





Abstract
Systemic inflammation has been suggested to be associated with Alzheimer’s-disease progression, although whether it is a cause or a downstream effect is still controversial. This study aims to assess the effect of systemic inflammatory regulators on Alzheimer’s disease within a bidirectional Mendelian-randomization design.
Genetic associations with Alzheimer’s disease were obtained from the largest and most up-to-date genome-wide association study (GWAS) (cases and proxy cases: 71 880; controls: 383 378) and with inflammatory regulators from two recent GWASs on the human proteome and cytokines. Estimates were obtained by inverse-variance weighting with sensitivity analyses using MR-Egger, weighted median and MR-PRESSO. Possible bias due to selective survival and competing risk was also considered.
None of 41 systemic inflammatory regulators was associated with risk of Alzheimer’s disease with consistent results in validation analysis. Conversely, Alzheimer’s disease was suggestively associated with five systemic inflammatory regulators, i.e. basic fibroblast growth factor, granulocyte-colony-stimulating factor, interferon gamma, interleukin-13 and interleukin-7.
The systemic inflammatory regulators considered did not appear to be associated with the risk of Alzheimer’s disease. Conversely, specific systemic inflammatory regulators may be downstream effects of Alzheimer’s disease or consequences of common factors causing both inflammation and Alzheimer’s disease.
The 41 systemic inflammatory regulators assessed were not associated with risk of Alzheimer’s disease.
Conversely, Alzheimer’s disease was associated with an increase in five of the assessed systemic inflammatory regulators.
Systemic inflammation may be a downstream effect of Alzheimer’s disease.
Introduction
Alzheimer’s disease is the most common type of dementia contributing to 60–80% of cases.1 Apart from amyloid beta and neurofibrillary tangles, inflammation could also play an important role in the pathogenesis of Alzheimer’s disease.2,3 Systemic inflammation has been suggested to accelerate Alzheimer’s-disease progression and is associated with blood-brain-barrier changes that may favour amyloid-beta accumulation in the brain.4,5 A study also found that systemic immune challenges alone may lead to Alzheimer’s-disease-like neurological symptoms.6 However, whether such systemic inflammation is a cause or a downstream effect of Alzheimer’s disease is still controversial and mixed results were found in meta-analysis of observational studies.7,8 However, these observational studies could be subject to unmeasured confounding, particularly by health status, making it difficult to distinguish causes from symptoms, and so they may not reflect the true causal effect.
In this case, Mendelian randomization (MR) can be a useful design for assessing the role of systemic inflammatory factors in Alzheimer’s disease. MR as an instrumental variable analysis utilizing genetic variants gives unconfounded estimates, because genetic variants are allocated randomly during conception well before illness onset. As such, MR may provide more robust evidence about causal effects.9 To assess whether systemic inflammatory factors are associated with Alzheimer’s disease and to assess the direction of association, we performed a two-sample bidirectional MR study using the most up-to-date genome-wide association studies (GWASs) of systemic inflammatory regulators and Alzheimer’s disease.10,11
Methods
MR and assumptions
This study is a bidirectional two-sample MR study using genetic instruments [single-nucleotide polymorphisms (SNPs)] predicting systemic inflammatory regulators and Alzheimer’s disease from the latest GWAS. We used a bidirectional design to assess the association of systemic inflammatory regulators with Alzheimer’s disease as well as to test whether Alzheimer’s disease causes systemic inflammatory regulators. MR relies on three assumptions, i.e. the genetic predictors of exposure are strongly associated with the exposure of interest; the genetic predictors of exposure are independent from confounders of the exposure–outcome relation; and the genetic predictors are only linked to the outcome through affecting the exposure of interest (i.e. the exclusion–restriction assumption).12
Genetic associations with systemic inflammatory regulators
Genetic predictors of cytokines and other systemic inflammatory regulators and genetic associations with systemic inflammatory regulators were obtained from the most recent GWAS of 41 systemic inflammatory regulators in 8293 Finnish individuals from the Cardiovascular Risk in Young Finns Study (mean age men: 37.4 years; women: 37.5 years) and the ‘FINRISK’ studies (mean age FINRISK1997 men: 48.3; women: 47.3 and FINRISK2002 men: 60.4; women: 60.1).10 Circulating inflammatory regulators were measured using Bio-Rad’s premixed Bio-Plex Pro Human Cytokine 27-plex Assay and 21-plex Assay, and a Bio-Plex 200 reader with Bio-Plex 6.0 software.10 The genetic associations were adjusted for age, sex, body mass index and the first 10 genetic principal components with genomic-control correction.10
