





Abstract
The xylose-fermenting yeasts (CTG clade yeasts, e.g. Scheffersomyces stipitis, Spathaspora passalidarum, Candida amazonensis and Candida jeffriesii) have the potential to be superior platforms for the conversion of lignocellulosic hydrolysate into fuel-grade ethanol and other chemical products. Here, a genetic expression system compatible with the genetic coding characteristics of CTG clade yeasts was constructed for use in xylose-fermenting yeasts. The pRACTH–gfpm plasmid based on an 18S rDNA shuttle vector was capable of stable integration into the genomes of a wide range of heterologous hosts. Green fluorescent protein was transformed and functionally expressed in S. stipitis, S. passalidarum, C. jeffriesii, C. amazonensis and Saccharomyces cerevisiae under control of the SpADH1 promoter and SpCYC1 terminator. Finally, the expression system was useful in multiple yeast hosts for construction of the plasmid pRACTH–ldh. Scheffersomyces stipitis, S. passalidarum, C. jeffriesii, C. amazonensis and S. cerevisiae were enabled to produce lactate from glucose or xylose by pRACTH-based expression of a heterologous lactate dehydrogenase. Among them, C. amazonensis (pRACTH–ldh) exhibited the highest lactate fermentation capacity, which reached a maximum of 44 g L−1 of lactate with a yield of 0.85 g lactate/g xylose.
INTRODUCTION
Lignocellulose is the largest source of renewable carbohydrate on earth. However, technically viable and commercial conversion of lignocellulosic biomass into fuels, lactate and other chemical products remains challenging due to the lack of robust microorganisms to efficiently ferment sugar mixtures via lignocellulosic hydrolysate (Xu, Singh and Himmel 2009; Abdel-Rahman, Tashiro and Sonomoto 2011; Zhong et al.2015). Xylose is the second most abundant sugar beside glucose in lignocellulosic hydrolysate, but native Saccharomyces cerevisiae, which can efficiently ferment glucose, fails to transform xylose. The well-defined life cycle, developed transformation protocols and available vector systems render S. cerevisiae a preferred organism for genetic research (Da Silva and Srikrishnan 2012), and therefore many recent improvements have focused on the genetic engineering of S. cerevisiae for xylose fermentation (Zhang et al.2015; de las Heras et al.2016). Engineering of the xylose metabolic pathway in S. cerevisiae enables xylose utilization. The engineering of S. cerevisiae harboring the xylose isomerase-based pathway significantly improved the xylose fermentation performance without the need for intensive downstream pathway engineering, with efficient conversion of xylose to ethanol with a yield of 0.45 g ethanol/g xylose (Lee, Jellison and Alper 2014). A recent report indicated that although S. cerevisiae expressed xylose isomerase, xylose cannot fully activate the fermentation process as well as glucose, which might lead to a glycolytic bottleneck and lower ethanol yield from xylose (Wasylenko and Stephanopoulos 2015). More efforts are needed to deal with these fundamental problems.
Development of an efficient xylose-fermenting strain is desirable to make full use of renewable biomass. Scheffersomyces stipitis (formerly Pichia stipitis) has a high native capacity for xylose fermentation, which is shared by most known native xylose-fermenting yeasts (e.g. Scheffersomyces sp., Spathaspora sp., Candida spp.) (Van Dijken et al.1986; Jeffries et al.2007; Cadete et al.2012). It shows very good growth characteristics with high tolerance of xylose (Wei et al.2015). The yeast Spathaspora passalidarum occurs in the gut of the wood-boring beetle Odontotaenius disjunctus (Nguyen et al.2006) and can simultaneously assimilate xylose, cellobiose and glucose aerobically. Moreover, it consumes xylose faster than glucose when both sugars are present. The xylose assimilation capacity of S. passalidarum is superior to S. stipitis and even recombinant S. cerevisiae (Long et al.2012). Candida amazonensis exhibited an excellent assimilation capacity of D-xylose to produce xylitol, with high xylitol yields on xylose (0.55–0.59 g/g) (Cadete et al.2012), and may be a promising industrial xylitol producer. Candida jeffriesii is a sister taxon of S. passalidarum (Nguyen et al.2006) that showed as fast a xylose consumption rate as S. passalidarum and faster assimilation capacity of inorganic nitrogen (i.e. urea and KNO3) than S. passalidarum (Fan et al.2015) in our previous experiments. These advantages of the native xylose-fermenting yeasts make them promising candidate strains for industrial bioethanol, lactate, xylitol and other chemical production from lignocellulosic biomass.
Few researchers have attempted to modify xylose-fermenting yeasts rationally, because the molecular manipulation of native xylose-fermenting yeasts is difficult, as with other members of the CTG clade of commensal fungi (Wohlbach et al.2011; Shi et al.2014). Even though S. stipitis was the most widely investigated host over past decades, few genetic tools have been developed. Most of the early S. stipitis genetic systems were based on auxotrophic strains (Jeffries 2008). Native gene expression elements (Laplaza et al.2006) rendered S. stipitis a relatively suitable host system for use of diversiform genetic tools. Recently, a genetic transformation protocol for S. passalidarum was established (Chen et al.2015). The CTG clade is a species group that translates CTG as serine instead of leucine (Santos et al.2011), and includes hundreds of known Candida species (including C. tropicalis, C. shehate and C. rugosa), Spathaspora species, and related species (S. stipitis and Debaryomyces hansenii) (Papon et al.2012). For quite some time, this particular codon usage has hampered the development of genetic tools to facilitate further metabolic engineering of the CTG clade.
