





Abstract
The eukaryotic biodiversity in historical air-dried samples of Dutch agricultural soil has been assessed by random sequencing of an 18S rRNA gene library and by denaturing gradient gel electrophoresis. Representatives of nearly all taxa of eukaryotic soil microbes could be identified, demonstrating that it is possible to study eukaryotic microbiota in samples from soil archives that have been stored for more than 30 years at room temperature. In a pilot study, 41 sequences were retrieved that could be assigned to fungi and a variety of aerobic and anaerobic protists such as cercozoans, ciliates, xanthophytes (stramenopiles), heteroloboseans, and amoebozoans. A PCR—denaturing gradient gel electrophoresis analysis of samples collected between 1950 and 1975 revealed significant changes in the composition of the eukaryotic microbiota.
Introduction
Monitoring the impact of global change on ecosystems is one of the major challenges for contemporary biology. While the study of communities of macroscopic, multicellular organisms is progressing using well-established approaches, analyses of the microbial domain of the various ecosystems are still rather fragmentary. In particular, our knowledge about the role and the dynamics of microbial eukaryotic communities in soil is very limited (Ekelund & Rønn, 1994; Anderson & Cairney, 2004; Bonkowski, 2004), although remarkable advances have been made recently using culture-independent, molecular approaches in aquatic environments (Moon-van der Staay et al., 2000, 2001; Lopez-Garcia et al., 2001; Dawson & Pace, 2002; Edgcomb et al., 2002a, b; Amaral Zettler et al., 2002).
Not a single molecular approach addressing eukaryotic biodiversity has as yet been published for historical, archived soil samples, which potentially could reveal changes of the eukaryotic soil microbiota over time. Such historical samples are available, for example, through the TAGA archive of soil, crop and manure samples that is maintained by Alterra B.V., Wageningen, The Netherlands. It has been shown previously that these samples can be used for molecular analyses of prokaryotic diversity (Dolfing et al., 2004), in particular of the spore-forming Bacillus group (Tzeneva et al., 2004). However, because the samples had been air-dried at 42°C, homogenized and stored at room temperature for several decades, it was unclear whether any intact eukaryotic DNA could persist in these samples. Here we describe a first attempt to analyse the molecular diversity of microbial eukaryotes in air-dried soil samples that document the land reclamation by drainage of a former sea bottom (Wieringermeer polder, The Netherlands) and its subsequent agricultural use over the years 1950–1975 (van Schreven & Harmsen, 1968; Terpstra, 1979). In this study, a clone library of eukaryotic SSU (small subunit) rRNA genes from a soil sample collected in 1975 was generated and analysed phylogenetically. Nearly the whole spectrum of eukaryotic soil microorganisms was represented in this sample, suggesting that it might be feasible to monitor changes in the composition of the eukaryotic soil microbiota over extended periods of time. A preliminary denaturing gradient gel electrophoresis (DGGE) analysis of eukaryotic 18S rRNA (SSU rRNA) gene fragments amplified from DNA extracted from soil samples collected between 1950 and 1975 demonstrated that it is possible to assess changes in the composition of the eukaryotic microbiota using air-dried soil samples that were collected decades ago.
Materials and methods
Sample collection and DNA extraction
The soil samples were from the top 0–25 cm layer of non-fertilized areas of an agricultural field in the Wieringermeer polder, The Netherlands, and were collected between 1942 and 1975. The samples were taken on 15-04-1942 (no eukaryotic amplification products obtained), 28-11-1950, 12-12-1951, 19-03-1966, 05-12-1973, and 16-10-1975. They were collected from the plough layer with an auger, and 40 cores per soil sample were taken. The samples were dried at 30–40°C, crushed, and sieved over 2 mm to remove plant residues (stubbles, roots), course grit and shells. They were then stored in the dark at ambient temperatures (∼15°C).