As a validation, we used the corresponding proteins as exposures, taken from a GWAS of the human proteome.13 More than 3600 proteins in 3301 individuals (mean age 43.7 years, 48.9% women) of European descent from the INTERVAL study were assessed.13 The GWAS adjusted for age, sex, duration between blood draw and processing, and the first three principal components of ancestry from multidimensional scaling.13
Genetic associations with Alzheimer’s disease
Genetic predictors of Alzheimer’s disease and genetic associations with Alzheimer’s disease were obtained from the most recent GWAS of Alzheimer’s disease.11 The study included 71 880 Alzheimer’s-disease cases and Alzheimer’s-disease-by-proxy cases based on parental diagnoses (mean age of onset: 64.3 years; 57.8% women) and 383 378 controls (mean age at last contact: 58.2; 53.9% women) of European ancestry from 4 consortia, including the Alzheimer’s-disease working group of the Psychiatric Genomics Consortium (PGC-ALZ), the International Genomics of Alzheimer’s Project, the Alzheimer’s Disease Sequencing Project (ADSP) and the UK Biobank.11 The genetic associations were adjusted for sex, batch (if applicable) and the first four ancestry principal components in the PGC-ALZ and ADSP cohorts.11 The PGC-ALZ cohort further adjusted for age in the association.11 The UK Biobank adjusted for age, sex, genotyping array, assessment centre and 12 ancestry principal components.11
Selection of genetic instruments
We used all SNPs that strongly and independently (R2 < 0.001) predicted exposures at genome-wide significance (P < 5 × 10–8). Since only 11 systemic inflammatory regulators had 3 or more independent SNPs at genome-wide significance, a higher cut-off (P < 5 × 10–6) was also used for obtaining SNPs predicting the systemic inflammatory regulators. Proxy SNPs (R2 > 0.9) from LDlink (https://ldlink.nci.nih.gov/) were used when the SNPs were not available for the outcome.14 Palindromic SNPs were removed since the GWASs of systemic inflammatory regulators did not provide the allele frequency so we could not be sure that these SNPs were aligned in the same direction for exposure and outcome. Instrumental strength for the SNP–exposure association was calculated as the average of SNP-specific F-statistics approximated by the square of the beta divided by the variance for the SNP–exposure association.15 Variance explained by the SNPs for Alzheimer’s disease was 7.1% and that for systemic inflammatory regulators ranged from 3.6% to 34%.10,11 Exposures with fewer than three independent SNPs were excluded.
Statistical analysis
In the main analysis, we obtained estimates from inverse-variance weighting meta-analysis with multiplicative random effects of the SNP-specific Wald estimates, which assumes balanced pleiotropy. Since it is difficult to verify the ‘exclusion–restriction’ assumption, sensitivity analyses with different assumptions were performed, including MR-Egger, weighted median and Mendelian Randomization Pleiotropy RESidual Sum and Outlier (MR-PRESSO).16–18 Given that the selective survival on the instrument and the competing risk of the outcome can invalidate genetic instruments by creating an additional open pathway from instrument to outcome, we assessed this possibility from the association of the selected SNPs with survival, proxied by age at recruitment to the UK Biobank.19 Considering the number of systemic inflammatory regulators tested, we used a Bonferroni correction. Results that were significant before but not after correction for multiple comparison were considered as suggestive. Power calculations were done for the main analyses using the approximation that the sample size for an MR study is the sample size for exposure on outcome divided by the r2 for instruments on exposure.20,21
Sensitivity analyses
Weighted median
The weighted median of SNP-specific estimates provides valid estimates when ≥50% of the information is contributed from valid SNPs.17
MR-Egger
MR-Egger regression provides valid estimates when the InSIDE (Instrument Strength Independent of Direct Effect) assumption holds, even when all SNPs are invalid.16 However, we focused on the consistency of the direction of the estimates rather than the significance of the estimates due to the low statistical power of MR-Egger. A significant MR-Egger intercept indicates the presence of directional pleiotropy and a potentially invalid inverse-variance weighting (IVW) estimate.
