





Abstract
Microbial communities in Calyptogena sediment and microbial mats of Sagami Bay, Japan, were characterized using 16S rRNA gene sequencing and lipid biomarker analysis. Characterization of 16S rRNA gene isolated from these samples suggested a predominance of bacterial phylotypes related to Gammaproteobacteria (57–64%) and Deltaproteobacteria (27–29%). The Epsilonproteobacteria commonly found in cold seeps and hydrothermal vents were only detected in the microbial mat sample. Significantly different archaeal phylotypes were found in Calyptogena sediment and microbial mats; the former contained only Crenarchaeota clones (100% of the total archaeal clones) and the latter exclusively Euryarchaeota clones, including the anaerobic oxidation of methane archaeal groups ANME-2a and ANME-2c. Many of these lineages are as yet uncultured and undescribed groups of bacteria and archaea. Phospholipid fatty acid analysis suggested the presence of sulphate-reducing and sulphur-oxidizing bacteria. Results of intact glyceryl dialkyl glyceryl tetraether lipid analysis indicated the presence of nonthermophilic marine planktonic archaea. These results suggest that the microbial community in the Sagami Bay seep site is distinct from previously characterized cold-seep environments.
Introduction
Cold-seep biological communities have been found in the ocean, on continental shelf/slope, in deep-sea trenches, in subduction zones, in mud volcanoes and in gas hydrates associated with fluid expulsion (Paull et al., 1984; Kennicutt et al., 1985; Kulm et al., 1986; Mayer et al., 1988; Sibuet et al., 1988; Embley et al., 1990; Li et al., 1999a, b; Fujiwara et al., 2001). These communities include mussels, clams and tubeworms, and are supported by microbial chemosynthesis that is fueled by the oxidation of reduced chemical species (methane, sulphur and sulphide) seeping from the seafloor. Microbial processes are thought to play an important role in supporting the chemosynthetic communities. In particular, sulphate reduction and methanogenesis are perhaps the dominant microbial processes in marine sediments, because of the rapid depletion of other electron acceptors and the overwhelming abundance of sulphate in seawater (D'Hondt et al., 2002). Both laboratory and field experiments have demonstrated the syntrophic partnership between methanogens and sulphate-reducing bacteria in anaerobic methane oxidation or ‘reverse methanogenesis’ (Hinrichs et al., 1999; Damaste et al., 2000; Boetius et al., 2000; Pancost et al., 2000; D'Hondt et al., 2002).
Sagami Bay is located in the northern convergence front along the Sagami Trough between the Japanese Islands and the Philippine Sea Plate. Biological communities of the vesicomyid clam, Calyptogena soyoae, were discovered in the seafloor of Sagami Bay in 1984 (Ohta et al., 1987). Colonies of the giant clam extend for about 5–7 km along the 1000 m isobath in the western part of Sagami Bay (Hashimoto et al., 1995). These communities are apparently supported by methane and hydrogen sulphide seeping out from fault lines in Sagami Bay (Hashimoto et al., 1995). The Sagami Bay site is perhaps globally the most comprehensively studied seep site, including both in situ and ROV-based geological, geophysical, geochemical and biological investigations, and is a natural laboratory for studying cold-seep biogeochemistry and microbiology for several reasons. The Sagami Bay region has a relatively higher thermal gradient (1.5°C per 20 cm Momma et al., 1998) than other cold-seep sites (e.g. Gulf of Mexico). The subsea bottom temperature inside the Calyptogena communities is, on average, 0.7–1°C higher than outside the communities (Kobayashi, 2002). Active subsurface venting and mixing between seawater, porewater and groundwater bring nutrient- and methane and hydrogen sulphide-rich water to the surface and near-surface of the sea bottom (Kobayashi, 2002). Frequently occurring tectonic events affect the temperature regime as well as the distribution of biological communities in the region (Kobayashi, 2002). Finally, geochemical measurements suggest that sulphate reduction and anaerobic methane oxidation are possible microbial processes that support the dense biological communities at the site (Masuzawa et al., 1992). However, no studies have been reported on the microbial communities in sediments and microbial mats associated with colonies of the Calyptogena communities.
In this study, we collected sediment and microbial mat samples from Sagami Bay, Japan. The analysis of lipid biomarkers and the characterization of 16S rRNA gene amplified from sediments and microbial mats allowed us to determine the microbial diversity and metabolic relationship inside the Calyptogena communities.
Materials and methods
Sample collection
Sediment and microbial mat samples were collected by a submersible Shinkai 2000 operated by the Japan Agency for Marine-Earth Science and Technology Center (JAMSTEC) in December 2001 (Dive 2K-1325, Cruise NT01-11) in Sagami Bay, off Hatsushima Island. The sediment sample was taken from inside the Calyptogena community at a depth of 1168 m (sample SBC, 35°00′N 139°14′E), and the bacterial mat sample was collected at the periphery of the Calyptogena community at a depth of 1174 m (sample SBM, 35°00′N 139°13′E) using sterilized sediment samplers. The samples were kept in liquid nitrogen (−180°C) immediately after collection.