The samples were provided from the TAGA archive. TAGA is an archive of soil, crop and manure samples that is maintained by Alterra B.V., Wageningen, The Netherlands. (Contact information: Philip A. I. Ehlert, Alterra B. V., P.O. Box 47, 6700 AA Wageningen, The Netherlands. E-mail: [email protected].) The archive contains about 250 000 soil samples from experiments performed in the period 1879–1998, plus data and information on the experiments from which the samples were taken. Land reclamation by drainage of the Wieringermeer former sea bottom was set up in 1930 and completed in 1940, followed by transformation into agricultural lands (Terpstra, 1979), interrupted by a flooding at the end of World War 2. Genomic DNA was isolated directly from 1 g of soil after bead-beating using the Fast DNA SPIN Kit (Q BIOgene, Cambridge, UK) according to the manufacturer's instructions. Usually 2–5 μg of DNA were recovered from 1 g of dried soil sample. If not otherwise mentioned, repeated PCR amplifications and DGGE runs have been performed.
Gene amplification, sequencing, and phylogenetic analysis
For the construction of clone libraries, the eukaryotic 18S (SSU) rRNA genes were amplified by PCR with the oligonucleotide primers targetting the conserved sequences close to the 5′ and 3′ termini of eukaryotic 18S rRNA genes (Moon-van der Staay et al., 2000) using DNA from the 1975 sample as template. The PCR product was inserted into pGEM-T Easy plasmid vector (Promega, Madison, WI), and Escherichia coli XL1 blue cells were transformed with the ligation mixture. 42 recombinant clones from the 18S rRNA gene library were selected randomly for sequencing. The initial comparison of the partial environmental sequences with those from Genbank using FASTA (Pearson & Lipman, 1988) and BLAST searches (Altschul et al., 1997) revealed a high phylogenetic diversity of the clones. All selected clones were sequenced completely. The CHECK_CHIMERA program of the Ribosomal Database Project (Cole et al., 2005), the BLAST searches and phylogenetic analyses of separate sequence domains identified one potential chimerical gene artefact, which was excluded from further phylogenetic analyses. Thus, only the non-chimeric 41 recombinant clones were used for the phylogenetic analyses.
Nucleotide sequences were aligned with sequences obtained from the GenBank database using the ClustalX program (Jeanmougin et al., 1998) and refined manually with the aid of the BioEdit program (Hall, 1999). The program Gblocks (Castresana, 2000) was used to identify regions of defined sequence conservation. The number of nucleotide positions used in the phylogenetic analyses is shown in the figure legends. A global eukaryotic phylogenetic tree was inferred by the neighbour-joining method with the PHYLIP package (Felsenstein, 2002). Evolutionary distances were calculated with the Kimura two-parameter model, with a transition/transversion ratio of 2.0. The monophyly of the clusters was assessed using bootstrap replicates. Bayesian analyses that evaluate posterior likelihood probabilities of clades were also performed for the detailed phylogenetic analyses of subgroups with the MRBAYES program version 3.1.1 (Huelsenbeck & Ronquist, 2001) using the GTR+I+G model (with four gamma-distributed rate categories plus invariant positions), which was selected based on the Akaike information criteria (AIC) using Modeltest 3.06 (Posada & Crandall, 1998). Markov-chain Monte Carlo from a random starting tree was initiated and run until the standard deviation of split frequencies reached below 0.01, and trees were sampled every 100 generations. The first 30% of the samples were discarded as ‘burnin’, and the rest of the samples, after the chain reached apparent stationarity, were used for inferring a Bayesian tree.
Clones with more than 99% sequence identity were considered to be identical, because the possibility that PCR errors (Kwiatowsky et al., 1991) and minor differences between the individual repeats of reiterated or amplified rRNA genes might be responsible for such variations cannot be excluded.
DGGE
18S rRNA gene-targetted PCR-DGGE fingerprinting was used to analyse the profiles of the eukaryotic community from Wieringermeer soil samples. PCR was performed with Taq polymerase (Life Technologies, Gaithersburg, MD). DNA was amplified in a Whatman Biometra Thermocycler (Göttingen, Germany) with eukaryotic primers Euk516-GC-forward and Euk 1A-reverse using the conditions for specific eukaryotic DGGE PCR as described earlier (Diez et al., 2001). The PCR products were separated by DGGE (Muyzer et al., 1993; Muyzer & Smalla, 1998). Electrophoresis was performed with 6% polyacrylamide gels (ratio of acrylamide to bisacrylamide, 37.5 : 1) at 60°C. A gradient of 15–45% of the denaturing chemicals (100% denaturing condition was defined as 7 M urea and 40% formamide) was used, gel electrophoresis was performed according to Heilig. (2002), and the gels were stained with AgNO3 according to the method of Sanguinetti. (1994). Gels were scanned at 400 d.p.i. and further analysed using the software Bionumerics 3.0 (Applied Maths BVBA, Sint-Martens-Latern, Belgium). The similarity between DGGE profiles was determined by calculating similarity indices of the densitometric curves of the profiles compared using the Pearson product-moment correlation (Häne et al., 1993; Zoetendal et al., 2001). The unweighted pair group method with arithmetic mean (UPGMA) algorithm was used as implemented in the analysis software for the construction of dendrograms.