The genomes of S. stipitis and S. passalidarum have had been sequenced because of the unusual xylose conversion enzymes of these species (Jeffries et al.2007; Wohlbach et al.2011). In addition, the genomes of several other valuable xylose-fermenting yeasts (i.e. Candida amaltose, Candida tenuis, Candida tropicalis and Debaryomyces hansenii) have also been completely deciphered over the past decade for the study of genetic and metabolic mechanisms underlying xylose fermentation. As a result, numerous genes as potential targets for metabolic engineering have been identified and warrant further investigation. However, full-scale exploitation of the information contained in xylose-fermenting yeast genome sequences requires sufficient tools for rapid genetic manipulation, and current genetic tools based on a particular host are hardly compatible with those available for other xylose-fermenting yeasts.
Therefore, to potentially overcome these limitations, we assessed the effectiveness of a multi-host vector system for xylose-fermenting yeast to not only give further insight into the basic biochemistry and genetics of xylose-fermenting yeast, but also provide a platform to evaluate optimal gene expression in the host, at least to some extent, especially for CTG clade source genes (Fusetti et al.1996; Mileto et al.1998). Here, we report the construction and evaluation of a multi-host vector system based on a single donor vector containing an S. passalidarum 18S rDNA fragment for direct integration into the genomic target, S. passalidarum-derived promoters and terminators for control of heterologous gene expression, and a modified hygromycin B resistance gene (hph) for mutant selection. This system was successfully applied in the xylose-fermenting yeasts S. passalidarum, S. stipitis, C. amazonensis and C. jeffriesii.
MATERIALS AND METHODS
Bacterial strains, medium and culture conditions
The strains used in this study are listed in Supplementary Table S1. Escherichia coli JM109 cells (Invitrogen Corp., Carlsbad, CA, USA) were used for the construction and propagation of all plasmids used in this study. Spathaspora passalidarum NRRL Y-27907 cells were kindly provided by Dr Meredith Blackwell (Louisiana State University, Baton Rouge, LA, USA) (Nguyen et al.2006). The wild-type strains of S. stipitis NRRL Y-7124 and C. jeffriesii NRRL Y-27738 were obtained from the Agricultural Research Service Culture Collection (NRRL, Peoria, IL, USA). Candida amazonensis CBS 12363 was obtained from the Centraalbureau voor Schimmel cultures (Delft, The Netherlands). The S. cerevisiae haploid strain ANGA1 (MAT a) was derived from S. cerevisiae CICIMY0086 (http://cicim-cu.sytu.edu.cn/) (Guo et al.2009). Lactobacillus plantarum CICIMB1444 was obtained from the China University Industrial Microbial Resources and Information Center (CICIM, WuXi, Jiangsu, China; http://cicim-cu.sytu.edu.cn/).
Escherichia coli was grown in lysogeny broth medium containing yeast extract (5 g L−1), peptone (10 g L−1), and NaCl (10 g L−1). Then agar (15 g L−1) was added to solidify the media and ampicillin (100 μg mL−1) was added for selection of transformants. Lactobacillus plantarum CICIMB1444 was grown in MRS broth composed of peptone (10 g L−1), beef extract (10 g L−1), yeast extract (5 g L−1), glucose (20 g L−1), Tween 80 (1 g L−1), dipotassium phosphate (2 g L−1), sodium acetate (5 g L−1), ammonium citrate (2 g L−1), magnesium sulfate (0.2 g L−1) and manganese sulfate (0.05 g L−1). Cultivation of E. coli and L. plantarum was performed at 37°C in a rotary shaker at 200 rpm for 12–14 h. Yeast strains were grown at 30°C in yeast extract peptone dextrose (YPD) media containing yeast extract (10 g L−1), peptone (20 g L−1), glucose (20 g L−1) and solid media agar (15 g L−1), while being shaken at 200 rpm. Hygromycin B was added to a final concentration of 100–900 μg L−1 for selection of recombinant strains. YPD-KMnO4-KBr plates were supplemented with KMnO4 (2 g L−1) and KBr (2.5 g L−1) as indicators of lactate-producing strains, since lactate is oxidized by KMnO4 and KBr promotes the reaction, which results in the formation of a clear transparent circle. YPD-50 and YPX-50 media supplemented with CaCO3 (30 g L−1) were used to promote lactate production, as described earlier (Ilmén et al.2007). Precultures were prepared by inoculating recombination strains into 30 mL of YPD-50 or YPX-50 medium in 100-mL flasks. The flasks were incubated at 30°C in a rotary shaker at 200 rpm for 18–24 h. Fermentation was performed with 50 mL of lactate production medium in 100-mL flasks, inoculated with an initial cell OD600 of 0.4∼0.5. After inoculation, the flasks were incubated in a rotary shaker with a rotation speed of 100 rpm, except for S. cerevisiae, which was fermented in a static culture.
Preparation of nucleic acids
DNA manipulation was carried out using standard techniques (Green and Sambrook 2012). Briefly, plasmid DNA was prepared from E. coli cultures using the AxyPrep Plasmid Miniprep Kit (Axygen Scientific, Inc., Union City, CA, USA) and polymerase chain reaction (PCR) products and restriction endonuclease-digested DNA fragments were eluted from agarose gels using the AxyPrep DNA Gel Extraction Kit. Genomic DNA of yeast was extracted using a procedure described elsewhere (Hoffman and Winston 1987). The Fast Digest restriction enzymes (Fermentas, Burlington, ON, Canada) and a commercial ligase kit (Takara Bio, Inc., Kusatsu, Japan) were used for vector construction. Digestion or ligation of the fragment was carried out using the manufacturer's instructions.
