





Abstract
MDR Mycobacterium tuberculosis clinical strains that cause large outbreaks, particularly among HIV-negative patients, are likely to have undergone the most successful compensatory evolution. Hence, mutations secondary to the acquisition of drug resistance are worthy of consideration in these highly transmissible strains. Here, we assessed the role of a mutation within rpoB, rpoB V615M, secondary to the rifampicin resistance-conferring mutation rpoB S531L, which is associated with a major MDR tuberculosis outbreak strain that evolved in an HIV-negative context in northern Tunisia.
Using BCG as a model organism, we engineered strains harbouring either the rpoB S531L mutation alone or the double mutation rpoB S531L, V615M. Individual and competitive in vitro growth assays were performed in order to assess the relative fitness of each BCG mutant.
The rpoB V615M mutation was found to be invariably associated with rpoB S531L. Structural analysis mapped rpoB V615M to the same bridge helix region as rpoB compensatory mutations previously described in Salmonella. Compared with the rpoB single-mutant BCG, the double mutant displayed improved growth characteristics and fitness rates equivalent to WT BCG. Strikingly, the rpoB double mutation conferred high-level resistance to rifampicin.
Here, we demonstrated the fitness compensatory role of a mutation within rpoB, secondary to the rifampicin resistance mutation rpoB S531L, which is characteristic of an MDR M. tuberculosis major outbreak strain. The finding that this secondary mutation concomitantly increased the resistance level to rifampicin argues for its significant contribution to the successful transmission of the MDR-TB strain.
Introduction
MDR-TB, defined as tuberculosis that is resistant to at least isoniazid and rifampicin, is a major concern for public health worldwide. According to the latest WHO estimates, 3.5% of new and 20.5% of previously treated TB cases had MDR-TB in 2013, corresponding to an estimated 480 000 people globally.1
The steady worldwide increase in MDR-TB contrasts with the expected reduced fitness often associated with drug resistance mutations in Mycobacterium tuberculosis.2–5 Indeed, as in almost any bacteria, drug resistance mutations in M. tuberculosis target important biological functions in the cell, thus imparting a fitness cost. The fitness cost usually refers to a reduction in relative bacterial growth in vivo6 or in vitro,7,8 inefficient transmission9 and/or changes in relative virulence.10–13
Among the antibiotic resistance mutations that could have multiple effects on the fitness of MDR M. tuberculosis clinical isolates are those associated with rifampicin, which is the most important first-line anti-TB drug.14,15 Rifampicin binds to, and inhibits, the β subunit of DNA-dependent RNA polymerase (rpoB) by obstructing the exit tunnel for nascent RNA, thus disrupting transcription.16,17 In M. tuberculosis clinical isolates, >70 different mutations conferring rifampicin resistance have been reported.18,19 In >95% of M. tuberculosis clinical isolates, these mutations are confined to an 81 bp core region of the rpoB gene (codons 507–534, Escherichia coli numbering), known as cluster I of the rifampicin resistance-determining region (RRDR). Mutations at positions 531, 526 and 516 are the most frequent among high-level rifampicin-resistant M. tuberculosis clinical isolates.18,20,21
Consistent with the essential role of RNA polymerase in transcription, rifampicin resistance mutations were shown to be associated with a fitness cost in M. tuberculosis.22–25 It is now well established that M. tuberculosis clinical strains restore their fitness by accumulating secondary site mutations to buffer the deleterious impact of rifampicin resistance mutations.26–28
MDR or XDR M. tuberculosis clinical isolates that cause large outbreaks, particularly in HIV-negative patients, should be viewed as the most successful example of compensatory evolution. Understanding the molecular basis underlying fitness compensation in these successful clinical isolates is critical to our understanding of their increased transmission phenotype.29–32 Here, we demonstrate the role in fitness cost compensation and increased rifampicin resistance of a secondary mutation within the rpoB gene of a severe MDR-TB outbreak strain bearing the rifampicin resistance mutation rpoB S531L.
Methods
Description of the MDR-TB outbreak
The MDR-TB outbreak strain under consideration was identified in the region of Bizerte, northern Tunisia, as early as 2001. The clinical and epidemiological features of this MDR-TB outbreak have been previously described in detail.33–35 Briefly, from September 2001 to June 2011, the outbreak involved 45 HIV-negative, non-institutionalized individuals (mean age 29.7 years; 88.9% male),9 20% of whom have died. All patients were first treated according to the national standardized first-line regimen, and once the MDR nature of the strain was established they were given second-line drugs.
The genotype of the outbreak strain was determined by spacer oligonucleotide typing (spoligotyping), which detects variability in the direct repeat (DR) region of M. tuberculosis. The spoligoprofile of the outbreak strain displayed the ST53 signature of the Haarlem family.33 Drug susceptibility testing coupled to mutational analysis suggested that primary transmission involved a strain that was simultaneously resistant to isoniazid, rifampicin, ethambutol and streptomycin.33 Mutational analysis further confirmed this finding as all outbreak strains bore the katG S315T, rpoB S531L, emB M306I and gidB R47W mutations. In the rpoB gene, all outbreak strains showed an additional mutation, rpoB V615M.
Full-length sequencing of the RNA polymerase genes of the MDR-TB outbreak strain
The full-length coding sequences of rpoA, rpoB and rpoC genes of the MDR-TB strain were PCR amplified. The primers used for PCR amplification and sequencing are provided in Table S1 (available as Supplementary data at JAC Online).