DNA extraction and purification and PCR amplification
Total DNA was extracted directly from the sediment samples using an Ultra Clean soil DNA kit (MO Bio Laboratories, Solana Beach, CA). PCR amplification was performed with a DNA Thermal Cycler model 9600 (Perkin-Elmer/Cetus Co., Foster City, CA) using a 50 μL PCR reaction mixture containing 50 mM KCl, 10 mM Tris-HCl (pH 8.3 at room temperature), 1.5 mM MgCl2, 0.2 mM of each dNTP, 0.2 mM of forward and reverse primers, 1.25 units of Taq-Ex DNA polymerase and 10–50 ng of DNA template, under the conditions recommended by the enzyme manufacturers (Takara Co., Kyoto, Japan). Thirty cycles of amplification were performed with template DNA denaturation at 95°C for 1.5 min, primer annealing at 55°C for 1.5 min and primer extension at 72°C for 1.5 min (DeLong, 1992). The oligonucleotide primers (DeLong, 1992) used to amplify 16S rRNA gene were bacterial primers Eubac27F (AGAGTTTGATCCTGGCTCAG) and Eubac1492R (GGTTACCTTGTTACGACTT), and archaeal primers Arch21F (TTCCGGTTGATCCYGCCGGA) and Arch958R (YCCGGCGTTGAMTCCAATT), where Y is C or T and M is A or C.
Cloning of PCR products
The PCR products were cloned into the TA Cloning Vector (Invitrogen, Carlsbad, CA). Escherichia coli transformants containing 16S rRNA gene inserts were identified by agarose gel electrophoresis in 1 × TAE [40 mM Tris-acetate, 1 mM EDTA (pH 8.0)].
Terminal restriction fragment length polymorphism analysis
To screen the 16S rRNA gene clones for grouping into identical clone types for DNA sequencing analysis, restriction fragment length polymorphism (RFLP) analysis, using restriction enzymes that recognized a 4 base pair (bp) restriction site, was performed, and RsaI (GT′AC) and MspI (C′CGG) were used in this experiment. To identify unique 16S rRNA gene and to select representative clones from each library, at least 50 bacterial and 50 archaeal clones from each sample were screened by RFLP. The restriction enzyme reactions were electrophoresed through a 3% agarose X gel (Nippon Gene, Toyama, Japan) in 1 × TAE. The selected cloned 16S rRNA gene fragments were then amplified and subsequently sequenced using the dideoxynucleotide chain-termination method with a model 3110 DNA Sequencer (Perkin-Elmer/Applied Biosystems Co., Foster City, CA), according to the manufacturer's instructions. The synthesized oligonucleotide primers are shown in Table 1.
Oligonucleotide primers used for 16S rRNA gene sequencing in this study
Numbers correspond to the Escherichia coli 16S rRNA gene sequence (accession number, JO1695).
Oligonucleotide primers used for 16S rRNA gene sequencing in this study
Numbers correspond to the Escherichia coli 16S rRNA gene sequence (accession number, JO1695).
The 16S rRNA genes for terminal RFLP analysis were amplified with PCR using Bac27F and 5′-Fluorescein Phosphoramidite (FAM)-labeled Bac927R for bacterial 16S rRNA genes, and Arch21F and FAM-labeled Arch958R for archaeal 16S rRNA genes, according to the procedure reported previously (Inagaki et al., 2002). Amplified rDNAs were subjected to agarose gel electrophoresis, and the labeled PCR products were purified using a Gel Spin DNA purification kit (MO Bio Laboratories). DNAs were precipitated with ethanol, centrifuged, and the pellets were resuspended in double-distilled water. The purified rDNAs were digested with HhaI at 37°C for 8 h. The terminal restriction fragments (TRFs) were analysed using a model 310 automated sequencer (Perkin-Elmer/Applied Biosystems) equipped with GENESCAN software version 3.1 (Perkin-Elmer/Applied Biosystems). The precise lengths of TRFs were determined by comparison with an internal size standard (GENESCAN-2500 ROX, Perkin-Elmer/Applied Biosystems) added to each digested sample. The electrophoresis conditions and the procedures followed were those suggested by the manufacturer.
Determining DNA sequences and phylogenetic relationships
The sequences of 16S rRNA genes were compared with DNA sequences in the DNA Data Base of Japan (DDBJ) using fasta within the genetyx-max/cd program (version 11.2.1, Software Co., Tokyo, Japan). Sequences were aligned and phylogenetic trees were constructed from a matrix of pairwise genetic distances by the maximum-parsimony algorithm and the neighbour-joining method using the clustal w program (Saitou & Nei, 1987). The amplified rDNA sequences reported in this paper have been deposited in the DDBJ, EMBL and GenBank nucleotide sequence databases (accession numbers AB188770–AB188809).
Lipid analysis
Total lipids were extracted using a modified Bligh and Dyer extraction method (Fang & Findlay, 1986). Approximately 0.1–0.7 g of freeze-dried sediment/mat sample was extracted in a test-tube with 5 mL of methanol, 2.5 mL of dichloromethane (DCM) and 2.2 mL of phosphate buffer (2 : 1 : 0.8). The extraction mixture was allowed to stand overnight in the dark at 4°C. The lipids were then partitioned by adding DCM and water, such that the final ratio of DCM to methanol to water was 1 : 1 : 0.9. The upper aqueous phase was discarded and the lower organic phase was decanted through a cellulose #4 filter into a test-tube. The solid residue retained on the filter was washed with 3 × 1 mL of DCM. The total lipid extract was dried under a gentle stream of nitrogen and was once again dissolved in hexane: OCM (70:30, v/v). Total lipids were separated into different lipid classes using miniature champagne columns (Supelco Inc., Bellefonte, PA). Neutral lipids, glycolipids and phospholipids were eluted with 4 mL of chloroform, acetone and methanol, respectively.