Mutation (CTG→TTG) of hph and egfp
The hph gene (GenBank accession no. V01499.1) was PCR-amplified using plasmid pRS303H (Taxis and Knop 2006) as a template with the primer pair HPH-F/HPH-R. The PCR program was as follows: initial denaturation, 95°C for 5 min; denaturation, 95°C for 45 s; annealing, 58°C for 30 s; extension, 72°C for 1 min and 16 s; amplification 30 cycles; elongation at 72°C for 5 min. The PCR products were purified and recovered before further use. A 1351-bp fragment, which included the hph open reading frame was obtained and cloned into a simple pMD19-T vector (Takara Bio, Inc.), resulting in plasmid pMD–HPH. With the same method, a 717-bp DNA fragment of egfp was also cloned from the template pQE60–EGFP (Qiagen, Inc., Valencia, CA, USA) to yield the plasmid pMD–EGFP. Then, the nine CTG codons present in the hph open reading frame and one codon in egfp were mutated to TTG codons using the QuikChangeLightning Multi Site-Directed Mutagenesis Kit (Agilent Technologies Corp., Santa Clara, CA, USA). According to the manufacturer's instructions, amplification and mutation of hph and egfp were performed using the primers listed in Supplementary Table S3, which resulted in the mutated genes hphm and gfpm, respectively.
Construction of the series of PR vectors
The basic vectors used in this study were created by cloning genetic elements from the genomes of S. passalidarum and S. stipitis. The 18S rDNA (GenBank accession no. DQ232894.1) cloned from S. passalidarum was used for genomic integration of the vector in yeast. The genetic elements encoding the promoter of the S. passalidarum genes TEF1 (SpTEF1P), XYL1.1 (SpXYL1.1P) and ADH1 (SpADH1p), as well as the promoter of the S. stipitis XYL1.1 (SsXYL1.1P), the terminator of the S. passalidarum XYL1.1 (SpXYL1.1T) and ADH1 (SpADH1T), the terminator of S. cerevisiae CYC1 (ScCYC1T) and the terminator of the S. passalidarum CYC1 (SpCYC1T) were amplified to control expression of the target genes. All of the functional fragment sequences used in this study are available from the National Center for Biotechnology Information (NCBI) database (http://www.ncbi.nlm.nih.gov/S. passalidarum GenBank accession no. AEIK00000000.1, S. stipitis GenBank accession no. NC_009068.1) or the European Molecular Biology Laboratory European Bioinformatics Institute database (http://www.ebi.ac.uk/). The primers used for PCR amplification of these fragment sequences are listed in Supplementary Table S3 with the restriction enzyme sites underlined. After PCR amplification and restriction enzyme digestion, DNA fragments were sequentially cloned into pMD19 T-simple vectors and the resulting plasmids are summarized in Supplementary Table S2. To construct the shuttle plasmid pAACTH–gfpm, 18S rDNA fragment of pRACTH–gfpm was replaced by the S. stipitis-derived autonomous replication sequence, ARS2.
The ldhL gene (GenBank accession no. AB177763.1) was amplified from L. plantarum genomic DNA by PCR with the primer pair ldh-F/ldh-R. The PCR products were digested with the restriction enzymes SalI and NotI before cloning into the respective sites of plasmid pRACTH–gfpm to generate the recombinant plasmid pRACTH–ldhL.
Transformation and stability assays
Saccharomyces cerevisiae and xylose-fermenting yeasts S. passalidarum, S. stipitis, C. jeffriesii and C. amazonensis were transformed by the lithium acetate method, as described previously (Gietz et al.1995). One microgram of linearized plasmid DNA was used for transformation. Cells were recovered by incubated at 30°C for 3 h in YPD medium to allow for the expression of the hphm gene and then spread onto YPD plates supplemented with 100–900 μg mL−1 of hygromycin B for selection of positive clones (100 μg mL−1 for C. jeffriesii, 900 μg mL−1 for C. amazonensis and 250 μg mL−1 for S. passalidarum, S. stipitis and S. cerevisiae). Successful integration of plasmids with the antibiotic marker was confirmed by direct colony PCR. To confirm the positive colony, 24 transformants were determined. Colony cells were pretreated by high power microwave for 3 min, and then used as a template. PCR was performed with the primer pair HPH-F/HPH-R to verify the presence of the HPHm fragment in genomic DNA. A positive colony was streaked on selective YPD plates to obtain a stable integration transformant. Plasmid stability was confirmed according to a method described earlier (Morlino et al.1999). Transformant colonies were grown in non-selective liquid YPD medium for 39–80 generations by re-inoculation of the cultures into fresh medium with successive rounds.
Analytical methods
Yeast strains were cultured in YPD medium until the exponential growth phase, harvested and suspended in normal saline to an appropriate cell density. The expression of enhanced green fluorescent protein (EGFP) in live cells was observed by fluorescence microscopy with a Nikon DXM1200C camera (Nikon Corp., Tokyo, Japan) and digital images were processed with Nikon ACT-1C acquisition software (Nikon Corp.). The absorbance of harvested cells at 600 nm (OD600) was measured with a spectrophotometer (UV-2100 UV/Vis spectrophotometer, UNICO Instruments Co., Ltd, Shanghai, China) to monitor yeast growth. The culture broths were centrifuged at 1 6500 g for 20 min, then filtrated with an aqueous membrane with a pore size of 0.22 μm. After appropriately diluting the supernatant (5- to 50-fold dilution), the glucose, xylose, lactate, ethanol and xylitol concentrations were analyzed by high-performance liquid chromatography with a Shodex SC1011 column (Showa Denko Co., Ltd, Tokyo, Japan), according to conditions described earlier (Guo et al.2011).