The amplification reaction mixture contained 20 ng of template genomic DNA, 10 μL of ×10 buffer (Qiagen), 10 μL of DMSO, 2 μL of 10 mM nucleotide mix (Amersham Biosciences), 2 μL of each primer (20 μM stock), 0.25 μL (1.25 U) of HotStar Taq DNA polymerase (Qiagen) and sterile nuclease-free water (Amersham Biosciences) in a total reaction volume of 50 μL. Cycling was carried out in a 2720 thermocycler (Applied Biosystems) with an initial denaturation step of 10 min at 96°C followed by 35 cycles consisting of 1 min at 95°C, 1 min at 60°C and 2 min at 72°C. Amplification ended with a final elongation step of 7 min at 72°C.
Amplicons were subjected to sequencing after treatment with Exonuclease I (Amersham Biosciences) and shrimp alkaline phosphatase (Amersham Biosciences). The reaction consisted of 1.5 μL of BigDye terminator cycle sequencing reagents, 4 μL of Big Dye terminator cycle sequencing buffer, 1 μL of 20 μM concentrations of primers and sufficient UltraPure Distilled DNase/RNase-Free Water (Gibco/Invitrogen) to make a 20 μL reaction. Cycle sequencing was performed using a 2720 thermocycler (Applied Biosystems) programmed for 25 cycles at 96°C for 10 s, 50°C for 5 s and 60°C for 4 min. The template DNA was ethanol precipitated, washed and subjected to automated sequencing on an ABI Prism 3130 genetic analyser (Applied Biosystems) according to the manufacturer's protocol. Both strands of each amplicon were sequenced from two independent PCR amplification reactions.
We also searched for MDR-TB outbreak-associated RNA polymerase mutations in a Tunisian collection of susceptible and MDR clinical isolates within and outside the outbreak region (Supplementary Data).
Phylogenomic distribution of the non-synonymous mutations identified in the RNA polymerase genes of the MDR-TB outbreak strain
All genomic data included in this study were retrieved from different databases, including the European Nucleotide Archive (ENA), The Broad Institute and NCBI (Supplementary Data). The genomic dataset includes complete genome sequences and single- and paired-end reads. The genome sequence of M. tuberculosis H37Rv strain (version NC_000962.3 in the NCBI database) was used as a reference strain. Complete genomes were aligned to the reference sequence using progressive Mauve version 2.3.0.36 Single- and paired-end reads were mapped using BWA aligner.37 Several Python modules were written to analyse the output files from the different aligner programs to identify specific and shared SNPs between all compared genomes. SNPs in PE/PGRS genes, mobile elements and those linked to insertion/deletion regions were excluded from our analysis. Intragenic SNPs were investigated for their effect on protein-coding genes to find non-synonymous and synonymous SNPs.
RaxML (version 8.1.3) was used for maximum likelihood (ML)-based estimates of the M. tuberculosis complex (MTBC) strains phylogeny and 1000 bootstrap replicates were performed to assess statistical support. The phylogenetic tree based on whole-genome data and sequence alignment of the concatenation of rpoB, rpoC and rpoA genes were combined using Gingr from the Harvest suite38 to visualize the distribution of the RNA polymerase-associated SNPs throughout the MTBC lineages.
Locating the mutated residues in the RNA polymerase complex
Positions of mutated residues were deduced from the crystal structure of Thermus thermophilus RNA polymerase, which was modelled with Swiss-Pdb Viewer version 4.1.0 (http://www.expasy.org/spdbv/).39 Coordinates were obtained from the Protein Data Bank (accession number 2O5I).
Generation of BCG mutants
Isogenic mutant BCG strains harbouring the rpoB mutation S531L alone or the double mutation rpoB S531L, V615M were generated by allelic exchange using the WT Pasteur BCG vaccine strain (Pasteur 1173P2) (Figure 1). For this purpose, a 1000 bp PCR fragment encompassing the rpoB single mutation rpoB S531L was PCR amplified using the primer pair TBB1 (5′-ATCCACACCGCAGACGTTG-3′) and TBB2 (5′-TGCATCACAGTGATGTAGTCG-3′). The amplicon was then subcloned into pCSn plasmid vector (Figure 1a), which contains the E. coli galactokinase (galK) gene, used as a counter-selectable marker.40 Plasmid pCSn was constructed in our laboratory using pCR2.1 plasmid vector (Invitrogen) as a backbone. In pCSn, the galK gene is expressed under the control of the mycobacterial β-lactamase promoter and the transcription terminator of ESAT-6. Freshly prepared electrocompetent BCG cells were electroporated (2.5 kV; 25 mF; 1000 Ω) with 250 ng each of pCSn constructs in a 0.2 cm cuvette. Then 3 mL of fresh 7H9-OADC-Tween 80 medium was added, and the electroporated BCG cells were incubated at 37°C for 7 days under agitation before plating on 7H10-OADC agar plates containing 0.5% 2-deoxy-galactose (2-DOG).