A portion of the phospholipid fraction was subjected to a mild alkaline trans-methylation procedure to produce fatty acid methyl ester. The fatty acid methyl esters were analysed on an Agilent 6890 gas chromatograph interfaced with an Agilent 5973N mass selective detector. Analytical separation of the compounds was accomplished using a 30 m × 0.25 mm (internal diameter, i.d.) DB-5 MS fused-silica capillary column (J&W Scientific, Folsom, CA). The column temperature was programmed from 50 to 120°C at 10°C min−1, and then to 280°C at 3°C min−1. Individual compounds were identified from their mass spectra. Response factors were obtained for each compound using duplicate injections of quantitative standards at five different concentration levels. Concentrations of individual compounds were obtained on the basis of the gas chromatography/mass spectrometry (GC/MS) response relative to that of an internal standard (C22 fatty acid ethyl ester), after correction for recovery efficiency using a C18 fatty acid ethyl ester. Method blanks were extracted with each set of samples, and were assumed to be free of contamination if chromatograms of the blanks contained no peaks. A standard containing known concentrations of 11 fatty acids was analysed daily by GC/MS to check analytical accuracy (>90% of true values). Replicate analyses (2 ×) of samples were performed to ensure reproducibility (variation of ≤10%).
The double-bond position and geometry of monounsaturated fatty acids were determined using the methods described by Dunkleblum. (1985). Fatty acids are designated by the total number of carbon atoms to the number of double bonds (i.e. a 16-carbon alkanoic acid is C16:0). The position of the double bond is indicated by a Δ number closest to the carboxyl end of the fatty acid molecule with the geometry of either cis (c) or trans (t). The cyclopropyl group is indicated by ‘cy’.
High-performance liquid chromatography-atmospheric pressure chemical ionization-mass spectrometry (HPLC-APCI-MS)
Glyceryl dialkyl glyceryl tetraether (GDGT) lipid analysis was performed on a combined phospholipid fraction using an HP1100 series liquid chromatograph-mass spectrometer (Hewlett-Packard, Palo Alto, CA) equipped with an auto-injector and Chemstation chromatography manager software. Separation was achieved on a Prevail Cyano column (2.1 × 150 mm, 3 μm; Alltech, Deerfield, IL), maintained at 30°C. Injection volumes varied from 1 to 5 μL. Tetraethers were eluted isocratically with 99% hexane and 1% propanol for 5 min, followed by a linear gradient to 1.8% propanol in 45 min. The flow rate was 0.2 mL min−1. After each analysis, the column was cleaned by back-flushing hexane – propanol [90 : 10, volume in volume (v/v)] at 0.2 mL min−1 for 10 min. Detection was achieved using APCI-MS of the eluent. Conditions for APCI-MS were as follows: nebulizer pressure, 60 psi; vaporizer temperature, 400°C; drying gas (N2) flow, 6 L min−1; drying gas temperature, 200°C; capillary voltage, −3 kV; corona, 5 μA (∼3.2 kV). As a result of the small amounts of lipids, selective ion monitoring was used to detect GDGTs, scanning the [M+H]+ ions of 1302, 1300, 1298, 1296 and 1292 with a dwell time of 237 ms.
Results
Electropherograms
Terminal RFLP analysis suggested a different microbial community structure between the Calyptogena sediment (inside the clam beds, SBC) and the microbial mat (outside the clam beds, SBM) (Fig. 1). The terminal RFLPs from the Calyptogena sediment had consistently fewer peaks than those from the bacterial mats. For example, peaks at 65, 169 and 552 bp were relatively low in the Calyptogena sediment (Fig. 1a). The 169- and 552-bp peaks, corresponding to the sulphate-reducing bacteria (Deltaproteobacteria group), were strong in the microbial sample (SBM). However, the samples shared the same peak with similar intensity at 350 bp, representing marine bacteria from the Gammaproteobacteria group. Clearly, the microbial mat supported a more diverse bacterial community, as suggested by terminal RFLP analysis.
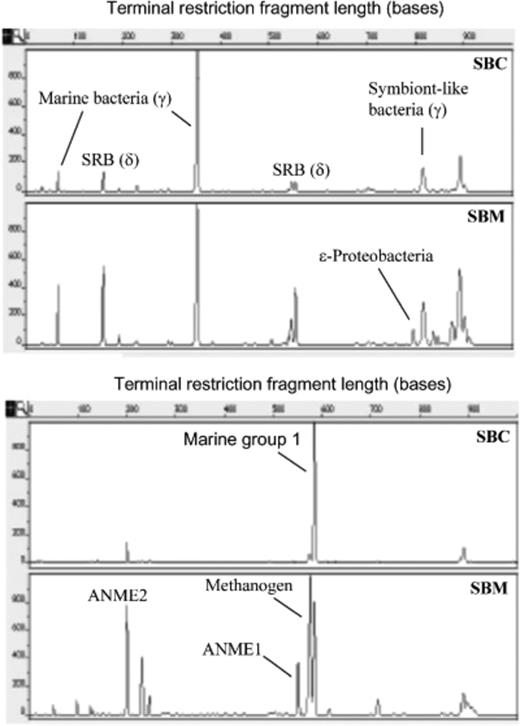
Electropherograms of bacterial (a) and archaeal (b) terminal restriction fragment length polymorphisms (RFLPs) of HhaI-digested 16S rRNA gene amplified from Sagami Bay Calyptogena sediment (SBC) and microbial mat (SBM). The lengths of the fragments (x-axis) and the relative fluorescence intensities of the peaks (y-axis) are indicated.