MR-PRESSO
MR-PRESSO was used to test for and remove horizontal pleiotropic outliers assuming ≥50% of the instruments were valid, have balanced pleiotropy and the InSIDE assumption holds.18
Selective survival
Given that Alzheimer’s disease typically occurs in old age, the participants need to have survived the exposure and competing risk of Alzheimer’s disease in order to be recruited, which could create bias.22 To assess this possibility, we examined whether the SNPs predicting systemic inflammatory regulators and Alzheimer’s disease were associated with survival, proxied by age at recruitment. The main and sensitivity analyses for pleiotropy were repeated after removing such SNPs. The association of these SNPs with age at recruitment was obtained from the UK Biobank GWAS, which included 361 194 participants from the UK.23 The associations with age at recruitment to the UK Biobank (http://www.nealelab.is/uk-biobank) were adjusted for the first 20 principal components, age, age2, sex, age × sex and age2 × sex. Considering the number of SNPs predicting Alzheimer’s disease and inflammatory regulators, we applied a Bonferroni correction when assessing the association of these SNPs with age at recruitment.
All analyses were performed using R Version 3.4.2 with the R package ‘TwosampleMR’, ‘MendelianRandomization’ and ‘MR-PRESSO’.18,24,25 We used publicly available summary data so no ethical approval is required.
Results
Genetically predicted systemic inflammatory regulators on risk of Alzheimer’s disease
Among the 41 available systemic inflammatory regulators, 11 had 3 or more independent genome-wide significant SNPs, whereas all 41 had 3 or more SNPs when using the higher cut-off (P < 5 × 10–6). All these SNPs were included in the analyses. F-statistics ranged from 18.8 to 295.8, suggesting that weak instruments bias may not be substantial. Details of the SNPs included are in Supplementary Tables 1 and 2, available as Supplementary data at IJE online.
Supplementary Table 3, available as Supplementary data at IJE online, and Figure 1 show that none of the 11 systemic inflammatory regulators predicted by genome-wide significant SNPs [i.e. macrophage inflammatory protein-1β (MIP1b; CCL4), TNF-related apoptosis-inducing ligand (TRAIL), vascular endothelial growth factor (VEGF), eotaxin, interleukin-12p70 (IL-12p70), interleukin-18 (IL-18), monocyte chemotactic protein-1 (MCP1; CCL2), platelet-derived growth factor BB (PDGFbb), stem cell growth factor beta (SCGFb), cutaneous T-cell attracting chemokine (CCL27; CTACK) and interleukin-16 (IL-16)] were associated with Alzheimer’s disease in any analyses before or after correcting for multiple comparisons. MR-PRESSO and the MR-Egger intercept did not identify any pleiotropic SNPs. When using SNPs obtained with the higher cut-off (P < 5 × 10–6), only the interleukin-1 receptor antagonist showed a suggestive inverse association with Alzheimer’s disease before correcting for multiple comparisons using IVW with inconsistent estimates from sensitivity analyses. MR-PRESSO and the MR-Egger intercept suggested pleiotropic SNPs may be present for PDGFbb, stem cell factor, granulocyte-colony-stimulating factor and growth-regulated oncogene-α but MR-PRESSO did not identify any outliers except for granulocyte colony-stimulating factor (GCSF), which showed no association after removing the outlier (Supplementary Table 4, available as Supplementary data at IJE online, and Figure 2). We had >80% power to detect Alzheimer’s-disease risk with an odds ratio of 1.02:1.06 per SD increase in systemic inflammatory regulators for the analyses using genome-wide significant SNPs, as shown in Supplementary Table 5, available as Supplementary data at IJE online.
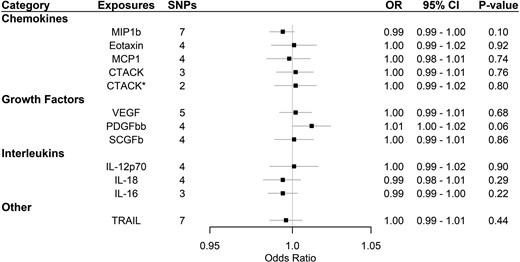
Association of systemic inflammatory regulators with Alzheimer’s disease using Mendelian randomization (with genome-wide significant SNPs). Odds ratio (OR) and 95% confidence interval (CI) represent the change in the odds ratio of Alzheimer’s disease per 1-SD increase in the systemic inflammatory regulator level. * indicates results after removing SNPs associated with age at recruitment. After correcting for multiple comparison, p-value < 0.05/11 = 0.0045 was considered as significant. CTACK, cutaneous T-cell attracting chemokine; IL, interleukin; MCP1, monocyte chemotactic protein-1; MIP1b, macrophage inflammatory protein-1β; PDGFbb, platelet-derived growth factor BB; SCGFb, stem cell growth factor beta; SNPs, single-nucleotide polymorphisms; TRAIL, TNF-related apoptosis-inducing ligand; VEGF, vascular endothelial growth factor.