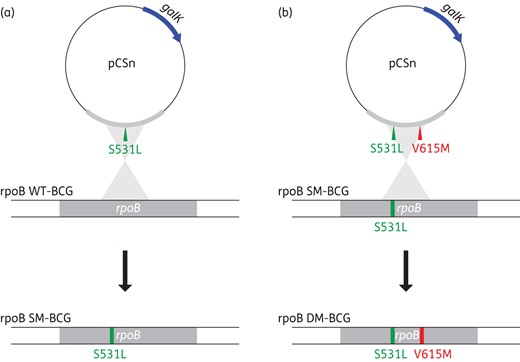
Schematic representation of the protocol used for the generation of a mutant BCG strain harbouring the rpoB S531L mutation (a) and its derived isogenic double mutant bearing the secondary mutation rpoB V615M, a hallmark of the Tunisian MDR-TB outbreak strain (b). This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Mutant BCG clones harbouring the rpoB S531L mutation were then selected by inoculating Löwenstein–Jensen (L–J) slants containing 40 mg/L rifampicin. The introduced mutation was confirmed by nucleotide sequencing. To obtain an isogenic BCG strain bearing the double rpoB mutation, allelic exchange was performed as described above, using the rpoB S531L single-mutant BCG strain as a recipient host (Figure 1b). The latter strain was electroporated with a pCSn vector containing a double-mutant amplicon (rpoB S531L, V615M), which was PCR amplified using the MDR-TB outbreak strain chromosomal DNA. PCR amplification followed by nucleotide sequencing was performed on rifampicin-resistant BCG colonies to identify double mutants.
In vitro growth kinetics of rpoB mutant BCG strains
Single colonies of WT or mutant BCG strains were picked from L–J slants and inoculated into 4 mL of Middlebrook 7H9 broth (BACTEC, Becton Dickinson Diagnostic Systems, Sparks, MD, USA) supplemented with OADC–0.5% glycerol. After a 2 week incubation at 37°C under agitation in the presence of glass beads, the culture was transferred to a sterile tube and allowed to sediment for 30 min. The cleared supernatant was transferred to a new tube, pulse-vortexed six times (∼1 s per pulse), then allowed to settle for 30 min. The cleared supernatant was collected, adjusted in 7H9 broth to an OD600 of 0.4 and preserved at −80°C as a 15% glycerol stock. BCG colonies were counted for the preserved vials by plating on 7H10. A total of 3 × 103 cfu were cultured in 20 mL of 7H9 medium and incubated at 37°C under agitation. At days 1, 3, 5, 7, 11, 13 and 15, a 1 mL sample of each culture was collected. For each timepoint sample, serial dilutions were prepared and plated on 7H10 agar with 10% OADC for 35 days, after which the cfu were determined.
In vitro competition experiments
The fitness cost of a resistant mutation was determined by direct competition against the WT BCG strain, essentially as previously described.22 Briefly, ODs of the WT and mutant BCG strains were first adjusted to the same value. Then 30 μL of the mutant and 10 μL of the WT BCG bacterial suspensions were mixed in 20 mL of 7H9 medium and incubated at 37°C under agitation. Tenfold serial dilutions were prepared from each mixed culture at days 1, 3, 7, 11 and 14 and plated on both drug-free and rifampicin (40 mg/L)-containing Middlebrook 7H10–10% OADC. Counting of the cfu was performed after an incubation of 35 days at 37°C. The cfu counts of the WT BCG strain were obtained by subtracting the cfu on drug-containing plates from the total cfu on drug-free plates.
In single and competition cultures, the doubling time was estimated from a plot of ln(cfu) = f(time), where the slope was ln2/doubling time (h). The relative fitness was the ratio of doubling time (WT)/doubling time (mutant).
Drug susceptibility testing
Drug susceptibility of BCG mutant strains and the MDR-TB Haarlem outbreak strain was tested in L–J medium using rifampicin concentrations of 40, 60, 80, 100, 120, 140 and 160 mg/L, essentially as previously described.41
Statistical analyses
Statistical differences between BCG strains were analysed by one-way ANOVA using GraphPad Prism version 4.02 with a P value <0.05.
Results
Mutational profile of the MDR-TB outbreak strain RNA polymerase genes
The full-length sequences of rpoA, rpoB and rpoC genes were determined from the chromosomal DNA of a representative MDR-TB Haarlem outbreak strain. Aside from the previously described rpoB mutations S531L and V615M,35,35 no additional amino acid substitutions could be identified in rpoB. A single mutation (G519E) was observed in rpoC, while rpoA was devoid of any mutation. Further PCR amplifications and sequencing targeting the three RNA polymerase mutations showed that they were common to six additional MDR-TB strains that were associated with the Tunisian outbreak (Table 1). Next, we searched for these mutations in a Tunisian strain collection of diverse genotypes, drug resistance profiles and geographical origins (Supplementary Data). The rpoB V615M mutation was found to be restricted to the outbreak strain and was invariably associated with the rifampicin-conferring mutation S531L. The rpoC G519E mutation was common to all the Haarlem strains of the outbreak region irrespective of their drug resistance profile, including the drug-susceptible pre-outbreak strain 23335 (Table 1 and Supplementary Data). This mutation was also found in 7 out of 14 (50%) Haarlem isolates from outside the outbreak region, and could also be associated, albeit less frequently, with other genotypes, such as the Latin American Mediterranean (LAM) (Table 1 and Supplementary Data).