The electropherogram of the archaeal DNA fragments suggested different patterns. The microbial mat housed the most diverse archaeal communities, including the groups performing anaerobic oxidation of methane (ANME-1 and ANME-2), marine group 1, methanogens and an unknown group. The Calyptogena sediment (SBC) had the highest peak at 582 bp, representing marine group 1 archaea (Masuzawa et al., 1992). ANME-1 and ANME-2 were abundant in the microbial mat and were below the detection limit in Calyptogena sediment.
Bacterial 16S rRNA gene sequence profiles
The analysis of the 29 bacterial clones revealed a great bacterial diversity (Fig. 2a). These clones were classified as 24 different sequences. These sequences were clustered into six bacterial phylogenetic groups (Fig. 2a). The majority of the clones from the Sagami Bay sediment/microbial mat samples were clustered within the phylum Proteobacteria. Of the clones within Proteobacteria, 52% were related to the Gammaproteobacteria. The second most dominant phylotype (24%) was sulphate-reducing bacteria in the Deltaproteobacteria. Three clones were related to the Cytophaga–Flavobacterium–Bacteriodes (CFB) group (Fig. 2a). These taxonomic groups are described in more detail below.


Phylogenetic trees showing the relationship of 16S rRNA gene sequences amplified from Sagami Bay Calyptogena sediment and microbial mat to those of representatives of bacteria (a) (CFB, Cytophaga–Flavobacterium–Bacteriodes group; G+, Gram-positive bacterial group) and archaea (b). The phenogram was constructed from a matrix of pairwise genetic distances by the maximum-parsimony algorithm and the neighbour-joining method. Representatives of the Gram-positive bacteria and Korarchaeota were used as the outgroup in the bacterial and archaeal trees, respectively. Bootstrap values greater than 50% are shown. Scale represents a 2% difference in sequence. Numbers in parentheses show clone numbers. Sample designations are the same as shown in Fig. 1. MG-1, marine group 1. SBC and SBM are Calyptogena sediment and microbial mat samples, respectively.
Alphaproteobacteria
This subdivision of the Proteobacteria was solely represented by the clone libraries isolated from the Calyptogena sediment (SBC). The sequence of SBC-118 clustering into the Alphaproteobacteria was observed to be affiliated with the family Rhodobacteraceae, which contains purple nonsulphur bacteria. The clones were specifically associated with the 16S rRNA gene sequences of Rhodobacter sphaeroides (Dryden & Kaplan, 1990). This group of bacteria contains some of the most metabolically diverse microorganisms known, being capable of growing in a wide variety of conditions, including photosynthetic, lithotrophic, aerobic and anaerobic. Rhodobacter sphaeroides has also been shown to detoxify a number of metal oxides (Nepple et al., 2000).
Gammaproteobacteria
Most Gammaproteobacteria clones were related to sulphur-oxidizing bacterial endosymbionts of bivalves at sulphidic mud flats (Cavanaugh, 1994). The Gammaproteobacteria clones constituted the largest segment of the respective bacterial clone libraries, representing 64% (seven clones) and 57% (eight clones) of the corresponding total clone libraries of samples SBC (Calyptogena sediment) and SBM (microbial mats), respectively. Ten sequences fell into this group. All of the sequences from the Calyptogena sediment (SBC-101, SBC-107 and SBC-126) are related to the thioautotrophic symbionts I and II found in the Japan Trench Maorithyas hadalis clams (Fujiwara et al., 2001), whereas the sequences identified in the microbial mat (SBM) are related to a sequence found in sediments of a Guaymas Basin vents site with demonstrated anaerobic methanotrophic microbial communities (Teske et al., 2002). One sequence (SBM-621) of the microbial mat exhibited high similarity (95.1%) to a sequence from the Guaymas Basin, whereas another sequence (SBM-612) showed 98.1% homology to Klebsiella oxytocagene isolated from an anaerobic sludge (Sawayama et al., 2000).
Deltaproteobacteria and phylogenetic diversity of sulphate-reducing bacteria
This was the second most dominant group of phylotypes (24% of total bacterial clones). Seven sequences of the Calyptogena sediment (SBC) and the microbial mat (SBM) samples were clustered in the Deltaproteobacteria. All sequences were affiliated with dissimilatory sulphate-reducing bacteria in modern or ancient marine habitats. Sequences SBC-119, SBC-102 and SBM-324 showed 97.6%, 78.7% and 95.9% homology to environmental sequences of Sva0103, Sva130a and Sva0669, respectively, which were previously identified from Arctic permanently cold marine sediments (Ravenschla et al., 1999). These sequences were also closely related to Desulfobulbus sp. (Ravenschla et al., 1999). Sequence SBC-121 exhibited 92.6% identity to sequences found from remnants of an ancient oceanic microbial habitat (Inagaki et al., 2005), which were also similar to those found in the Eel River methane seeps (Boetius et al., 2000; Orphan et al., 2001) and in cold seeps in the Japan Trench (Li et al., 1999a). These Deltaproteobacteria clones came from samples that yielded either ANME-1 or ANME-2 archaeal clones (see below), suggesting a possible syntrophic relationship between sulphate reducers and anaerobic methane-oxidizing archaea that are involved in reverse methanogenesis.