RESULTS
Determination of antibiotic minimal inhibitory concentration for yeasts
In order to develop an efficient marker of antibiotic resistance, a pilot experiment of spot assays was conducted using three antibiotics to preliminarily assess the inhibitory efficiency of the markers on the growth of the tested xylose-fermenting yeast. Then, liquid tests of different gradients of hygromycin B supplementation were further carried out to assess reliability for post-transformation screening. The sensitivity and resistance of the yeast to hygromycin B, zeocin and G418 on YPD plates are shown in Table 1. At a concentration of 100 μg mL−1 on YPD plates, the three tested antibiotics inhibited the growth of S. passalidarum and C. jeffriesii, suggesting that the tested antibiotics were good candidates to select for transformants. Scheffersomyces stipitis was more sensitive to hygromycin B and zeocin relative to G418, and showed growth inhibition at a concentration of 2000 μg mL−1. Candida amazonensis was sensitive to zeocin, hygromycin B and G418 at concentrations of 1200, 900 and 400 μg mL−1, respectively. A previous study reported that zeocin induced an adaption response in some low eukaryotes and frequent use increased the rate of mutagenesis for Chlamydomonas reinhardtii (Chankova et al.2007). These results made zeocin not the most suitable antibiotic resistance marker. Besides, analysis of the sequences of hph and neo revealed that the open reading frames contained nine and 11 CTG codons, respectively. To reduce the extent of manipulation by site-directed mutagenesis, hygromycin B was chosen as a marker of antibiotic resistance.
Sensitivity and resistance analysis of tested yeast cells to zeocin, hygromycin B and G418. Growth of the yeast strain was inhibited at the concentration shown.
| Zeocin | Hygromycin | G418 | |
|---|---|---|---|
| Strain | (μg mL−1) | B (μg mL−1) | (μg mL−1) |
| S. stipitis | 100 | 200 | 2000 |
| S. passalidarum | 100 | 100 | 100 |
| C. amazonensis | 1200 | 900 | 400 |
| C. jeffriesii | 100 | 100 | 100 |
| Zeocin | Hygromycin | G418 | |
|---|---|---|---|
| Strain | (μg mL−1) | B (μg mL−1) | (μg mL−1) |
| S. stipitis | 100 | 200 | 2000 |
| S. passalidarum | 100 | 100 | 100 |
| C. amazonensis | 1200 | 900 | 400 |
| C. jeffriesii | 100 | 100 | 100 |
Sensitivity and resistance analysis of tested yeast cells to zeocin, hygromycin B and G418. Growth of the yeast strain was inhibited at the concentration shown.
| Zeocin | Hygromycin | G418 | |
|---|---|---|---|
| Strain | (μg mL−1) | B (μg mL−1) | (μg mL−1) |
| S. stipitis | 100 | 200 | 2000 |
| S. passalidarum | 100 | 100 | 100 |
| C. amazonensis | 1200 | 900 | 400 |
| C. jeffriesii | 100 | 100 | 100 |
| Zeocin | Hygromycin | G418 | |
|---|---|---|---|
| Strain | (μg mL−1) | B (μg mL−1) | (μg mL−1) |
| S. stipitis | 100 | 200 | 2000 |
| S. passalidarum | 100 | 100 | 100 |
| C. amazonensis | 1200 | 900 | 400 |
| C. jeffriesii | 100 | 100 | 100 |
The minimum concentration of hygromycin B required for selection of transformants was further determined. Briefly, yeast cells were cultured in liquid YPD with the concentration of hygromycin B adjusted to 100–900 μg mL−1 and the growth were measured after 48 h. As shown in Fig. 1, the most suitable screening concentrations of hygromycin B for S. stipitis, S. passalidarum, C. jeffriesii, S. cerevisiae and C. amazonensis were 200, 200, 100, 200 and 1200 μg mL−1, respectively. The growth of S. stipitis, S. passalidarum, C. jeffriesii and S. cerevisiae was completely inhibited (OD600 < 1.0) at these concentrations.
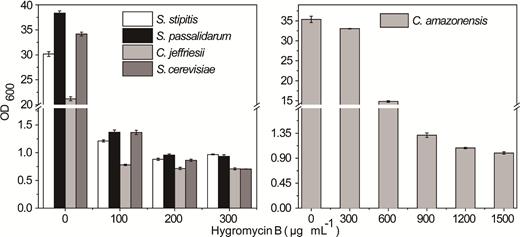
The growth condition of tested yeasts shown in media with different concentrations of hygromycin B. Histograms represent the effect of different concentrations of hygromycin B on the tested yeasts.
Design and construction of the pRTH integrated vector
Although S. stipitis vector systems have been established in several labs, most are unsuitable for use in other xylose-fermenting yeast (Laplaza et al.2006; Jeffries 2008). To construct a multi-host vector system, a series of pR-integrated vector systems based on a simple pMD19-T vector were established containing 18S rDNA, a selection marker for hygromycin B, and an expression cassette. The general design of the pR integration plasmid is depicted in Fig. 2.
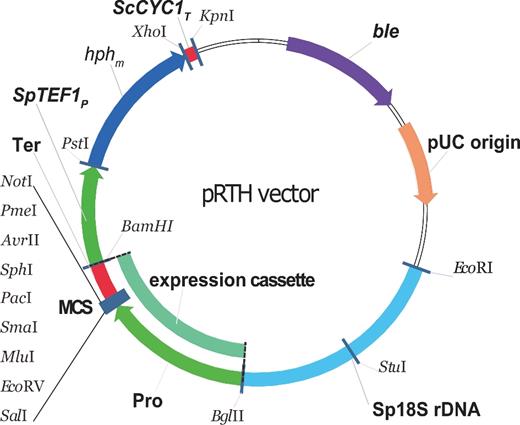
Design of pRTH vector system. The pRTH vector were established containing pMD19-T vector skeleton, 18S rDNA, a selection marker for hygromycin B and an expression cassette. The expression cassette module constituted a synthetic multiple cloning site and different origin promoter and terminator to control the expression of target genes. The pUC origin enables it to proliferate in an E. coli system. Abbreviations: ble, β-lactamase gene region; hphm, the nine CTG codons of hph that were changed to TTG; MCS, multiple cloning site; Pro and Ter represent different origin promoter and terminator for control expression of the target genes; ScCYC1T, the terminator of the S. cerevisiae CYC1; Sp18S rDNA, S. passalidarum 18S rDNA fragment for direct integration into the genomic target; SpTEF1P, the promoter of the S. passalidarum TEF1.