Nature and frequency of rpoB and rpoC mutations found in M. tuberculosis clinical isolates within and outside the MDR-TB outbreak regiona
| M. tuberculosis clinical isolates | n | rpoC mutation G519E (%) | rpoB mutations (%) | ||||
|---|---|---|---|---|---|---|---|
| S531L and V615M | S522L | S531L | S531T | Q510R and H526L | |||
| Within the outbreak region | |||||||
| MDR-TB outbreak Haarlem strain | 7 | 100 | 100 | — | — | — | — |
| MDR-TB isolates unrelated to the outbreak | 3 | — | — | — | — | — | 100 |
| Drug-susceptible Haarlem isolates unrelated to the outbreak | 24 | 100 | — | — | — | — | — |
| Non-Haarlem drug-susceptible isolates | 6 | 16.66 | — | — | — | — | — |
| Outside the outbreak region | |||||||
| Drug-susceptible and non MDR Haarlem isolates unrelated to the outbreak | 14 | 50 | — | — | — | — | — |
| Drug-susceptible non Haarlem isolates | 3 | 33.33 | — | — | — | — | — |
| Non-Haarlem MDR isolates | 7 | — | — | 14.28 | 28.57 | 14.28 | — |
| M. tuberculosis clinical isolates | n | rpoC mutation G519E (%) | rpoB mutations (%) | ||||
|---|---|---|---|---|---|---|---|
| S531L and V615M | S522L | S531L | S531T | Q510R and H526L | |||
| Within the outbreak region | |||||||
| MDR-TB outbreak Haarlem strain | 7 | 100 | 100 | — | — | — | — |
| MDR-TB isolates unrelated to the outbreak | 3 | — | — | — | — | — | 100 |
| Drug-susceptible Haarlem isolates unrelated to the outbreak | 24 | 100 | — | — | — | — | — |
| Non-Haarlem drug-susceptible isolates | 6 | 16.66 | — | — | — | — | — |
| Outside the outbreak region | |||||||
| Drug-susceptible and non MDR Haarlem isolates unrelated to the outbreak | 14 | 50 | — | — | — | — | — |
| Drug-susceptible non Haarlem isolates | 3 | 33.33 | — | — | — | — | — |
| Non-Haarlem MDR isolates | 7 | — | — | 14.28 | 28.57 | 14.28 | — |
aDetails of the M. tuberculosis clinical isolates are provided in Supplementary Data.
Nature and frequency of rpoB and rpoC mutations found in M. tuberculosis clinical isolates within and outside the MDR-TB outbreak regiona
| M. tuberculosis clinical isolates | n | rpoC mutation G519E (%) | rpoB mutations (%) | ||||
|---|---|---|---|---|---|---|---|
| S531L and V615M | S522L | S531L | S531T | Q510R and H526L | |||
| Within the outbreak region | |||||||
| MDR-TB outbreak Haarlem strain | 7 | 100 | 100 | — | — | — | — |
| MDR-TB isolates unrelated to the outbreak | 3 | — | — | — | — | — | 100 |
| Drug-susceptible Haarlem isolates unrelated to the outbreak | 24 | 100 | — | — | — | — | — |
| Non-Haarlem drug-susceptible isolates | 6 | 16.66 | — | — | — | — | — |
| Outside the outbreak region | |||||||
| Drug-susceptible and non MDR Haarlem isolates unrelated to the outbreak | 14 | 50 | — | — | — | — | — |
| Drug-susceptible non Haarlem isolates | 3 | 33.33 | — | — | — | — | — |
| Non-Haarlem MDR isolates | 7 | — | — | 14.28 | 28.57 | 14.28 | — |
| M. tuberculosis clinical isolates | n | rpoC mutation G519E (%) | rpoB mutations (%) | ||||
|---|---|---|---|---|---|---|---|
| S531L and V615M | S522L | S531L | S531T | Q510R and H526L | |||
| Within the outbreak region | |||||||
| MDR-TB outbreak Haarlem strain | 7 | 100 | 100 | — | — | — | — |
| MDR-TB isolates unrelated to the outbreak | 3 | — | — | — | — | — | 100 |
| Drug-susceptible Haarlem isolates unrelated to the outbreak | 24 | 100 | — | — | — | — | — |
| Non-Haarlem drug-susceptible isolates | 6 | 16.66 | — | — | — | — | — |
| Outside the outbreak region | |||||||
| Drug-susceptible and non MDR Haarlem isolates unrelated to the outbreak | 14 | 50 | — | — | — | — | — |
| Drug-susceptible non Haarlem isolates | 3 | 33.33 | — | — | — | — | — |
| Non-Haarlem MDR isolates | 7 | — | — | 14.28 | 28.57 | 14.28 | — |
aDetails of the M. tuberculosis clinical isolates are provided in Supplementary Data.
Phylogenetic distribution of the RNA polymerase mutations associated with the Tunisian MDR-TB outbreak strain
To get a better insight into the frequency and phylogenetic distribution of the RNA polymerase mutations associated with the MDR-TB outbreak strain, we reconstructed a phylogenetic tree based on 102 genomes from drug-susceptible, rifampicin-resistant and MDR M. tuberculosis clinical strains distributed worldwide and belonging to seven MTBC lineages (Supplementary Data). To visualize the distribution of mutations in the RNA polymerase genes, we combined each genome of the phylogenetic tree with its corresponding rpoA, rpoB and rpoC sequences. As shown in Figure 2, the rpoC G519E mutation was associated with, and restricted to, all genomes of a lineage that included Haarlem strains, as well as the reference strain CDC155142 and the outbreak C strain.43 Hence, rpoC G519E is a common feature of this lineage and was associated with either rifampicin-susceptible or rifampicin-resistant strains. As expected, rpoB S531L, the most frequently encountered rifampicin resistance-conferring mutation among clinical M. tuberculosis isolates, was associated with several rifampicin-resistant strains from diverse lineages. By contrast, the rpoB V615M mutation was absent from all genome sequence data used in the phylogenomic analysis, thus confirming its restriction to the Tunisian MDR-TB outbreak strain.