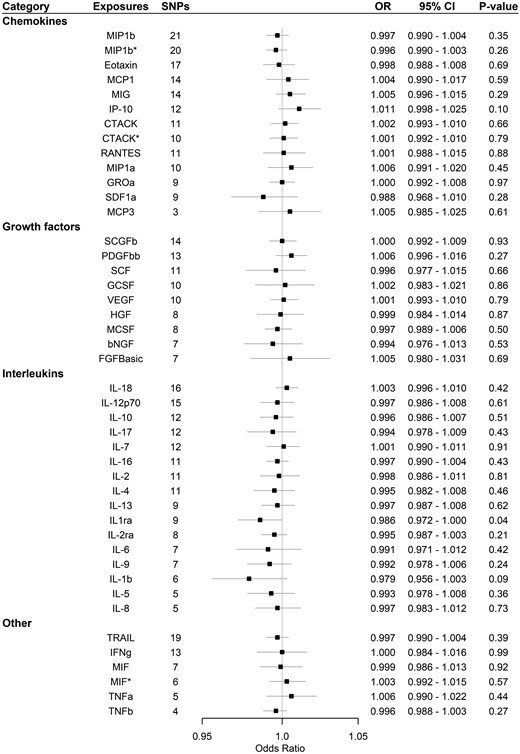
Association of systemic inflammatory regulators with Alzheimer’s disease using Mendelian randomization (with SNPs reaching p < 5 × 10–6). Odds ratio (OR) and 95% confidence interval (CI) represent the change in the odds ratio of Alzheimer’s disease per 1-SD increase in the systemic inflammatory regulator level. * indicates results after removing SNPs associated with age at recruitment. After correcting for multiple comparison, p-value < 0.05/41 = 0.0012 was considered as significant. bNGF, beta nerve growth factor; CTACK, cutaneous T-cell attracting chemokine; FGFBasic, basic fibroblast growth factor; GCSF, granulocyte colony-stimulating factor; GROa, growth-regulated oncogene-α; HGF, hepatocyte growth factor; IFNg, interferon gamma; IL, interleukin; IP, interferon-gamma-induced protein 10; MCP1, monocyte chemotactic protein-1; MCP3, monocyte-specific chemokine 3; MCSF, macrophage colony-stimulating factor; MIF, macrophage-migration inhibitory factor; MIG, monokine induced by interferon gamma; MIP1a, macrophage inflammatory protein-1α; MIP1b, macrophage inflammatory protein-1β; PDGFbb, platelet-derived growth factor BB; RANTES, regulated on Activation, Normal T Cell Expressed and Secreted; SCF, stem cell factor; SCGFb, stem cell growth factor beta; SDF1a, stromal cell-derived factor-1 alpha; SNPs, single-nucleotide polymorphisms; TNFa, tumour necrosis factor alpha; TNFb, tumour necrosis factor beta; TRAIL, TNF-related apoptosis-inducing ligand; VEGF, vascular endothelial growth factor.
After assessing survival, one SNP (rs55764737) predicting CTACK was found to be associated with age at recruitment after Bonferroni correction p-value (0.05/number of SNPs used in the analysis) for the analyses using genome-wide significant SNPs, whereas the same SNPs for CTACK, one SNP predicting MIF (rs78098071) and one SNP predicting MIP1b (rs17138331) were also associated with age at recruitment using a higher cut-off (5 × 10–6). The results were similar before and after removing these SNP, as shown in Supplementary Tables 3 and 4, available as Supplementary data at IJE online, and Figures 1 and 2.
Twenty-nine of the 41 systemic inflammatory regulators in the GWAS of cytokines were available in the proteome GWAS for validation analysis and all had three or more SNPs. One SNP for CCL3 (rs113937085), one SNP for CCL7 (rs149906452) and one SNP for KITLG (rs17416509) were not available for the outcome without proxy SNPs so these SNPs were removed from the analyses. F-statistics for the available SNPs ranged from 21.7 to 50.5, suggesting that weak instrument bias may not be substantial (Supplementary Table 6, available as Supplementary data at IJE online).
None of the systemic inflammatory regulators in the validation analyses was associated with Alzheimer’s disease other than interleukin-10 (IL10) as shown in Supplementary Table 7, available as Supplementary data at IJE online, and Figure 3. IL-10 was inversely associated with Alzheimer’s disease in IVW but no association was found in sensitivity analyses. No association was found after correcting for multiple comparison. One SNP (rs9652103) for interferon-gamma-induced protein 10 (IP-10/CXCL10) was associated with age at recruitment and the results were similar before and after removing that SNP.