A 1550-bp sequence of 18S rDNA derived from S. passalidarum was cloned to a simple pMD19-T vector to enable integration of foreign DNA into the genomic sequences of selected hosts. The E. coli derived hph gene was chosen to confer resistance to hygromycin B in the transformants. Nine CTG codons were identified in the open reading frame of the hph gene, for use for an alternative yeast genetic code. They may result in the expression of a non-active resistance protein in CTG clade species (Hara et al.2000), for which the CTG codon was found to give a serine rather than a leucine residue. To favor expression in CTG clade xylose-fermenting yeasts, the nine CTG codons of the hph gene were replaced with TTG codons and adapted to hphm for selection of transformants. The functional elements SpTEF1P and ScCYC1T were introduced to express the hphm gene. The SpTEF1P, SpADH1P and SpXYL1.1P promoters, and the ScCYC1T and SpCYC1T terminators were cloned and inserted to control transcription of the expression cassette. The sole CTG codon present in the EGFP gene was changed to TTG. To verify the effectiveness of the genetic elements, the gfpm gene was inserted between the promoter and terminator sequences. pRA–gfpm was used as the control plasmid without the 18S rDNA fragment. The resulting plasmids are summarized in Supplementary Table S2 and were linearized by the StuI restriction enzyme, then transformed into S. stipitis and S. passalidarum via the lithium acetate method to confirm integration of heterogeneous genes in the genome. PCR analysis was performed using the primer pair HPH-F1 and HPH-R for amplification of 1.1-kb fragments of HPH-ScTEF1P to confirm incorporation of the plasmid into the genomes of all yeast hosts. Transformants harboring the hphm cassette were able to grow on a hygromycin B plate, which indicated the effectiveness of site-directed mutagenesis of the nine CTG codons of the hph gene and the hphm for use as a resistance marker for construction of integration vectors in CTG clade yeast. Independent of the yeast species, transformation frequencies between 15 and 50 transformants per microgram linearised plasmid DNA were achieved (Table 2). In contrast, no positive transformants were detected when control plasmid pRA–gfpm was employed (Table 2). More than 85% of the transformants were hph gene positive.
The effect of pRTH series plasmids on S. stipitis and S. passalidarum. Symbols: green fluorescence was not observed (–), green fluorescence was observed (+). Abbreviation: S. pas, S. passalidarum; S. sti, S. stipitis.
| Transformants/μg (DNA) | EGFP fluorescence | |||
|---|---|---|---|---|
| plasmids | S. pas | S. sti | S. pas | S. sti |
| pRA–gfpm | 0 | 0 | – | – |
| pRSXXTH–gfpm | 48 | 34 | – | + |
| pRACTH–gfpm | 50 | 16 | + | + |
| pRAXTH–gfpm | 31 | 46 | + | – |
| pRXCTH–gfpm | 44 | 18 | + | + |
| pRXXTH–gfpm | 46 | 30 | + | – |
| pAACTH–gfpm | 0 | 62 | – | + |
| Transformants/μg (DNA) | EGFP fluorescence | |||
|---|---|---|---|---|
| plasmids | S. pas | S. sti | S. pas | S. sti |
| pRA–gfpm | 0 | 0 | – | – |
| pRSXXTH–gfpm | 48 | 34 | – | + |
| pRACTH–gfpm | 50 | 16 | + | + |
| pRAXTH–gfpm | 31 | 46 | + | – |
| pRXCTH–gfpm | 44 | 18 | + | + |
| pRXXTH–gfpm | 46 | 30 | + | – |
| pAACTH–gfpm | 0 | 62 | – | + |
The effect of pRTH series plasmids on S. stipitis and S. passalidarum. Symbols: green fluorescence was not observed (–), green fluorescence was observed (+). Abbreviation: S. pas, S. passalidarum; S. sti, S. stipitis.
| Transformants/μg (DNA) | EGFP fluorescence | |||
|---|---|---|---|---|
| plasmids | S. pas | S. sti | S. pas | S. sti |
| pRA–gfpm | 0 | 0 | – | – |
| pRSXXTH–gfpm | 48 | 34 | – | + |
| pRACTH–gfpm | 50 | 16 | + | + |
| pRAXTH–gfpm | 31 | 46 | + | – |
| pRXCTH–gfpm | 44 | 18 | + | + |
| pRXXTH–gfpm | 46 | 30 | + | – |
| pAACTH–gfpm | 0 | 62 | – | + |
| Transformants/μg (DNA) | EGFP fluorescence | |||
|---|---|---|---|---|
| plasmids | S. pas | S. sti | S. pas | S. sti |
| pRA–gfpm | 0 | 0 | – | – |
| pRSXXTH–gfpm | 48 | 34 | – | + |
| pRACTH–gfpm | 50 | 16 | + | + |
| pRAXTH–gfpm | 31 | 46 | + | – |
| pRXCTH–gfpm | 44 | 18 | + | + |
| pRXXTH–gfpm | 46 | 30 | + | – |
| pAACTH–gfpm | 0 | 62 | – | + |
Apart from S. passalidarum (pRSXXTH–gfpm), all of the S. passalidarum transformants expressed the hphm and gfpm genes (Table 2), which demonstrated the functionality of the predicted promoters and terminators. Scheffersomyces stipitis and S. passalidarum, which harbored the pRACTH–gfpm and pRXCTH–gfpm cassette, both showed green fluorescence. Thereby we confirmed that the transcription elements SpADH1P, SpXYL1.1P and SpCYC1T were effectively expressed in S. stipitis and S. passalidarum. We speculate that these elements will prove useful to scientific community.