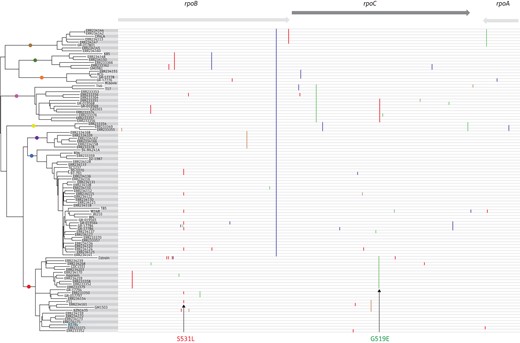
Maximum likelihood phylogenetic tree based on the genome sequence data of 102 M. tuberculosis strains (see Supplementary Data) and their corresponding mutational profile of rpoB, rpoC and rpoA genes (vertical coloured lines). Mutations associated with the Tunisian MDR-TB outbreak strain are indicated with arrows. Note the absence of the rpoB V615M mutation from all the genomes used in the phylogenetic reconstruction.
The rpoB V615M mutation maps to a region that interacts with the bridge helix of RNA polymerase
The fact that rpoB V615M was invariably associated with rpoB S531L and proved to be restricted to the Tunisian MDR-TB outbreak strain raised the possibility that it might be involved in fitness cost compensation, inasmuch as the rpoC G519E mutation seemed to be of phylogenetic significance. Based on the RNA polymerase crystal structure of T. thermophilus,44,45 the V615M mutation was mapped to a region of the RNA polymerase β-subunit that interacts with the bridge helix of the β′-subunit (Figure 3), a flexible structure that interacts with the DNA template strand.46,47 Of note, the compensatory mutations rpoB R637C and rpoB H673Y previously identified in Salmonella were also shown to be associated with the bridge helix.48
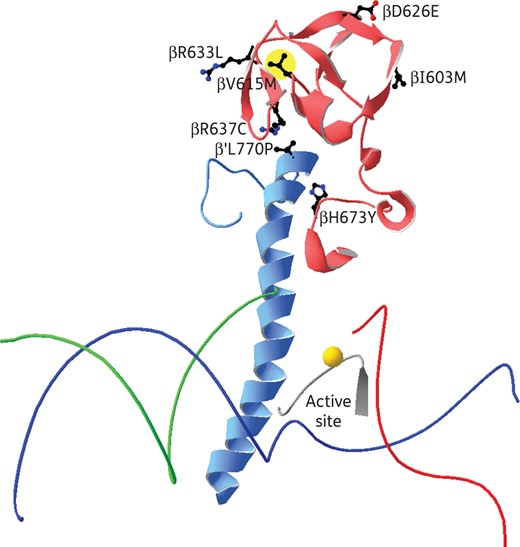
Illustration of the central part of the RNA polymerase of T. thermophilus, PDB code 2O5I. The template strand of DNA is shown in blue, the non-template strand in green and the RNA in red, and the Mg2+ ion in the active centre is marked by a gold sphere. The bridge helix of the β′ subunit is shown in pale blue and the part of the β subunit that interacts with the bridge helix in pale red. All side chains in the β and β′ subunits that are mutated are shown and labelled. The rpoB V615M mutation is highlighted in yellow.
In BCG, the rpoB V615M mutation efficiently compensated the fitness defect incurred by rpoB S531L
To unambiguously demonstrate the fitness compensation role of rpoB V615M, we constructed a BCG strain carrying the rpoB S531L mutation alone, from which we derived its isogenic double mutant by introducing the second rpoB mutation, rpoB V615M. As shown in Figure 4(a), the mutant BCG harbouring rpoB S531L yielded fewer colonies with smaller size in L–J slants compared with its isogenically derived double mutant, which bears the additional rpoB V615M mutation. This finding was confirmed in liquid cultures, as the single mutant exhibited ∼2-fold slower growth kinetics in single culture assays (doubling time 54.15 h) (Figure 4b; Table 2) compared with the rpoB double mutant and WT BCG strains, which showed similar kinetics with comparable doubling times (23.49 versus 22.72 h). Similar results were observed in growth competition assays, with a slight advantage in growth kinetics of the double mutant over the WT BCG (Table 2). Consistent with the observed doubling time values, the relative fitness of the single rpoB mutant BCG strain was significantly lower compared with its derived double mutant in both single and competition culture assays (0.41 versus 0.96 and 0.45 versus 0.95, respectively; P < 0.05) (Table 2). In single and competition culture assays, the relative fitness of the rpoB double mutant was comparable to that of WT BCG (0.96 versus 0.99 and 0.95 versus 0.93) (Table 2 and Figure 5). Taken together, these findings indicate that the fitness cost incurred by rpoB S531L is fully restored by rpoB V615M.