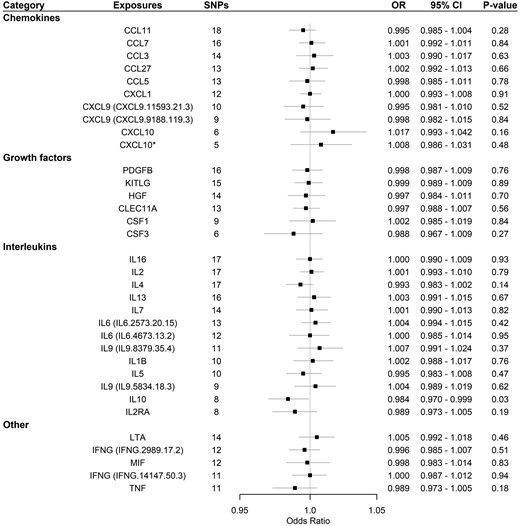
Association of systemic inflammatory regulators with Alzheimer’s disease using Mendelian randomization (validation proteome GWAS, with SNPs reaching p < 5 × 10–6). More than one set of data were available for some exposures. Their corresponding IDs were provided. Odds ratio (OR) and 95% confidence interval (CI) represent the change in the odds ratio of Alzheimer’s disease per 1-SD increase in the systemic inflammatory regulator level. * indicates results after removing SNPs associated with age at recruitment. After correcting for multiple comparison, p-value < 0.05/29 = 0.0017 was considered as significant. CCL3, macrophage inflammatory protein-1α; CCL5, regulated on Activation, Normal T Cell Expressed and Secreted; CCL7, monocyte-specific chemokine 3; CCL11, Eotaxin-1; CCL27, cutaneous T-cell attracting chemokine; CELC11A, stem cell growth factor beta; CSF1, macrophage colony-stimulating factor 1; CSF3, granulocyte colony-stimulating factor; CXCL1, growth-regulated oncogene-α; CXCL9, monokine induced by interferon gamma; CXCL10, r interferon-gamma-induced protein 10; HGF, hepatocyte growth factor; IFNg, interferon gamma; IL, interleukin; KITLG, KIT ligand; LTA, lymphotoxin-alpha; MIF, macrophage-migration inhibitory factor; PDGFB, platelet-derived growth factor subunit B; SNPs, single-nucleotide polymorphisms; TNF, tumour necrosis factor.
Genetically predicted Alzheimer’s disease on systemic inflammatory regulator levels
We extracted 27 SNPs strongly and independently predicting Alzheimer’s disease. For the analyses with monocyte-specific chemokine 3 (MCP3; CCL7) and tumour necrosis factor beta (TNFb), 3 and 4 SNPs were not available in the outcome GWAS, respectively, with no proxy SNPs available, leaving 24 and 23 SNPs for analysis. The variance of Alzheimer’s disease explained by these SNPs was 7.1%.11 The F-statistic was 163.4, suggesting that weak instruments bias may not be severe. Details of these SNPs are presented in Supplementary Table 8, available as Supplementary data at IJE online.
Supplementary Table 9, available as Supplementary data at IJE online, and Figure 4 show that Alzheimer’s disease had a suggestive association with increased basic fibroblast growth factor (FGFBasic), GCSF, interferon gamma (IFNg), interleukin-7 (IL-7) and interleukin-13 (IL-13) using IVW, weighted-median and MR-Egger analysis. A suggestive positive association was also found for interleukin-2 (IL-2) and tumour necrosis factor alpha (TNFa) using IVW. A suggestive association with decreased IL-18 was found using IVW. No association was found after correcting for multiple comparisons. The MR-Egger intercept for MCP1, Regulated on Activation Normal T Cell Expressed and Secreted (RANTES, CCL5) and VEGF, and MR-PRESSO for SCGFb suggested that there may be pleiotropic SNPs before correction for multiple comparisons, but MR-PRESSO did not identify any pleiotropic SNPs. We had 80% power to detect a change of 0.116–0.363 standard deviations in systemic inflammatory regulator levels (Supplementary Table 5, available as Supplementary data at IJE online).