Epsilonproteobacteria
Two sequences (SBM-524 and SBM511) were obtained in the microbial mat sample. SBM-524 was similar to Thiomicrospira denitrificans, a type of thiosulphate-oxidizing, nitrate-reducing microorganism. Thiomicrospira denitrificans was originally isolated from marine sediment of the Dutch Wadden Sea (Timmer-Ten Hoor, 1975). SBM-511 was similar to the epibiont of Alvienella pompejana, a polychaetous annelid found at hydrothermal vents in the East Pacific Rise, which is also a sulphide oxidizer (Cary et al., 1975). No sequences of the Epsilonproteobacteria were detected in the Calyptogena sediment.
The Cytophaga–Flavobacterium–Bacteriodes (CFB) phylum
The Cytophagales are mostly aerobic chemoorganoheterotrophs specialized for polymer degradation and commonly found in cold marine sediments (Ravenschla et al., 1999). Clones of the CFB phylum were obtained exclusively from the microbial mat (SBM) (Fig. 2a). Two sequences were found, one of which (SBM-613) was closely related to Cytophaga fermentans. This is a facultative anaerobic bacterium which was isolated from a marine mud sample (Bachmann, 1955) and, recently, in an intertidal meadow off Western Europe (Cifuentes et al., 2000). The other sequence (SBM-610) was closely related to that found in sediments of the Guaymas Basin vent site (Teske et al., 2002).
Archaeal 16S rRNA gene sequence profiles
A total of 34 archaeal clones was obtained from SBC and SBM samples. Seventeen sequences were selected from the clones by RFLP analysis and sequenced (Fig. 2b). Phylogenetic analysis showed that nine sequences belonged in the domain Crenarchaeota and eight sequences in Euryarchaeota. The Crenarchaeota domain was well represented by sequences from both samples (Fig. 2b). SBC-A120, SBM-A514 and SBM-A201 were closely related to the unidentified archaeon Ar12-16 (98.7% similarity), which was identified from symbiotic archaeal communities in marine sponges (Lee et al., 2003), and to marine group 1 (SBAR 5 and SBAR12), sequences of bacterioplankton obtained from coastal waters of North America (DeLong et al., 1992). Sequences SBM-A310 and SBM-A618 were similar to a sequence found in deep-sea sediments from the Japan Trench (Ryu et al., 2004). Sequence SBC-A110 from the Calyptogena sediment was almost identical (99.2% homology) to sequence HMMVPog-37 found at the Haakon Mosby mud volcano (Loesekann et al., 2003). One of the sequences in the microbial mat (SBM-A602) was phylogenetically nearly identical (99.1%) to that identified in the Nankai Trough subseafloor sediments (Loesekann et al., 2003).
The Euryarchaeota domain was dominated by sequences from the microbial mat (100% of all Euryarchaeota clones). There were eight sequences found in the Euryarchaeota domain. The microbial mat (SBM) yielded clones of uncultured, anaerobic, methane-oxidizing archaeal group ANME-2 (Orphan et al., 2000; Hinrichs et al., 1999; Lanoil et al., 2001; Teske et al., 2002; Newberry et al., 2004). SBM-A307 and SBM-A320, and SBM-A302, were assigned to the subgroups ANME-2a and ANME-2c, respectively, of Methanosarcinales, represented by the Eel River sequence clones Eel-36a2A5 and BA2H11fin (Hinrichs et al., 1999; House et al., 2001). The total number of clones of these two groups constituted 36% of all SBM archaeal clones. The other sequence (SBM-A608) was closely related to marine tropical euryarchaeote VIDL-2 (Kellogg, 2004).
Lipid profiles
The fatty acids detected included saturated, monounsaturated, polyunsaturated, cyclopropane, hydroxyl, terminal and mid-methyl-branched fatty acids (Table 2). The phospholipid fatty acid (PLFA) profiles were dominated by monounsaturated fatty acids, with concentrations ranging from 48% to 64% of the total fatty acids. The most abundant monounsaturated fatty acids were 16:1Δ9 and 18:1Δ11. These two fatty acids constituted 33–41% of the total. A number of 16:1 fatty acids were detected with double-bond locations at Δ8, Δ9 and Δ11. Terminally branched fatty acids consisted of the iso and anteiso isomers of C15 and C17 and the iso C14 and C16 fatty acids. The proportions of these fatty acids varied from 5.4% to 8.2%. The only mid-methyl-branched fatty acid detected was 10Me16:0 (0.5–3.0% of total fatty acids). Also detected was a saturated cyclopropane fatty acid cy17:0. Polyunsaturated fatty acids present in the sediments and microbial mats included 18:2Δ9,12, 18 : 3, 20 : 5 and 22 : 6. The microbial mat sample (SBC) contained a higher concentration of total fatty acids (456.6 nmol g−1) than did the Calyptogena sediment (89.9 nmol g−1) (Table 2). Assuming that 1 pmol of PLFA is equivalent to 2.5 × 104 cells (Balkwill et al., 1988), the microbial biomass was estimated to be 2 × 109 and 1 × 1010 cells g−1 dry weight in Calyptogena sediment and microbial mats, respectively.