Growth characteristics parameters of rpoB S531L single-mutant BCG and its isogenically derived double mutant (rpoB S531L and rpoB V615M) compared with WT BCG
| Strain | Mutation | MIC (mg/L) | Single culturea | Competition culturea | ||||
|---|---|---|---|---|---|---|---|---|
| doubling Time (h) | no. of divisions | relative fitness | doubling time (h) | no. of divisions | relative fitness | |||
| rpoB WT BCG | none | 10 | 22.72 | 15.84 | 0.99 | 24.42 | 14.62 | 0.930 ± 0.02 |
| rpoB SM BCG | S531L | 40 | 54.15 | 6.17 | 0.4113 ± 0.087 | 53.67 | 6.707 | 0.455 |
| rpoB DM BCG | S531L and V615M | >160 | 23.49 | 14.4 | 0.96 ± 0.05 | 23.18 | 15.53 | 0.95 ± 0.03 |
| Strain | Mutation | MIC (mg/L) | Single culturea | Competition culturea | ||||
|---|---|---|---|---|---|---|---|---|
| doubling Time (h) | no. of divisions | relative fitness | doubling time (h) | no. of divisions | relative fitness | |||
| rpoB WT BCG | none | 10 | 22.72 | 15.84 | 0.99 | 24.42 | 14.62 | 0.930 ± 0.02 |
| rpoB SM BCG | S531L | 40 | 54.15 | 6.17 | 0.4113 ± 0.087 | 53.67 | 6.707 | 0.455 |
| rpoB DM BCG | S531L and V615M | >160 | 23.49 | 14.4 | 0.96 ± 0.05 | 23.18 | 15.53 | 0.95 ± 0.03 |
aData shown represent the average of four independent replicate experiments. Standard errors for relative fitness are indicated. In single and competition cultures, the doubling time was estimated from a plot of ln(cfu) = f(time), where the slope is ln2/doubling time (h). The relative fitness is the ratio of doubling time (WT)/doubling time (mutant).
Growth characteristics parameters of rpoB S531L single-mutant BCG and its isogenically derived double mutant (rpoB S531L and rpoB V615M) compared with WT BCG
| Strain | Mutation | MIC (mg/L) | Single culturea | Competition culturea | ||||
|---|---|---|---|---|---|---|---|---|
| doubling Time (h) | no. of divisions | relative fitness | doubling time (h) | no. of divisions | relative fitness | |||
| rpoB WT BCG | none | 10 | 22.72 | 15.84 | 0.99 | 24.42 | 14.62 | 0.930 ± 0.02 |
| rpoB SM BCG | S531L | 40 | 54.15 | 6.17 | 0.4113 ± 0.087 | 53.67 | 6.707 | 0.455 |
| rpoB DM BCG | S531L and V615M | >160 | 23.49 | 14.4 | 0.96 ± 0.05 | 23.18 | 15.53 | 0.95 ± 0.03 |
| Strain | Mutation | MIC (mg/L) | Single culturea | Competition culturea | ||||
|---|---|---|---|---|---|---|---|---|
| doubling Time (h) | no. of divisions | relative fitness | doubling time (h) | no. of divisions | relative fitness | |||
| rpoB WT BCG | none | 10 | 22.72 | 15.84 | 0.99 | 24.42 | 14.62 | 0.930 ± 0.02 |
| rpoB SM BCG | S531L | 40 | 54.15 | 6.17 | 0.4113 ± 0.087 | 53.67 | 6.707 | 0.455 |
| rpoB DM BCG | S531L and V615M | >160 | 23.49 | 14.4 | 0.96 ± 0.05 | 23.18 | 15.53 | 0.95 ± 0.03 |
aData shown represent the average of four independent replicate experiments. Standard errors for relative fitness are indicated. In single and competition cultures, the doubling time was estimated from a plot of ln(cfu) = f(time), where the slope is ln2/doubling time (h). The relative fitness is the ratio of doubling time (WT)/doubling time (mutant).
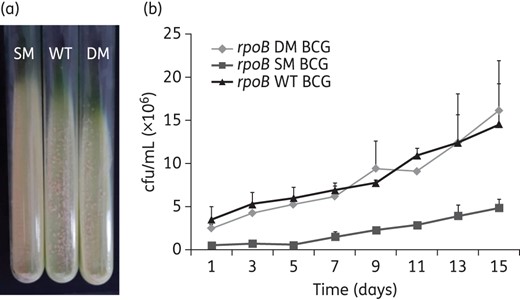
Growth characteristics of the rpoB S531L single-mutant BCG and its isogenically derived double mutant (rpoB S531L and rpoB V615M) compared with WT BCG. (a) Mutant and WT BCG strains growing in L–J slants. (b) Growth kinetics of mutant and WT BCG strains in 7H9 liquid medium. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
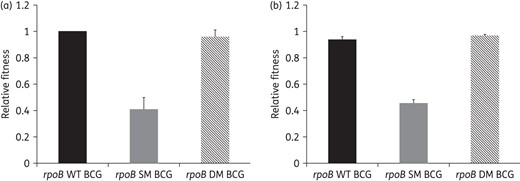
Relative fitness of the rpoB S531L single-mutant BCG and its isogenically derived double mutant (rpoB S531L and rpoB V615M) compared with WT BCG in single (a) and competition (b) cultures.