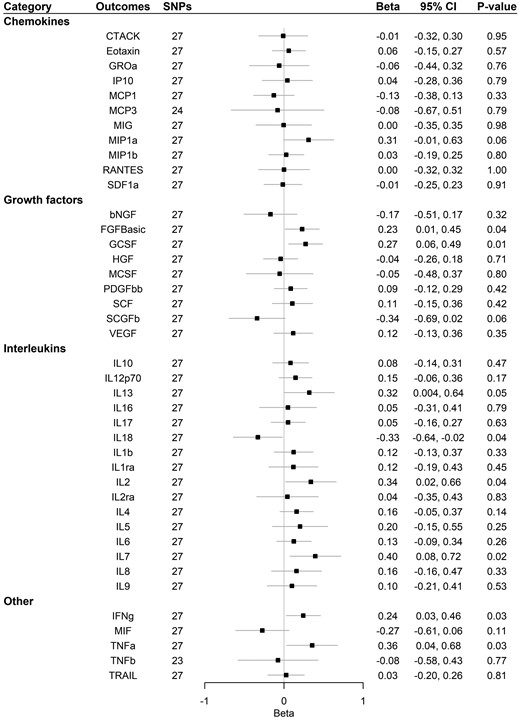
Association of Alzheimer’s disease with systemic inflammatory regulators using Mendelian randomization. Beta and 95% confidence interval (CI) represent the change in the SD of inflammatory regulators per log odds increase in Alzheimer’s disease. After correcting for multiple comparison, p-value < 0.05/41 = 0.0012 was considered as significant. bNGF, beta nerve growth factor; CTACK, cutaneous T-cell attracting chemokine; FGFBasic, basic fibroblast growth factor; GCSF, granulocyte colony-stimulating factor; GROa, growth-regulated oncogene-α; HGF, hepatocyte growth factor; IFNg, interferon gamma; IL, interleukin; IP, interferon-gamma-induced protein 10; MCP1, monocyte chemotactic protein-1; MCP3, monocyte-specific chemokine 3; MCSF, macrophage colony -stimulating factor; MIF, macrophage-migration inhibitory factor; MIG, monokine induced by interferon gamma; MIP1a, macrophage inflammatory protein-1α; MIP1b, macrophage inflammatory protein-1β; PDGFbb, platelet-derived growth factor BB; RANTES, regulated on Activation, Normal T Cell Expressed and Secreted; SCF, stem cell factor; SCGFb, stem cell growth factor beta; SDF1a, stromal cell-derived factor-1 alpha; SNPs, single-nucleotide polymorphisms; TNFa, tumour necrosis factor alpha; TNFb, tumour necrosis factor beta; TRAIL, TNF-related apoptosis-inducing ligand; VEGF, vascular endothelial growth factor.
One SNP predicting Alzheimer’s disease (rs113260531) was associated with age at recruitment after Bonferroni correction at p-value (0.05/27 = 0.002). After removing that SNP, results were similar (Supplementary Table 10, available as Supplementary data at IJE online, and Figure 5).
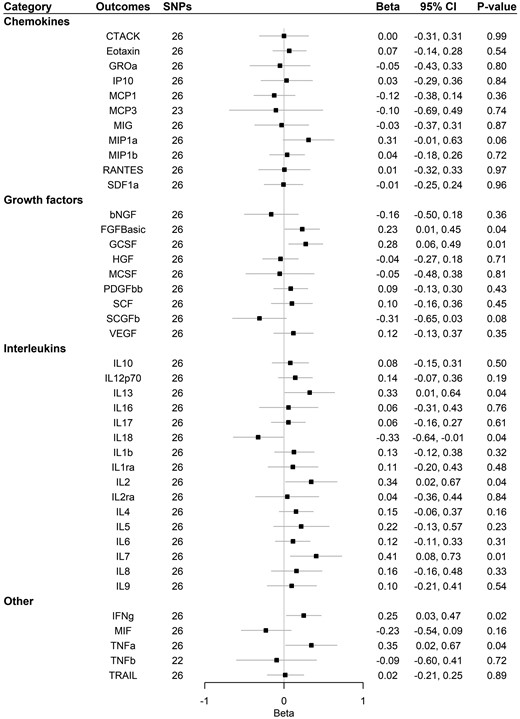
Association of Alzheimer’s disease with systemic inflammatory regulators using Mendelian randomization after removing SNPs associated with age at recruitment. Beta and 95% confidence interval (CI) represent the change in the SD of inflammatory regulators per log odds increase in Alzheimer’s disease. After correcting for multiple comparison, p-value < 0.05/41 = 0.0012 was considered as significant. bNGF, beta nerve growth factor; CTACK, cutaneous T-cell attracting chemokine; FGFBasic, basic fibroblast growth factor; GCSF, granulocyte colony-stimulating factor; GROa, growth-regulated oncogene-α; HGF, hepatocyte growth factor; IFNg, interferon gamma; IL, interleukin; IP, interferon-gamma-induced protein 10; MCP1, monocyte chemotactic protein-1; MCP3, monocyte-specific chemokine 3; MCSF, macrophage colony-stimulating factor; MIF, macrophage-migration inhibitory factor; MIG, monokine induced by interferon-gamma; MIP1a, macrophage inflammatory protein-1α; MIP1b, macrophage inflammatory protein-1β; PDGFbb, platelet-derived growth factor BB; RANTES, regulated on Activation, Normal T Cell Expressed and Secreted; SCF, stem cell factor; SCGFb, stem cell growth factor beta; SDF1a, stromal cell-derived factor-1 alpha; SNPs, single-nucleotide polymorphisms; TNFa, tumour necrosis factor alpha; TNFb, tumour necrosis factor beta; TRAIL, TNF-related apoptosis-inducing ligand; VEGF, vascular endothelial growth factor.