Wide-range application of the pRACTH–gfpm vector system
To further demonstrate the functionality of the multifunctional vector system, the plasmid pRACTH–gfpm, in which the EGFP gene was under the control of the SpADH1 promoter and the SpCYC1 terminator, was linearized by StuI restriction and used to transform S. cerevisiae and the xylose-fermenting yeasts S. stipitis, S. passalidarum, C. amazonensis and C. jeffriesii. Clones of all of the tested yeasts grew on plates containing hygromycin B, and colony PCR showed that transformation frequencies of between 10 and 50 transformants per microgram linearized plasmid DNA and more than 85% positive transformants were achieved, which confirmed that the hphm cassette was functional in all of the tested yeasts. The integrated pRACTH–gfpm copy number of S. stipitis, S. passalidarum, C. jeffriesii, C. amazonensis and S. cerevisiae recombinants ranged from 1 to 12, 5 to 40, 3 to 27, 2 to 8, and 5 to 15, respectively (details are shown in Supplementary Material S1).
The potential presence of free plasmids in the transformants was tested by re-transformation of E. coli with the total DNA of the tested yeast, but no E. coli transformants were obtained. In contrast, the transformation frequencies of control plasmid pAACTH–gfpm reached more than 50 transformants per microgram total DNA of the S. stipitis (pAACTH–gfpm). These results demonstrated that no free plasmids with the pUC ori sequence were present in the selected yeast transformants and further confirmed that the plasmid pRACTH–gfpm was indeed integrated into the genomes of the yeast hosts. Integration of the pRACTH–gfpm cassette into the 18S rDNA region of the chromosomal sequences was certified by colony PCR, and details are shown in Supplementary Material S2.
For determination of heterologous gene expression in the respective hosts, we selected the intracellular EGFP as a reporter protein to confirm promoter activity of the transformants. In all of the transformants, intracellular accumulation of EGFP was visualized by fluorescence microscopy, which showed that all of the transformants efficiently produced the EGFP and the protein was distributed homogeneously within the cytoplasm (Fig. 3), but completely excluded from vacuoles, as is expected from EGFP translation products without a secretion leader sequence. These results demonstrated that the expression elements of the SpADH1 promoter and SpCYC1 terminator were functional in all of the tested yeast, indicating a wide range of applications in xylose-fermenting yeast and S. cerevisiae. Next, the stability of the pRACTH–gfpm cassette was investigated in corresponding hosts under non-selection conditions. The results are shown in Table 3. After proliferation of about 80 generations, the integrated pRACTH–gfpm cassette was found to still be stable in the genomes of S. passalidarum, S. stipitis and C. amazonensis. Candida jeffriesii has a longer growth cycle and reproduced for about 39 generations during 120 h of cultivation and the stability of the cassette reached 94.05 ± 0.16%. The pRACTH–gfpm cassette was not as stable in S. cerevisiae as in xylose-fermenting yeasts. After about 64 generations, only 35.21 ± 1.43% of the S. cerevisiae clones were found to harbor the integrated cassette in the genome. Based on these results, we concluded that the pRACTH–gfpm cassette was mitotically stable and was integrated into the genome of xylose-fermenting yeast hosts.
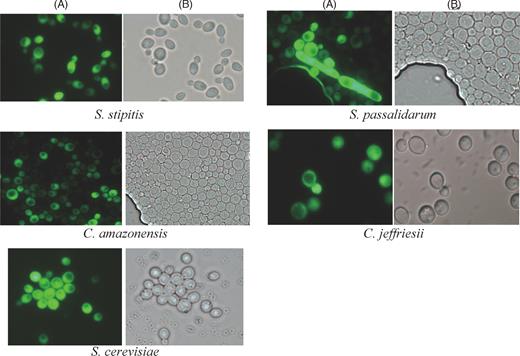
Detection of recombinant GFP-producing yeast cells by fluorescence microscopy. Transformants were cultured for 18 h in YEPD medium at 30°C. The cells were washed twice with 0.9% NaCl and subsequently used for fluorescence analysis. A, GFP fluorescence; B, transmission.
The stability of plasmid pRACTH–gfpm in tested yeast cells. Values are the average of three independent experiments ± standard deviation.
| Yeast | Generation (n) | Plasmid stability (%) |
|---|---|---|
| S. passalidarum | 80 | 98.16 ± 0.27 |
| S. stipitis | 77 | 99.48 ± 0.52 |
| C. amazonensis | 75 | 96.09 ± 0.24 |
| C. jeffriesii | 39 | 94.05 ± 0.16 |
| S. cerevisiae | 64 | 35.21 ± 1.43 |
| Yeast | Generation (n) | Plasmid stability (%) |
|---|---|---|
| S. passalidarum | 80 | 98.16 ± 0.27 |
| S. stipitis | 77 | 99.48 ± 0.52 |
| C. amazonensis | 75 | 96.09 ± 0.24 |
| C. jeffriesii | 39 | 94.05 ± 0.16 |
| S. cerevisiae | 64 | 35.21 ± 1.43 |
The stability of plasmid pRACTH–gfpm in tested yeast cells. Values are the average of three independent experiments ± standard deviation.
| Yeast | Generation (n) | Plasmid stability (%) |
|---|---|---|
| S. passalidarum | 80 | 98.16 ± 0.27 |
| S. stipitis | 77 | 99.48 ± 0.52 |
| C. amazonensis | 75 | 96.09 ± 0.24 |
| C. jeffriesii | 39 | 94.05 ± 0.16 |
| S. cerevisiae | 64 | 35.21 ± 1.43 |
| Yeast | Generation (n) | Plasmid stability (%) |
|---|---|---|
| S. passalidarum | 80 | 98.16 ± 0.27 |
| S. stipitis | 77 | 99.48 ± 0.52 |
| C. amazonensis | 75 | 96.09 ± 0.24 |
| C. jeffriesii | 39 | 94.05 ± 0.16 |
| S. cerevisiae | 64 | 35.21 ± 1.43 |
Engineering yeast for lactate production
The vector system was further found to be useful in multiple yeast hosts by construction of the plasmid pRACTH–ldhL, which contains the L-lactate dehydrogenase gene (ldhL) derived from L. plantarum. The plasmid pRACTH–ldhL was transformed into S. stipitis, S. passalidarum, C. jeffriesii, C. amazonensis and S. cerevisiae to form a lactate metabolism pathway. Dozens of positive clones were initially tested for lactate production on YPD-KMnO4-KBr plates (Fig. 4). Several colonies with larger transparent zones were picked out for further fermentation testing. All of the recombination xylose-fermenting yeasts were cultivated in YPX-50 medium to determine lactate and by-product production in a 100-mL flask by batch fermentation. The fermentation results are shown in Fig. 5. Among them, C. amazonensis (pRACTH–ldhL) exhibited the highest production of lactate, which reached a maximum of 44 g L−1 of lactate with 51.54 g L−1 of xylose with a yield of lactate on xylose up to 0.85 g g−1 (Fig. 5a). In addition to lactate, C. amazonensis (pRACTH–ldhL) accumulated 4.64 g L−1 of xylitol with no accumulation of ethanol (Fig. 5a). Candida jeffriesii (pRACTH-ldhL) produced 21.60 g L−1 of lactate and 12.01 g L−1 of ethanol, and 48.62 g L−1 of xylose with yields of lactate and ethanol of 0.44 g g−1 and 0.25 g g−1 on xylose, respectively (Fig. 5b). Spathaspora passalidarum (pRACTH–ldhL) (Fig. 5c) and S. stipitis (pRACTH-ldhL) (Fig. 5d) exhibited similar fermentation properties: 12.39 and 14.67 g L−1 of lactate, 18.27 and 13.08 g L−1 of ethanol produced by about 50 g L−1 of xylose, respectively. Because there is no xylose metabolic pathway in S. cerevisiae, we choose glucose to verify the function of the PRACTH–ldhL cassette. Saccharomyces cerevisiae (pRACTH–ldhL) showed poor production of lactate, with a maximum concentration of only 8.81 g L−1 after 36 h of cultivation, with accumulation of 19.49 g L−1 of ethanol (Fig. 6). Scheffersomyces stipitis, S. passalidarum, C. jeffriesii, C. amazonensis and S. cerevisiae were used as control strains, and no lactate was detected in the medium (Fan et al.2015). These results demonstrated that lactate production was successfully induced by transformation of engineered cells with the recombinant plasmid pRACTH–ldhL.
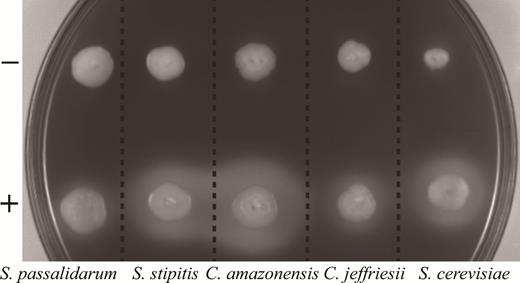
Identification of L-lactate-producing transformants by transparent circle zone. +, transformants; –, wild type. Transparent circles represent the capacity of recombinant yeast for lactate production.
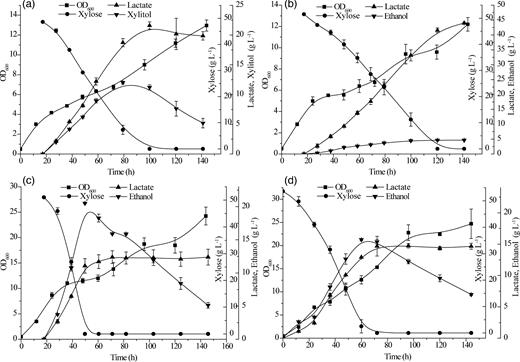
The xylose fermentation performance of four recombinant yeasts for L-lactate production. C. amazonensis (a), C. jeffriesii (b) S. passalidarum (c), S. stipitis (d). All error bars indicate ±SD, n = 3.
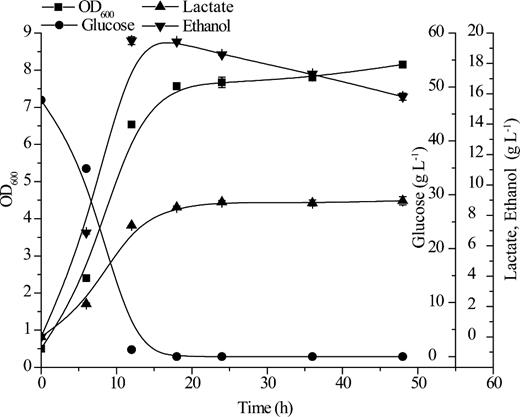
The glucose fermentation performance of S. cerevisiae (pRACTH–ldhL) for L-lactate production. OD600 (square), glucose (circle), L-lactate (upward triangle), ethanol (downward triangle). All error bars indicate ±SD, n = 3.
DISCUSSION
The xylose-fermenting yeasts (S. passalidarum, S. stipitis, C. amazonensis, C. jeffriesii, etc.) have potential as superior platforms for conversion of lignocellulosic hydrolysate into fuel-grade ethanol and other useful chemical products. However, they performed poorly in the presence of hydrolysate inhibitors, exhibiting low ethanol tolerance. In addition, with S. passalidarum, another problem is the presence of hexoses (mainly glucose and mannose) that compete with or inhibit xylose utilization (Harner et al.2015). These limitations have restricted their application in biorefining. Genetic improvement strategies (adaptation, random mutagenesis, protoplast fusion, genome shuffling) were used to improve the performance of native pentose fermenting yeasts in lignocellulosic hydrolysates (Harner et al.2015). It was reported that ethanol tolerance of S. stipitis could be improved by adaptation (Watanabe et al.2011). The classical improvement strategies may result in undesirable mutation. Therefore, precise modifications of specific pathways are required before industrial production with xylose-fermenting yeast can be realized.
However, the lack of relevant multi-host expression systems of these yeast species retards further development. To address this issue, we designed and constructed a stable multi-host integrative vector system for use in S. passalidarum, S. stipitis, C. amazonensis and C. jeffriesii. As a prerequisite, there must be an available vector that contains the targeted elements and suitable functional fragments for each of the tested candidates. rDNA targeting has been described in some yeast species, including S. cerevisiae (Rossolini et al.1992) and Yarrowia lipolytica (Le Dall, Nicaud and Gaillardin 1994), which are able to co-integrate multiple copies of plasmids carrying heterologous genes into the ribosomal DNA in a single transformation step (Klabunde et al.2002). Homogeneity among the 18S rDNA sequences identified for S. passalidarum, C. jeffriesii, S. stipitis and S. cerevisiae (there was no sequence for 18S rDNA of C. amazonensis in the NCBI database) was compared using the Basic Local Alignment Search Tool available from the NCBI website (http://blast.ncbi.nlm.nih.gov/Blast.cgi), which revealed identities among these species of 100.00%, 99.17%, 97.70% and 94.98%, respectively. The tested sequences were also highly conserved among other yeasts as well. Due to the high conservation of 18S rDNA, direct integration into the genomes of a broad range of heterologous hosts is feasible, as indicated by the results of the present study.
Although the pRACTH–gfpm vector was efficiently integrated into the S. cerevisiae genome, the stability was lower than that of xylose-fermenting yeast. However, the impact of heterogeneous elements on the mitotic instability of S. cerevisiae 18S rDNA has not been sufficiently investigated, and thus further research is warranted. Even without conditions of resistance selection, the pRACTH–gfpm vector exhibited excellent stability in xylose-fermenting yeast, indicating its potential as a powerful genetic toolkit for industrial applications.
The further development of suitable selectable markers and reporter genes for the genetic manipulation of this yeast is indispensable. Elucidation of the genetic systems of xylose-fermenting yeast species, such as S. stipitis (Jeffries 2008), is based on metabolic markers. While auxotrophic markers include unwanted phenotypic effects, such as growth defects or the presence of sequence homology between the marker and the genome, such defects can be reversed by the use of antibiotic resistance markers, which are commonly used to identify transformants (Papon et al.2012). As an advantage, this system theoretically allows the transformation of all sensitive strains, especially wild-type strains. However, only recently, vector systems based on resistance selection markers have been widely applied in xylose-fermenting yeast. Kawaguchi et al. (1989) initially revealed a non-universal genetic mechanism that employed the CTG codon in the yeast Candida cylindracea to code for a serine residue, rather than leucine. Afterward, a growing number of Candida and related species were found to adopt such a particular codon usage (Ohama et al.1993; Sugita and Nakase 1999; Laplaza et al.2006). To address this problem, the selectable marker and reporter gene sequences must be modified to favor their use in CTG clade yeast. Previous studies have reported that hygromycin appears to generally inhibit metabolism of Candida species at concentrations of 400–1000 mg mL−1 in rich or minimal media. The codon-modified HPH cassette was the first to be adapted to convey drug resistance to Candida species and has since been used for efficient transformation of wild-type strains of C. tropicalis, C. oleophila, C. albicans and C. guilliermondii (Papon et al.2012). EGFP is one of the most widely used fluorescent tags and frequently used as a reporter in molecular and cellular biology (Palm and Wlodawer 1999). The substitution of the TTG codon for the CTG codon in the EGFP gene is required for functional expression in CTG clade yeast (Morschhauser, Michel and Hacker 1998). Thus, we chose modified HPH and EGFP as a resistance marker and reporter gene, respectively.
Promoters and terminators serve as key elements in construction of efficient vectors. Previous studies reported strong constitutive activity of Arxula adeninivorans-derived TEF1 promoter in all yeast species tested so far (Terentiev et al.2004). To promote expression of the hphm gene in CTG clade yeast, we used the S. passalidarum-derived TEF1 promoter, which was found to elicit hygromycin B drug resistance in each of the five yeast species tested (Table 2). Transformants of S. stipitis (pRSXXTH–gfpm), which harbors the SsXYL1.1P–gfpm–SpXYL1.1T cassette, produced green fluorescence, in contrast to S. passalidarum (pRSXXTH–gfpm), indicating that the latter failed to express EGFP. These results indicate that the SsXYL1.1 element was specific to S. stipitis, but could potentially be exploited in other native hosts. In most cases, researchers are concerned about the impact of the promoter for expression of target genes, while ignoring the role of the terminator. In the process of constructing pR series plasmids, the gfpm gene under control of the SsXYL1.1P, SpADH1P or SpXYL1.1P promoter and the SpCYC1T terminator was found to successfully express green fluorescence in S. stipitis. However, when the terminator was switched to SpXYL1.1T, S. stipitis failed to show green fluorescence (Table 2). These results suggest that the choice of a suitable terminator should also be taken into account (Papon et al.2012).
Ultimately, the pRTH series plasmids are versatile systems and the transcriptional regulatory elements of the expression cassette can be easily exchanged with one another to test or screen for the activities of promoters and terminators of xylose-fermenting yeast to gain further insight into basic genetics and reconstruct biosynthetic pathways to create an even more powerful genetic toolkit for engineering of xylose-fermenting yeast.
SUPPLEMENTARY DATA
Supplementary data are available at FEMSYR online.
FUNDING
This work was supported by the Science Found for Distinguished Young Scholars of Jiangsu province, China (BK20140002).
Conflict of interest. None declare.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}