The double mutation rpoB S531L, V615M is associated with high-level resistance to rifampicin
Because mutations in rpoB are essentially involved in rifampicin resistance, we compared the resistance level of the rpoB single-mutant BCG strain to its isogenically derived double mutant. Strikingly, as for the original M. tuberculosis MDR outbreak strain, the double rpoB mutant BCG showed an MIC value >160 mg/L, which is by far higher than the MIC value of 40 mg/L displayed by the single mutant (Table 2). Hence, the rpoB secondary mutation rpoB V615M contributed to increased rifampicin resistance levels.
Discussion
It has long been thought that MDR-TB strains could not successfully expand in the community due to their reduced fitness, which is the direct consequence of the combined cost of multiple mutations in their drug resistance genes.2–5 This assumption stems from in vitro studies, which clearly showed the growth defect of MDR M. tuberculosis clinical isolates, and from in vivo observations, reporting that almost all MDR-TB outbreaks have been found to be associated with HIV-positive patients.4 The increased transmission of MDR-TB strains among HIV-positive individuals has been attributed to the absence of a potent immune response rather than a true epidemic potential of the involved strain. Hence, it is reasonable to believe that MDR-TB strains that emerge and expand in an HIV-negative context should have evolved the most successful compensatory evolution, and should thus deserve particular attention. Such an MDR-TB outbreak strain was identified in northern Tunisia and involved 45 non-institutionalized, apparently immunocompetent, young individuals (mean age 29.7 years). By causing such a large outbreak, in the face of a competent immune response, we reasoned that this MDR-TB strain must be highly fit, thus prompting a detailed study. Here, we focused on its RNA polymerase mutations in order to understand its compensatory evolution.
Full-length sequencing analysis of rpoB, rpoC and rpoA, coupled to phylogenetic studies, identified the outbreak-restricted rpoB V615M mutation as potentially involved in fitness compensation. This mutation was shared by all MDR-TB strains recovered from the 45 outbreak cases, being invariably associated with rpoB S531L, suggesting that it is secondary to it. The rpoB V615M mutation proved very rare. To our knowledge it has been described only for one clinical isolate from China,49 in association with rpoB S531L, as for the Tunisian MDR-TB outbreak strain. The MDR-TB strain also harboured a mutation within rpoC, G519E, whose role in fitness cost compensation is very unlikely since it was common to drug-susceptible and drug-resistant strains and occurred prior to rpoB S531L, being identified in the drug-susceptible pre-outbreak strain. Another rpoC mutation in the same position (G519D) has been previously identified in an independent study and was deemed not to be involved in fitness cost compensation.26
Generation of a BCG strain bearing the single rpoB S531L mutation and its isogenically derived double mutant, into which rpoB V615M was introduced, unambiguously demonstrated the role of the rpoB V615M in the full restoration of the fitness defect incurred by rpoB S531L. This frequently encountered rifampicin resistance-conferring mutation is located in the RNA polymerase channel where the DNA/RNA hybrid complex is located. The serine residue forms a hydrogen bond to the peptide backbone of rpoB leucine residue at position 533, stabilizing the local structure of the exit tunnel.45 A mutation at this position from a serine to a leucine will have two effects: (i) the hydrogen bond to the peptide backbone will be lost; and (ii) a non-polar bump will form in the exit channel pointing towards the newly synthesized RNA four bases away from the catalytic centre. Therefore, it is reasonable to believe that this mutation will change the properties of the exit channel close to the catalytic centre.
Another rifampicin resistance mutation, rpoB R529C, has been described previously to have similar effects on the RNA polymerase structure of Salmonella typhimurium. It was shown that the fitness cost caused by this mutation could be compensated for by different groups of RNA polymerase mutations.48 One of these groups, consisting of the mutations rpoB R637C, rpoB H673Y and rpoC L770P, was shown to be associated with the bridge helix of the β′ subunit. The bridge helix is a flexible structure that interacts with the DNA template strand and is thought to move it through the RNA polymerase and position it in the active centre.46,47 Since mutations in the bridge helix can increase the elongation rate,50,51 it was thought that the compensatory mutations exert their function by influencing the dynamic properties of the bridge helix.48
The compensatory mutation described in this study, rpoB V615M, is located in the same region of rpoB as the compensatory mutations rpoB R637C and rpoB H673Y. The fitness costs of the two rifampicin resistance mutations rpoB S531L and rpoB R529C are probably caused by similar structural changes. It is therefore reasonable to believe that the compensatory mutations act by similar mechanisms. The importance of this region for the compensation of the fitness cost of rifampicin resistance mutations can be seen in further clinical M. tuberculosis isolates from China.49 Several isolates have been identified with two mutations in the rpoB gene. Each of these strains carried a common rifampicin resistance mutation and additionally a secondary, non-resistance mutation that is located in the same region as rpoB V615M (Table 3). This supports the hypothesis that modulations of the properties of the bridge helix can compensate for the fitness cost caused by different rifampicin resistance mutations. Additional rpoB mutations outside the bridge helix region (rpoB E761D, rpoB L731P) are also suspected to be involved in fitness cost compensation.30,32
List of isolates with putative compensatory mutations that affect the properties of the bridge helix
| Rifampicin resistance mutation | Secondary RNA polymerase mutation | Organism | Compensationa | Reference |
|---|---|---|---|---|
| rpoB Q513P | rpoB R633L | M. tuberculosis | ND | 49 |
| rpoB H526D | rpoB D626E | M. tuberculosis | ND | 49 |
| rpoB H526R | rpoB D626E | M. tuberculosis | ND | 49 |
| rpoB R529C | rpoB R637C | S. typhimurium | yes | 48 |
| rpoB R529C | rpoB H673Y | S. typhimurium | yes | 48 |
| rpoB R529C | rpoC L770P | S. typhimurium | yes | 48 |
| rpoB S531L | rpoB I603M | M. tuberculosis | ND | 49 |
| rpoB S531L | rpoB V615M | M. tuberculosis | yes | this study |
| Rifampicin resistance mutation | Secondary RNA polymerase mutation | Organism | Compensationa | Reference |
|---|---|---|---|---|
| rpoB Q513P | rpoB R633L | M. tuberculosis | ND | 49 |
| rpoB H526D | rpoB D626E | M. tuberculosis | ND | 49 |
| rpoB H526R | rpoB D626E | M. tuberculosis | ND | 49 |
| rpoB R529C | rpoB R637C | S. typhimurium | yes | 48 |
| rpoB R529C | rpoB H673Y | S. typhimurium | yes | 48 |
| rpoB R529C | rpoC L770P | S. typhimurium | yes | 48 |
| rpoB S531L | rpoB I603M | M. tuberculosis | ND | 49 |
| rpoB S531L | rpoB V615M | M. tuberculosis | yes | this study |
ND, not determined.