Discussion
In this bidirectional MR, we did not find evidence that systemic inflammatory regulators were causally associated with risk of Alzheimer’s disease. Conversely, our results suggested Alzheimer’s disease may be causally associated with some systemic inflammatory regulators, suggesting causes of Alzheimer’s disease may lie upstream.
Previous systematic reviews and meta-analysis of observational studies have shown Alzheimer’s disease to be associated with some inflammatory regulators, such as IL-6, TNFa, IL-1beta, transforming growth factor beta, IL-12 and IL-18.8,26,27 However, observational studies may be subject to confounding and cannot always distinguish symptoms from causes. Using genetically predicted systemic inflammatory regulators on Alzheimer’s disease, our results are in line with previous randomized–controlled trials showing null effects of non-steroidal anti-inflammatory drugs on cognitive decline in older adults with a family history of Alzheimer’s disease or on progression of pre-symptomatic Alzheimer’s disease,28,29 although short treatment time and treatment aimed at preventing progression rather than onset may explain the null results. Our results were also consistent with previous MR studies showing no association of C-reactive protein (a marker of systemic inflammation) and certain cytokines (IL-18, IL-1ra, IL-6) with risk of Alzheimer’s disease.30,31 Systemic inflammatory regulators could be produced by many cell types but are largely produced by helper T cells and macrophages.32 Previous studies also suggested helper T cells are highly activated in Alzheimer’s-disease patients compared with those in healthy controls.33 However, whether the activation of T cells causes or lies downstream of Alzheimer’s disease is still unclear. For the association of Alzheimer’s disease with inflammatory regulators, our positive associations with IL-2, INFg and TNFa are consistent with previous meta-analysis of observational studies whereas the inverse association with IL-18 is new.7 Our results suggest that systemic inflammation may be a downstream consequence of Alzheimer’s disease.
Previous studies in humans and mice have suggested that FGF-Basic, GCSF, IFNg, IL-7 and IL-13 may have favourable effects on reducing amyloid beta, such as increasing microglia activation for amyloid-beta uptake and decreasing amyloid-beta deposition.34–39 The increase in these inflammatory regulators found here may be a natural defence mechanism to counteract increasing amyloid beta and damaged neurons in the brain in the progression of Alzheimer’s disease. Alternatively, they could be consequences of common factors causing both inflammation and Alzheimer’s disease.
Studies investigating the correlation of inflammatory regulators in blood and cerebrospinal fluid (CSF) are limited. One study in patients with probable Alzheimer’s disease found a positive association of inflammatory regulators in blood and CSF.40 Conversely, mixed results were found in asymptomatic middle-aged to old adults and in patients with other diseases.41–43 As such a change in the systemic inflammatory regulator levels may not necessarily reflect changes in CSF, more evidence is needed about the correlation between compartments in healthy and diseased conditions. Although our study did not support the hypothesis that systemic inflammatory regulators may be a causal factor of Alzheimer’s disease among the regulators we assessed, the role of other systemic inflammatory regulators and neuroinflammation may be different. In addition, we only tested the effect of systemic inflammatory regulators on the risk of Alzheimer’s disease; we did not test their effects on disease progression because methods to do so are not currently available. Further investigation of the association between other systemic inflammatory regulators and neuroinflammation with Alzheimer’s-disease onset and all systemic inflammatory regulators with disease progression would be valuable in understanding Alzheimer’s disease.