Concentration (nmol g−1 dry weight, percentage in parentheses) of phospholipid fatty acids isolated from the Sagami Bay Calyptogena sediment (SBC) and microbial mat (SBM) samples
| Compound | SBC | SBM |
| i14:0 | 0.3 (0.4) | 2.1 (0.5) |
| 14:1 | 0.0 (0.0) | 0.8 (0.2) |
| 14:0 | 1.9 (2.4) | 21.2 (4.6) |
| i15:0 | 1.3 (1.6) | 7.8 (1.7) |
| a15:0 | 2.3 (2.9) | 12.8 (2.8) |
| 15:0 | 0.4 (0.6) | 2.6 (0.6) |
| i16:0 | 0.2 (0.2) | 1.6 (0.4) |
| 16:1Δ9c | 1.1 (1.3) | 4.6 (1.0) |
| 16:1Δ9t | 21.3 (27.0) | 111.2 (24.3) |
| 16:1Δ11c | 1.1 (1.4) | 21.6 (4.7) |
| 16:1Δ11t | 3.9 (4.9) | 88.6 (19.4) |
| 10Me16:0 | 2.4 (3.0) | 2.2 (0.5) |
| i17:0 | 0.2 (0.3) | 1.2 (0.3) |
| a17:0 | 2.2 (2.8) | 1.2 (0.3) |
| 17:1Δ9 | 0.8 (1.0) | 4.5 (1.0) |
| cy17:0 | 1.3 (1.7) | 0.6 (0.1) |
| 17:0 | 0.5 (0.6) | 2.0 (0.4) |
| 18:3 | 0.0 (0.0) | 2.9 (0.6) |
| 18:2 | 1.2 (1.5) | 10.3 (2.3) |
| 18:1Δ9 | 4.3 (5.5) | 13.6 (3.0) |
| 18:1Δ11 | 11.3 (14.3) | 39.2 (8.6) |
| 18:0 | 5.1 (6.4) | 19.6 (4.3) |
| 19:0 | 0.1 (0.1) | 0.1 (0.0) |
| 20:5 | 0.9 (1.1) | 5.5 (1.2) |
| 20:1Δ7 | 1.8 (2.3) | 5.3 (1.2) |
| 20:1Δ13 | 0.8 (1.1) | 2.9 (0.6) |
| 20:0 | 0.2 (0.3) | 2.0 (0.4) |
| 22:6 | 0.6 (0.8) | 1.5 (0.3) |
| Compound | SBC | SBM |
| i14:0 | 0.3 (0.4) | 2.1 (0.5) |
| 14:1 | 0.0 (0.0) | 0.8 (0.2) |
| 14:0 | 1.9 (2.4) | 21.2 (4.6) |
| i15:0 | 1.3 (1.6) | 7.8 (1.7) |
| a15:0 | 2.3 (2.9) | 12.8 (2.8) |
| 15:0 | 0.4 (0.6) | 2.6 (0.6) |
| i16:0 | 0.2 (0.2) | 1.6 (0.4) |
| 16:1Δ9c | 1.1 (1.3) | 4.6 (1.0) |
| 16:1Δ9t | 21.3 (27.0) | 111.2 (24.3) |
| 16:1Δ11c | 1.1 (1.4) | 21.6 (4.7) |
| 16:1Δ11t | 3.9 (4.9) | 88.6 (19.4) |
| 10Me16:0 | 2.4 (3.0) | 2.2 (0.5) |
| i17:0 | 0.2 (0.3) | 1.2 (0.3) |
| a17:0 | 2.2 (2.8) | 1.2 (0.3) |
| 17:1Δ9 | 0.8 (1.0) | 4.5 (1.0) |
| cy17:0 | 1.3 (1.7) | 0.6 (0.1) |
| 17:0 | 0.5 (0.6) | 2.0 (0.4) |
| 18:3 | 0.0 (0.0) | 2.9 (0.6) |
| 18:2 | 1.2 (1.5) | 10.3 (2.3) |
| 18:1Δ9 | 4.3 (5.5) | 13.6 (3.0) |
| 18:1Δ11 | 11.3 (14.3) | 39.2 (8.6) |
| 18:0 | 5.1 (6.4) | 19.6 (4.3) |
| 19:0 | 0.1 (0.1) | 0.1 (0.0) |
| 20:5 | 0.9 (1.1) | 5.5 (1.2) |
| 20:1Δ7 | 1.8 (2.3) | 5.3 (1.2) |
| 20:1Δ13 | 0.8 (1.1) | 2.9 (0.6) |
| 20:0 | 0.2 (0.3) | 2.0 (0.4) |
| 22:6 | 0.6 (0.8) | 1.5 (0.3) |
Concentration (nmol g−1 dry weight, percentage in parentheses) of phospholipid fatty acids isolated from the Sagami Bay Calyptogena sediment (SBC) and microbial mat (SBM) samples
| Compound | SBC | SBM |
| i14:0 | 0.3 (0.4) | 2.1 (0.5) |
| 14:1 | 0.0 (0.0) | 0.8 (0.2) |
| 14:0 | 1.9 (2.4) | 21.2 (4.6) |
| i15:0 | 1.3 (1.6) | 7.8 (1.7) |
| a15:0 | 2.3 (2.9) | 12.8 (2.8) |
| 15:0 | 0.4 (0.6) | 2.6 (0.6) |
| i16:0 | 0.2 (0.2) | 1.6 (0.4) |
| 16:1Δ9c | 1.1 (1.3) | 4.6 (1.0) |
| 16:1Δ9t | 21.3 (27.0) | 111.2 (24.3) |
| 16:1Δ11c | 1.1 (1.4) | 21.6 (4.7) |
| 16:1Δ11t | 3.9 (4.9) | 88.6 (19.4) |
| 10Me16:0 | 2.4 (3.0) | 2.2 (0.5) |