aSecondary mutation experimentally shown to compensate the fitness cost caused by the rifampicin resistance mutation.
List of isolates with putative compensatory mutations that affect the properties of the bridge helix
| Rifampicin resistance mutation | Secondary RNA polymerase mutation | Organism | Compensationa | Reference |
|---|---|---|---|---|
| rpoB Q513P | rpoB R633L | M. tuberculosis | ND | 49 |
| rpoB H526D | rpoB D626E | M. tuberculosis | ND | 49 |
| rpoB H526R | rpoB D626E | M. tuberculosis | ND | 49 |
| rpoB R529C | rpoB R637C | S. typhimurium | yes | 48 |
| rpoB R529C | rpoB H673Y | S. typhimurium | yes | 48 |
| rpoB R529C | rpoC L770P | S. typhimurium | yes | 48 |
| rpoB S531L | rpoB I603M | M. tuberculosis | ND | 49 |
| rpoB S531L | rpoB V615M | M. tuberculosis | yes | this study |
| Rifampicin resistance mutation | Secondary RNA polymerase mutation | Organism | Compensationa | Reference |
|---|---|---|---|---|
| rpoB Q513P | rpoB R633L | M. tuberculosis | ND | 49 |
| rpoB H526D | rpoB D626E | M. tuberculosis | ND | 49 |
| rpoB H526R | rpoB D626E | M. tuberculosis | ND | 49 |
| rpoB R529C | rpoB R637C | S. typhimurium | yes | 48 |
| rpoB R529C | rpoB H673Y | S. typhimurium | yes | 48 |
| rpoB R529C | rpoC L770P | S. typhimurium | yes | 48 |
| rpoB S531L | rpoB I603M | M. tuberculosis | ND | 49 |
| rpoB S531L | rpoB V615M | M. tuberculosis | yes | this study |
ND, not determined.
aSecondary mutation experimentally shown to compensate the fitness cost caused by the rifampicin resistance mutation.
To our knowledge, our study is the first that has experimentally proved the compensating role of a secondary mutation in the rpoB gene of a clinical M. tuberculosis strain (Table 3). In this study, we evaluated the role of this secondary rpoB mutation as it occurred in the outbreak strain, being associated with rpoB S531L. The fact that introduction of this secondary mutation was accompanied by a high MIC of rifampicin raises the possibility that it might be individually involved in rifampicin resistance. However, it is also tempting to speculate that the rpoB V615M-mediated increased MIC in association with rpoB S531L is rather a consequence of the modulation of the bridge helix properties.
The overall impact of drug resistance-conferring mutations is the result of a complex interplay of several factors, including differences in the strain's genetic background, the initial fitness cost of drug-resistance-conferring mutations, the presence of additional antibiotic resistance mutations and the ability to acquire compensatory mutations.4,23,24 In a recent study it has been suggested that the success of rifampicin resistance mutations in clinical M. tuberculosis isolates may be dependent not on a low initial fitness cost, but rather on the results of three factors: (i) a high rifampicin MIC; (ii) a relatively low initial fitness cost; and (iii) the ability to acquire compensatory mutations that alleviate the costs of resistance.28 The successful association of the rpoB S531L and rpoB V615M mutations described herein provides an edifying, naturally occurring example in line with the above suggestions. In this respect, one can reasonably argue that the combined effect on fitness and rifampicin resistance of the double rpoB mutation could have significantly contributed to the successful transmission of the Tunisian MDR-TB outbreak strain.
Funding
This work was supported by grants from the Tunisian Ministry of Higher Education and Scientific Research (LR11IPT01), as well as EU 7th Framework Program (EUMEDNETvsTB, project n° 245 872).
Transparency declarations
None to declare.
Acknowledgements
The authors thank Senda Zarrouk and Neila Khabouchi for their assistance with nucleotide sequencing.
References
Author notes
Present address: Department of Microbiology, Unit for Genome Dynamics, Oslo University Hospital, PO Box 10 4950, Nydalen, NO-0424 Oslo, Norway.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}