Despite the MR design being less susceptible to confounding than other observational studies, limitations exist. First, we assumed that the genetic instruments strongly predicted the exposures, which was supported by high F-statistics and by using genome-wide significance SNPs, where possible. Second, the genetic instruments were assumed to be independent from the confounders of the exposure–outcome association, which was shown in a previous study.44 Third, the ‘exclusion–restriction’ assumption assumes that the genetic instruments are only linked with the outcome through affecting the exposure of interest. This was assessed in two ways. Pleiotropic genetic instruments, which are SNPs associated with other phenotypes directly or indirectly associated with the outcome, were detected by using the MR-Egger intercept and MR-PRESSO, which showed that the presence of pleiotropic SNPs was minimal. Selective survival, which may create a spurious link between SNPs and outcome, was assessed from the association of the SNPs with survival, proxied by age at recruitment. Results were consistence before and after removing these SNPs, which may provide more confidence in the results. Fourth, the genetic associations with cytokines were adjusted for body mass index (BMI), which may be susceptible to collider bias. However, these genetic associations were obtained from people aged 37.5 years, for whom the BMI is unlikely to have been affected by Alzheimer’s disease, making any collider bias from this adjustment unlikely. Fifth, genetic associations may differ between the populations in the exposure and outcome GWASs, and may be affected by population stratification. These were minimized by utilizing genetic associations from studies mainly of people of European ancestry with genomic control. Sixth, our results were obtained from European populations, which may be different from other populations, although the underlying biological associations between systemic inflammatory regulators and Alzheimer’s disease should be similar across populations. Seventh, compared with observational studies, MR requires much larger sample sizes considering that the variance in phenotypes explained by SNPs could be small. Low power may be the reason for null results rather than a true null. However, our study had 80% power to detect Alzheimer’s disease risk with an odds ratio of 1.02–1.06 per SD increase in the systemic inflammatory regulator levels and a change of 0.116–0.363 standard deviation in systemic the inflammatory regulator levels for analyses using genome-wide significant SNPs. Eighth, measurement error of systemic inflammatory regulators may have limited the power and the identification of exposures predicting SNPs. We also used a higher cut-off (5 × 10–6) to obtain more SNPs for analysis in addition to the use of SNPs reaching genome-wide significance. Ninth, we only investigated the association of systemic inflammation, whereas the effect of neuroinflammation on risk of Alzheimer’s disease may be different. However, there is currently no large GWAS available for neuroinflammation. Genetic predictors for relevant gene expression in brain were also limited probably due to a small sample size. The availability of high-quality GWASs on neuroinflammation in the future would be valuable. Tenth, genetic association may be different due to differences in age between the populations in the GWASs of cytokines and the GWASs of Alzheimer’s disease. However, whether such a canalization effect exists is unknown and genetic association with cytokines is unlikely to differ by age. Finally, since the population in the GWAS for cytokines was relatively young and most participants had not reached the usual age of diagnosing Alzheimer’s disease, the effect of Alzheimer’s disease on cytokines found in this study may only be representing the effect of preclinical Alzheimer’s disease on the level of cytokines, but the effect of later-life Alzheimer’s disease on these cytokines may not be reflected. This may be supported by the long preclinical phase of Alzheimer’s disease45 and any changes due to Alzheimer’s disease may have occurred before patients are diagnosed with Alzheimer’s disease.
Our MR study did not find evidence for the hypothesis that systemic inflammatory regulators may be causally associated with the risk of Alzheimer’s disease. Conversely, Alzheimer’s disease may be causally associated with certain systemic inflammatory regulators. These results suggest that systemic inflammatory regulators may be downstream effects of Alzheimer’s disease or consequences of common factors causing both inflammation and Alzheimer’s disease. Further analysis of the association of upstream factors and other systemic inflammatory regulators and neuroinflammation on Alzheimer’s disease would provide more evidence concerning the aetiology of Alzheimer’s disease.
Supplementary Data
Supplementary data are available at IJE online.
Author Contributions
Both authors designed the study and interpreted the results. C.H.C.Y. conducted the analyses and wrote the first draft of the article with critical feedback and revision from C.M.S. Both authors gave approval for the final version of the article for publication.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial or not-for-profit sectors.
Acknowledgements
Data for systemic inflammatory regulators and Alzheimer’s disease are publicly available from published studies.10,11 Data on systemic inflammatory regulators have been contributed by the Computational Medicine Research Team and have been downloaded from http://computationalmedicine.fi/data#Cytokine_GWAS. Data on Alzheimer's disease summary statistics have been contributed by Danielle Posthuma's group and have been downloaded from https://ctg.cncr.nl/software/summary_statistics. Data on age at recruitment have been contributed by UK Biobank Ben Neale files and have been downloaded from http://www.nealelab.is/uk-biobank. We used publicly available summary data where no ethical approval was required. We would also like to thank Miss Ting Zhang for checking our analyses.
Conflict of interest
None declared.
References
Alzheimer's Association.
THE NEALE LAB. GWAS RESULTS.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}