| i17:0 | 0.2 (0.3) | 1.2 (0.3) |
| a17:0 | 2.2 (2.8) | 1.2 (0.3) |
| 17:1Δ9 | 0.8 (1.0) | 4.5 (1.0) |
| cy17:0 | 1.3 (1.7) | 0.6 (0.1) |
| 17:0 | 0.5 (0.6) | 2.0 (0.4) |
| 18:3 | 0.0 (0.0) | 2.9 (0.6) |
| 18:2 | 1.2 (1.5) | 10.3 (2.3) |
| 18:1Δ9 | 4.3 (5.5) | 13.6 (3.0) |
| 18:1Δ11 | 11.3 (14.3) | 39.2 (8.6) |
| 18:0 | 5.1 (6.4) | 19.6 (4.3) |
| 19:0 | 0.1 (0.1) | 0.1 (0.0) |
| 20:5 | 0.9 (1.1) | 5.5 (1.2) |
| 20:1Δ7 | 1.8 (2.3) | 5.3 (1.2) |
| 20:1Δ13 | 0.8 (1.1) | 2.9 (0.6) |
| 20:0 | 0.2 (0.3) | 2.0 (0.4) |
| 22:6 | 0.6 (0.8) | 1.5 (0.3) |
| Compound | SBC | SBM |
| i14:0 | 0.3 (0.4) | 2.1 (0.5) |
| 14:1 | 0.0 (0.0) | 0.8 (0.2) |
| 14:0 | 1.9 (2.4) | 21.2 (4.6) |
| i15:0 | 1.3 (1.6) | 7.8 (1.7) |
| a15:0 | 2.3 (2.9) | 12.8 (2.8) |
| 15:0 | 0.4 (0.6) | 2.6 (0.6) |
| i16:0 | 0.2 (0.2) | 1.6 (0.4) |
| 16:1Δ9c | 1.1 (1.3) | 4.6 (1.0) |
| 16:1Δ9t | 21.3 (27.0) | 111.2 (24.3) |
| 16:1Δ11c | 1.1 (1.4) | 21.6 (4.7) |
| 16:1Δ11t | 3.9 (4.9) | 88.6 (19.4) |
| 10Me16:0 | 2.4 (3.0) | 2.2 (0.5) |
| i17:0 | 0.2 (0.3) | 1.2 (0.3) |
| a17:0 | 2.2 (2.8) | 1.2 (0.3) |
| 17:1Δ9 | 0.8 (1.0) | 4.5 (1.0) |
| cy17:0 | 1.3 (1.7) | 0.6 (0.1) |
| 17:0 | 0.5 (0.6) | 2.0 (0.4) |
| 18:3 | 0.0 (0.0) | 2.9 (0.6) |
| 18:2 | 1.2 (1.5) | 10.3 (2.3) |
| 18:1Δ9 | 4.3 (5.5) | 13.6 (3.0) |
| 18:1Δ11 | 11.3 (14.3) | 39.2 (8.6) |
| 18:0 | 5.1 (6.4) | 19.6 (4.3) |
| 19:0 | 0.1 (0.1) | 0.1 (0.0) |
| 20:5 | 0.9 (1.1) | 5.5 (1.2) |
| 20:1Δ7 | 1.8 (2.3) | 5.3 (1.2) |
| 20:1Δ13 | 0.8 (1.1) | 2.9 (0.6) |
| 20:0 | 0.2 (0.3) | 2.0 (0.4) |
| 22:6 | 0.6 (0.8) | 1.5 (0.3) |
Because of the small amount of samples, no GDGT lipids were initially detected in any of the samples by HPLC-APCI-MS. Thus, the polar lipid fractions of the samples were combined and four GDGT lipids were detected by HPLC-APCI-MS (Fig. 3) [caldarchaeol, crenarchaeol, GDGT-1cy and GDGT-2cy (cy=cyclopentane ring)], suggesting that both samples contained these two lipids.
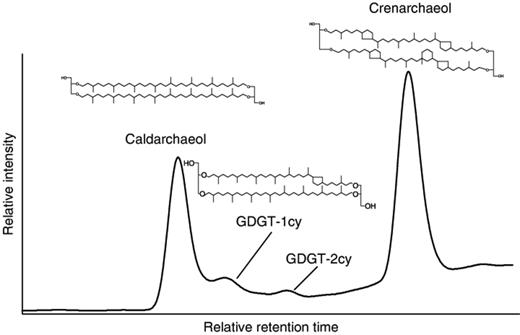
High-performance liquid base peak chromatogram of archaeal tetraether lipids detected in the Sagami Bay samples. GDGT-1cy and GDGT-2cy are glyceryl dialkyl glyceryl tetraether with one and two cyclopentane rings, respectively.
Discussion
Since the first discovery of cold-seep biological communities in Sagami Bay in 1984, various investigations have been conducted with the aim to determine the geological and geochemical environment (Kobayashi, 2002), benthic communities (Hashimoto et al., 1995) and microbial communities (Urakawa et al., 1999). Our study is the first to use lipid biomarkers and molecular 16S rRNA gene analysis to characterize microbial communities in the Sagami Bay seep site. The results of microbial diversity studies were consistent between three different approaches: terminal RFLP analysis, phylogenetic analysis and lipid biomarker analysis.
Microbial diversity identified on the basis of 16S rRNA gene
We found a diverse range of bacteria in the Sagami Bay sediment and microbial mat. Proteobacteria made up the majority (86%) of the clone libraries. The most abundant clones found in the samples were those belonging to the Gammaproteobacteria, closely related to Maorithyas hadalis symbiont II in cold seeps of the Japan Trench (Fujiwara et al., 2001), and those in the hydrothermally active sediments of the Guaymas Basin (Teske et al., 2002), suggesting that there are similarities in microbial communities between deep and shallow cold seeps (Sagami Bay vs. Japan Trench) and between cold seeps and hydrothermal vents. These clones were abundant in the sediment of the Calyptogena communities (64% of the total bacterial clones of the sample) as well as the microbial mat (39%). Clones similar to the Deltaproteobacteria (sulphate reducers) were equally abundant in the Calyptogena sediment and microbial mat. The predominance of Proteobacteria in the Sagami Bay sediment is similar to that in other deep-sea sites, e.g. Nankai Trough (Newberry et al., 2004), Japan Trench (Li et al., 1999a) and the Gulf of Mexico (Lanoil et al., 2001; Mills et al., 2003). Clones related to the Epsilonproteobacteria were detected in the microbial mat, but not in the Calyptogena sediment (Fig. 2a). Generally, rDNA from Epsilonproteobacteria is absent in normal deep-sea sediment (Urakawa et al., 2000), but is abundant in hydrothermal vents and cold-seep sites (Urakawa et al., 1999, 2000). The Epsilonproteobacteria have been found to be widespread in deep-sea hydrothermal vents associated with microbial mats (Miroshnickenko, et al., 2002), in vent waters and particles (Huber et al., 2003) and as epibionts of hydrothermal invertebrates (Cary et al., 1997). Epsilonproteobacteria isolated from hydrothermal vents can grow both autotrophically and heterotrophically. Those isolated from vent polychaetes grew lithoautotrophically using sulphur as electron acceptor and hydrogen as energy source (Miroshnickenko et al., 2002). The four Epsilonproteobacteria isolates from hydrothermal vent chimney samples were heterotrophic sulphur reducers and used formate as energy and carbon source (Campbell et al., 2003). Presumably, the Epsilonproteobacteria provide a source of autotrophic production for the vent microbial communities (Campbell et al., 2003). They were also found to be abundant in the Japan Trench and Nankai Trough cold-seep sediments (Inagaki et al., 2002; Li et al., 1999a, b), and in the Gulf of Mexico gas hydrate mounds (Mills et al., 2003). 16S rRNA gene sequences obtained from the Japan Trench and Nankai Trough sediments were similar to those of the Arcobacter-related endosymbionts or Alvinella pompejana epibionts of polychaete worms (Li et al., 1999a, b). A similar sequence (SBM511) was also detected in the microbial mat of the Sagami Bay cold-seep site, suggesting that epibiotic microflora are probably widely distributed in the ocean under hot (hydrothermal vents) and cold (seeps) conditions. The results also support the conclusion of Cary. (1997) that these bacterial symbionts do not have an obligate requirement for A. pompejana. Sequence SBM524 detected in the microbial mat of Sagami Bay is similar to Thiomicrospira denitrificans from several hydrothermal habitats (Huber et al., 2003). Furthermore, isolates closely related to T. denitrificans, that are also able to oxidize sulphide with nitrate, have been obtained from petroleum-contaminated groundwater (Watanabe et al., 2000), suggesting that these organisms possess diverse oxidizing capabilities and may play an important role in sulphur cycling in the cold-seep and hydrothermal vent environments (Distel et al., 1988).
There was limited archaeal diversity in samples from Sagami Bay. This finding was similar to that observed in other cold-seep and gas hydrate sites: Nankai Trough (Reed et al., 2002), Gulf of Mexico (Lanoil et al., 2001), the Eel River Basin (Hinrichs et al., 1999) and the Cascadia Margin (Bidle et al., 1999). The Sagami Bay site is different from other cold-seep sites in that most archaeal phylotypes are dissimilar to cultured organisms, suggesting the presence of novel archaea at the site. Significant differences were observed in the distribution of Crenarchaeota and Euryarchaeota clones between the Calyptogena sediment and the microbial mats. The Calyptogena sediment contained 40% of the Crenarchaeota clones, which were almost identical to symbiotic archaea detected in marine sponges (Lee et al., 2003). The detection of sequences similar to marine group 1 archaea found in the coastal waters of North America (DeLong et al., 1992) indicates that the planktonic archaea are widely distributed in the oceans, in shallow as well as deep waters, and in both aerobic and anaerobic conditions.
The microbial mat contained 100% of the Euryarchaeota clones, most of which were closely related to archaea performing anaerobic oxidation of methane found in various areas (e.g. Santa Barbara Basin, Guaymas Basin, the Eel River Basin, etc.). Furthermore, there were significantly more different phylotypes of archaea in microbial mats than in Calyptogena sediment (15 and two phylotypes, respectively). The different archaeal populations may reflect the different environmental conditions associated with microbial mats and Calyptogena communities. These findings corroborate those of Kobayashi (2002), who showed that microbial mats flourish in areas of active venting of anaerobic, CH4-rich fluids. PLFA analysis confirmed that the bacterial biomass was consistently higher in the mats than in the Calyptogena sediment (see below).
Microbial biomass and community structure based on lipids
The microbial biomass found in the Sagami Bay sediment and microbial mats is comparable with or slightly higher than that found in the Barbados Trench cold-seep sediment (109 cells per g dry weight Guezennec & Fiala-Medioni, 1996) and the Nankai Trough Calyptogena communities (7.9 × 109 cells per g dry weight Li et al., 1999b). The relatively high microbial biomass could be a result of the expulsion of large volumes of the nutrient-enriched (e.g. NH4+) fluids to the seafloor from the land (Ohta et al., 1987; