman IgG (555788; BD Biosciences, San Jose, CA) and antimouse IgG (A3673, Sigma Chemical Co., St. Louis, MO) and the signal was developed with o-phenylenediamine and H2O2. Data are reported as the OD at 490 nm.
Results
TSH receptor autoantibodies in Graves' disease
Clinically relevant, pathogenic TSHR autoantibodies detected by either the TSH binding inhibition (TBI) assay or by bioassay selectively recognize the active, not the inactive TSHR A-subunit form. However, the question arises as to whether sera from Graves' patients also contain autoantibodies to the inactive A-subunit form. To address this issue, we tested 34 Graves' sera in our freezer bank for both forms of autoantibodies. As expected with a panel of anonymous sera not characterized for the clinical status of their donors, TBI values varied widely from negative (<10% TSH binding inhibition) to 100% (total inhibition of TSH binding) (Figure 2A). TBI activity was clearly present in the majority (28/34) of samples, with only two being negative and four providing values in the borderline range (10–15% TSH binding inhibition). As negative controls, all nine sera from normal individuals without a history of autoimmune thyroid disease also lacked TBI activity (Figure 2B). As a positive control in the TBI assay we included serum from a mouse immunized with adenovirus expressing the human TSHR A-subunit (Figure 2C).
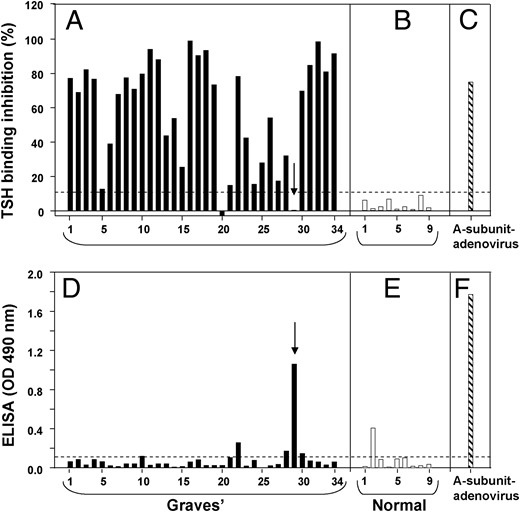
Pathogenic autoantibodies in Graves' sera do not recognize 'inactive' TSHR A-subunits.
Pathogenic autoantibodies in Graves' sera detected in the TSH binding inhibition (TBI) assay are neutralized by active, not inactive, A-subunits (5). A panel of 34 unselected Graves' sera (A) and nine sera from individuals without a history of autoimmune thyroid disease (B) were tested for TBI activity. TBI activity was clearly present in most samples, with only two being unequivocally negative (<10% TSH binding inhibition; horizontal dashed line). Four sera provided values in the borderline range (10–15% TSH binding inhibition). C, serum from a mouse immunized with adenovirus expressing the human TSHR A-subunit. The same sera were tested by ELISA for antibodies to purified, inactive A-subunits; 34 Graves' sera (D) and 9 control sera (E). The threshold for ELISA positivity (OD 0.1) is indicated by the horizontal dashed line. The arrows in Panels A and D identify Graves' serum No. 29 that was TBI negative, but positive by ELISA for inactive A-subunits. F, The same serum as in Panel C included as a positive control (using an antimouse IgG second antibody).
Remarkably, on testing the same 34 Graves' sera by ELISA using highly purified, inactive human TSHR A-subunits as antigen, the great majority were unreactive, generating OD values < 0.1 (Figure 2D). Graves' serum No. 22 was equivocally positive (OD, 0.26), within the range of values obtained with sera from the normal individuals (Figure 2E). Only one of the 34 Graves' sera (No. 29) clearly recognized inactive A-subunits by ELISA (OD, 1.06), as did one of the control sera (OD, 0.41). Of note, Graves' serum No. 29, clearly positive on ELISA, was negative in the TBI assay. The same serum from the mouse immunized with adenovirus expressing A-subunit (Figure 2C) was included as a positive control in the inactive A-subunit ELISA (using an antimouse IgG second antibody) (Figure 2F). The efficacy of the antihuman IgG was confirmed in the same ELISA using purified human thyroid peroxidase (TPO) and sera (1:100 dilution) from two patients with TPO autoantibodies (OD, 1.38 and 0.85, respectively, not shown in Figure 2).
TSH receptor antibodies in an induced mouse model of Graves' disease
In numerous previous studies, we observed that immunization of mice of different strains with adenovirus coding for the isolated human TSHR A-subunit generated pathogenic antibodies measured in both the TBI assay and nonpathogenic antibodies detected by ELISA using wells coated with purified, inactive human A-subunits. Because of importance of these findings for comparison with the data for patients with Graves' disease (Figure 2), we plotted data from 29 CXB mice that developed varying levels of TSHR antibodies detected in the TBI assay (28–84% inhibition of radiolabeled TSH binding) (Figure 3A), as well as six control mice that did not develop TBI antibodies (Figure 3B). Most the TBI-positive sera (20/29) generated a strong signal (OD > 0.5) in the ELISA for inactive TSHR A-subunits (Figure 3C). On ELISA, none of the control sera lacking TBI activity generated OD values > 0.1 (Figure 3D).
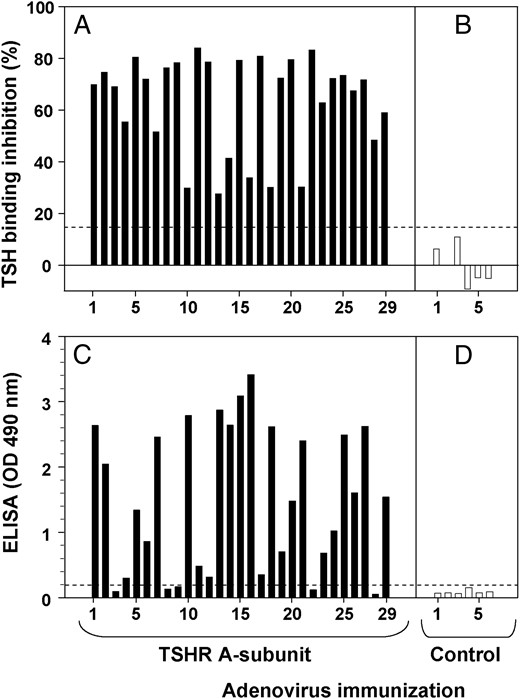
Pathogenic TSHR antibodies induced in mice by TSHR A-subunit DNA immunization also recognize inactive A-subunits.
Sera from 29 CXB recombinant inbred mice from a previous study (11) were randomly selected for TBI positivity following immunization with adenovirus coding for the human TSHR A-subunit (A). TBI values for sera from six mice injected with control adenovirus are shown in Panel B. Values above the dashed horizontal line (15% TSH binding inhibition) indicate unequivocal TBI positivity. The same sera were tested by ELISA for antibodies to purified, inactive A-subunits (C and D). The threshold for ELISA positivity (OD 0.1) is indicated by the horizontal dashed line.
Discussion
Pathogenic, TSHR autoantibodies in Graves' disease are of very high affinity, have discontinuous, conformational epitopes, and their detection (by TBI or bioassay) requires the use of native TSHR antigen. The shed A-subunit rather than the TSH holoreceptor is primarily responsible for affinity maturation of these highly potent autoantibodies (1, 9). A potential tool to provide insight into the pathogenesis of this process is that two different conformational forms of recombinant TSHR A-subunits generated in mammalian cells can be separately purified (5). The addition of nanogram quantities of one A-subunit form (termed active) to Graves' sera can fully neutralize the TBI or TSAb activity whereas A-subunits purified by mouse mAb 3BD10 are devoid of this activity (inactive) (5). The two A-subunit forms have identical primary amino acid sequences and their properties are destroyed by denaturation (5), indicating their native quality and that the difference between them resides in their conformation.
Until recently, our concept was that active and inactive TSHR A-subunits comprised two isomeric forms of the monomer (5). However, as mentioned above (Introduction), new information from the crystal structure of the 3BD10 Fab reveals that the A-subunit monomer cannot represent both forms (7). This observation enabled us to address the question of whether the shed A-subunit immunogen in Graves' disease is a monomer or a multimer. If a monomer, Graves' sera should contain autoantibodies to both active and inactive A-subunits. The present data exclude this possibility. None of the 28 Graves' sera that were unequivocally positive for TBI activity contained antibodies to inactive A-subunits as measured by ELISA (Figure 2). The inability to detect autoantibodies in Graves' sera to inactive A-subunits cannot be explained by a low TSHR autoantibody titer because similarly high TBI values induced in mice by A-subunit adenovirus immunization were associated with extremely high levels of antibodies to inactive A-subunits (Figure 3). Of interest, the single Graves' serum strongly positive for autoantibodies to inactive A-subunits was TBI negative, supporting the concept that a divergence from pathogenic to nonpathogenic antibodies that may lead to remission (10). However, in an anonymous bank of sera, no clinical information is available for this individual patient. Future clinical studies on patients with this rare autoantibody profile would be of value.
It is of heuristic value to compare the relative balance of TSHR antibodies to active and inactive A-subunits that have been observed under different circumstances (Table 1). Graves' disease is the only condition in which TSHR antibodies are solely to the active A-subunit. TSHR antibodies induced in mice by DNA vectors coding for the isolated A-subunit are directed against both active and inactive A-subunits (for example 10, 12–14) as are antibodies arising spontaneously in a transgenic mouse expressing the isolated human A-subunit in the thyroid when back-crossed on to NOD.H2h4 background (15) In contrast, conventional immunization of mice with TSHR A-subunit protein plus adjuvant generates only nonpathogenic antibodies that only recognize inactive A-subunits (for example 16).
Profile of TSHR Antibodies to Active and Inactive A-Subunits Observed in Different Conditions
Profile of TSHR Antibodies to Active and Inactive A-Subunits Observed in Different Conditions
In conclusion, we suggest a possible explanation for these disparate findings. There is strong evidence that the TSH holoreceptor on the thyrocyte surface exists as a multimer (for example 17, 18). Therefore, it is possible that the A-subunits shed from these holoreceptors are multimers, or multimers form shortly thereafter, for example in draining regional lymph nodes. Unlike A-subunits shed from the holoreceptor, DNA vectors and the thyroid-targeted transgene express the isolated A-subunit, which is more likely to exist primarily as a monomer, hence generating antibodies to both active and inactive A-subunit forms.
Acknowledgments
This work was supported by National Institutes of Health Grant DK19289.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- mAb
monoclonal antibody
- TBI
TSH binding inhibition
- TPO
thyroid peroxidase
- TSHR
TSH receptor
- TSAb
thyroid stimulating autoantibodies.

{kind=link}
{kind=link}
{kind=link}