Results and discussion
DNA from an air-dried soil sample collected in 1975 was used to generate an 18S (SSU) rRNA gene library. After DNA sequencing of 41 randomly chosen clones, the data were analysed phylogenetically as described in Materials and Methods. A total of 24 clades, which represent the majority of all microbial eukaryotic taxa known to be present in soil, could be identified. Eight of these clades matched with at least 99% sequence identity to published sequences of known eukaryotic microorganisms: two from cercozoans (WIM2, WIM57), four from fungi (WIM52, WIM38, WIM14, WIM108), and two from green algae (WIM12, WIM107).
The eukaryotic diversity in the soil sample from 1975 is displayed in a phylogenetic tree (Fig. 1). In addition to the above-mentioned clones, WIM103 showed a best (but lower than 99%) match with known sequences from xanthophytes. Only one (novel) ciliate sequence (WIM26) was found, which differed by more than 1% from the 18S rRNA sequences from known ciliates. This might correctly reflect the low abundance and diversity of ciliates in certain agricultural soils described earlier (Ekelund et al., 2002; Robinson et al., 2002). However, since a remarkable diversity of ciliates has been described for a variety of soils using a culture-dependent approach allowing the recovery of living ciliates from resting cysts (Foissner, 1999), the low number of different distinct ciliate species identified here might be a consequence of the failure to extract DNA from resting ciliate cysts. A PCR bias against the amplification of ciliate sequences is unlikely, because we know from extended studies (Moon-van der Staay et al., 2002; Foissner et al., 2003; Foissner et al., 2004) that our primers are well suited to amplifying ciliate rRNA genes. We cannot, of course, exclude odd losses of protist DNA as a result of the treatment and storage of the samples; however, the recovery of 14 fungal clones argues against such losses, since our results from the 30-year-old sample are comparable to those obtained from ‘native’ soil samples (Schadt et al., 2003; Anderson & Cairney, 2004; Lawley et al., 2004). Nine of our clones, represented by WIM14, WIM38, WIM52, WIM108, matched with known fungal sequences of Ascomycetes or Basidiomycetes. Notably, also two novel clades (WIM27, WIM48, represented by six clones) were clustering within the fungal lineage. They showed sequence identities of only 87–89% to known eukaryotic 18S rRNA gene sequences. Since many novel fungal large subunit (LSU) rRNA gene sequences have been described recently in soil samples, and, similarly, many novel fungal 18S (SSU) rRNA gene sequences from freshwater sediments (Lefranc et al., 2005; Luo et al., 2005), the characterization of the diversity of fungal lineages in both contemporary and ancient soil samples is a challenging task for future analyses.
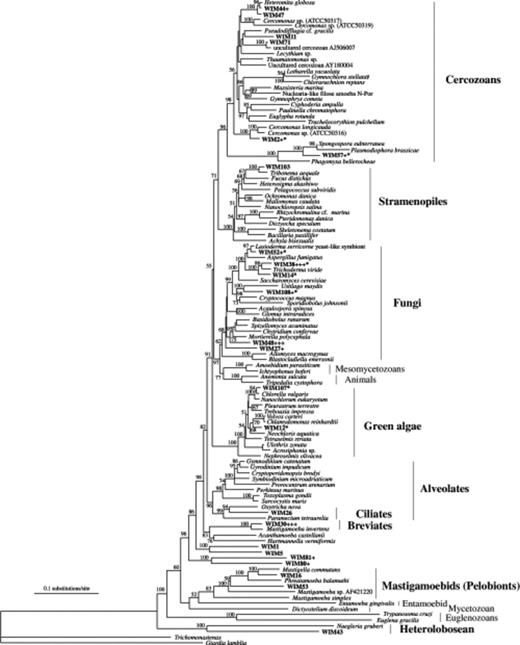
Neighbour-joining tree based on nuclear 18S (small subunit) rRNA gene sequences using 1615 positions. Sequences beginning with ‘WIM’ correspond to those retrieved from the Wieringermeer polder soil sample. The sequences matching known sequences with less than 99% sequence identity are marked with an asterisk. Closely related clones from our library with more than 99% sequence identity are indicated as a plus (+) next to the clone identifier. The number of pluses (+) indicates the number of closely related sequences. Numbers at nodes represent the bootstrap percentages from 100 replicates. Values below 50% are not shown.
Nine 18S rRNA clones, comprising six distinct clades, could be assigned to cercozoans. These flagellates are of major ecological importance in soil environments (Foissner, 1991; Ekelund & Rønn, 1994; Ekelund et al., 2001), and also in marine and freshwater habitats. They constitute a morphologically very diverse taxon, which was only recently recognized to be monophyletic (Cavalier-Smith, 1998). Recent culture-independent approaches have reported many novel cercozoan sequences (Dawson & Pace, 2002; Stoeck & Epstein, 2003; Bass & Cavalier-Smith, 2004), suggesting that the diversity of this important group of protists could be much higher than anticipated previously. A more detailed phylogenetic analysis of our clones (not shown) suggested that the clones WIM44, WIM47, and WIM71 belonged to the Heteromitidae. Clone WIM11 appeared to represent a sister clade to a cluster comprising Pseudodifflugia cf. gracilis and the environmental clone LKM48. One clone, WIM57, matched with the sequence of Polymyxa betae (Phytomyxea), a well-known plant pathogen. Therefore, the recovery of six very diverse cercozoan clades, out of the total of 41 clones analysed in this pilot study, suggests that it will be feasible to monitor complex changes within the cercozoan community over time or in response to different crop-management or fertilization regimes – even in historical samples of air-dried soil.
As many as 12 sequences belonging to 7 distinct clades were derived from unikont amoebozoans, but could not be placed unequivocally in our global 18S rRNA tree (Fig. 1). Therefore, we performed an additional phylogenetic analysis using Bayesian inference allowing among-site rate variation for these groups, which exhibit very different evolutionary rates. These phylogenetic analyses revealed the phylogenetic positions of the clades WIM30, WIM1, WIM5, WIM80, WIM81, WIM16, and WIM53 within the unikont amoebozoans (Fig. 2). Clone WIM30 clusters with Mastigamoeba invertens, but it is more closely related to M. invertens than the clade consisting of the known environmental sequences BOLA187 and BOLA366 (Berney et al., 2004; Cavalier-Smith, 2004). Clone WIM80 clusters with the environmental clones RT5iin21 and RT5iin44 (Amaral Zettler et al., 2002), and clone WIM81 clusters with the environmental clone LEMD267 (Dawson & Pace, 2002), suggesting a sister-group relationship with Filamoeba nolandi. The clones RT5iin21 and RT5iin44 were recovered from an extremely acidic river, and clone LEMD267 from anoxic freshwater sediments. The discovery of the closely related clones WIM80 and WIM81 in agricultural soil suggests that this lineage of microbial eukaryotes has a much broader ecological distribution than anticipated. Clones WIM1 and WIM5 appear as sister group to the lineages comprising the myxogastrids, the Gephyramoeba sp., and F. nolandi. Moreover, our analyses revealed two novel sequences (WIM16, WIM53) that could be assigned to the pelobiont mastigamoebids (Patterson, 1999; Edgcomb et al., 2002a, b). The sequences of these clones are very long, about 2.5 kb and 2.6 kb, as characteristic for pelobionts. Clones WIM16 and WIM53 cluster with Mastigella commutans and Mastigamoeba simplex, respectively.
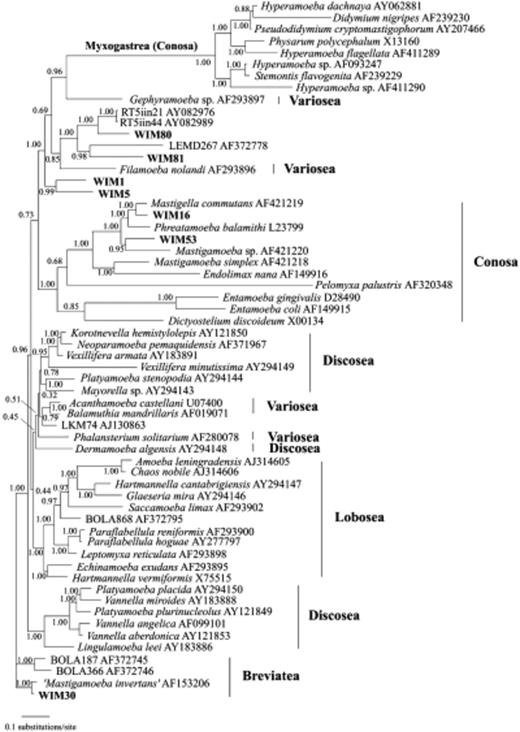
Bayesian tree of amoebozoans based on nuclear small subunit rRNA gene sequences using 1510 positions. The classification is based on the most recently revised system (Cavalier-Smith, 2003). Numbers at nodes represent the posterior probability.
Finally, we identified a clone, WIM43, that clusters with Stachyamoeba sp. (Fig. 3). Although the sequence identity between the two sequences is only about 85%, WIM43 clusters significantly with sequences from the Heterolobosea (Discicristata, Excavata), which are known to be common in soil (O'Kelly et al., 2003).
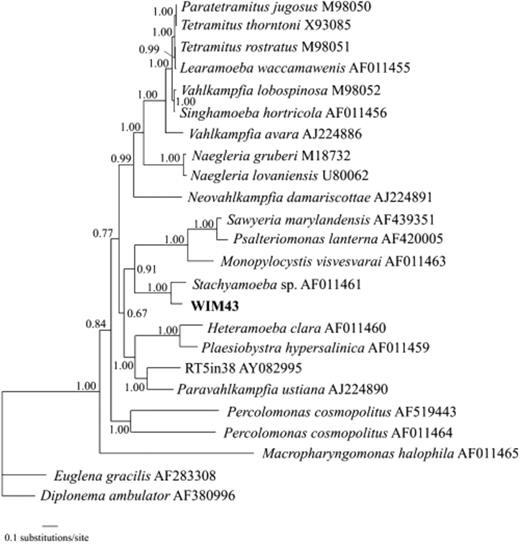
Bayesian tree of heteroloboseans based on nuclear small subunit rRNA gene sequences using 1698 positions. Numbers at nodes represent the posterior probability.
Thus, it is possible to identify a broad spectrum of representatives of all major microbial eukaryotes in samples of 30-year-old soil that has been heated to 42°C, air-dried and stored at room temperature. In a previous study, both culture-dependent and culture-independent (DGGE) studies had revealed substantial changes in the bacterial communities of the Bacillus group from 1942 to 1975 (Tzeneva et al., 2004). Using the same DNA as a template for PCR with approved primers for eukaryotic SSU rRNA genes, we obtained PCR products from all samples except the oldest one, which was collected in 1942. Separation of these DNAs with the aid of DGGE revealed considerable variations in the pattern of bands over the years (Fig. 4a). DGGE with amplicons of representative clones from the 1975 SSU rRNA gene library, which has been analysed in some detail and described above, strongly suggested that many of them match with major bands of the corresponding DGGE gel (Fig. 4a, lane 1975).
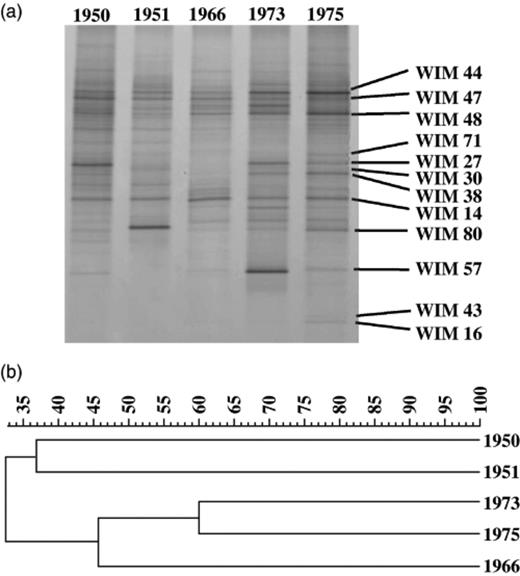
(a) Small subunit rRNA gene-targetted PCR–denaturing gradient gel electrophoresis profile of the eukaryotic community in air-dried soil samples from various years originating from Wieringermeer polder. 1950, 1951, 1966, 1973, 1975 indicate the year of sampling. The numbered bands on the 1975 profile indicate matching clones from the corresponding rRNA library. (b) UPGMA cluster analysis of the similarity indexes calculated for the various denaturing gradient gel electrophoresis profiles.
Currently, it remains unclear whether these changes in the DGGE patterns reflect actual changes in the composition of the eukaryotic microbiota or merely ‘noise’ arising from the inherent limitations in the sampling and sample conservation of the TAGA archive. The previous analysis of the DGGE profiles of the prokaryotic Bacillus group, however, argued in favour of authentic changes in time (Tzeneva et al., 2004). Calculation of the similarity indices of the densitometric curves of the profiles of the eukaryotic DGGE patterns displayed in Fig. 4(a) also supports the interpretation that the observed variations in time are not the consequence of random deviations caused by sampling problems or storage effects (Fig. 4b). Therefore, it is likely that a broad spectrum of eukaryotic rRNA gene fragments can survive in dried, archived soil samples for decades.
It has been shown that eukaryotic DNA can survive in Antarctic Holocene sediments for prolonged periods of time (Coolen et al., 2004). In addition, environmental DNA bound to marine sands allowed the construction of highly complex shotgun and PCR-based 18S rRNA gene libraries. Approximately 10% of the clones contained inserts of eukaryotic origin (Naviaux et al., 2005). In general, DNA bound to aquatic sediments is present in much higher concentrations than in the water column (Corinaldesi et al., 2005). Soil can contain as much as 1.9 microgram of free DNA per 1g of dried sample (Niemeyer & Gessler, 2002), which is in good agreement with the 2–5 μg of DNA per 1 g of dried soil sample recovered in our study. Note that our samples had been treated by bead-beating before DNA isolation, allowing also the recovery of DNA from resting cysts, which had been demonstrated in a previous study focussed on the recovery of DNA from spore-forming Bacillus species (Tzeneva et al., 2004). Therefore, we cannot discriminate between free DNA and DNA derived from cysts. Notwithstanding, there is a wealth of evidence that DNA bound to soil particles is much better protected against degradation than DNA in solution (Trevors, 1996; Coolen & Overmann, 1998; England et al., 1998; Crecchio et al., 2005). The TAGA samples of the present study had been air-dried at 30–40°C, crushed, sieved, and stored at ambient temperatures over extended periods of time. Obviously, such a treatment does allow the survival of eukaryotic DNA. Because our samples had been homogenized, sieved and thoroughly mixed, the recovered DNA is likely to be representative for the sampling site. Indeed, the spectrum of organisms identified in our study reasonably reflects the diversity of protists known from recent studies on freshwater eukaryotes (Slapeta et al., 2005).
In conclusion, we have shown that it is possible and feasible to analyse the composition of eukaryotic soil microbiota even in historical samples of air-dried and homogenized soil that were collected over extended periods of time and stored for more than 30 years at room temperature. Even the moderate number of clones analysed so far has allowed the identification of representatives of nearly all taxa of eukaryotic soil microbes, suggesting that the collection and the treatment of the soil samples did not destroy all the eukaryotic DNA in these samples. Although we cannot exclude odd losses of DNA or biases in PCR amplification of the various protists, the diversity of soil microbes clearly shows that it is possible to analyse ancient soil communities using historical samples. Even our limited, explorative approach revealed the presence of quite a number of hitherto unknown protist rRNA sequences. It seems feasible that real-time PCR using primers directed against a variety of genes will allow the quantification of particular protists in such samples. In fresh samples, fluorescence in situ hybridization using specific probes derived from the historical libraries of soil samples will facilitate the unequivocal identification of hitherto unknown protists, or, alternatively, the absence of well-known ‘indicator’ species, which might be indicative of local or global environmental changes.
Acknowledgements
This work was partly supported by a grant to the Laboratory of Microbiology, Wageningen University, from the European Union 5th framework project ‘Exploration of Genomic and Metabolite Diversity of a Novel Group of Abundant Soil Bacteria’ (BACREX-project QLK3-2000–01678). We would like to thank Phillip Ehlert, TAGA (Alterra B.V., Wageningen, The Netherlands), who provided access to the soil collection, and detailed information on the sample background that was essential for sample selection.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}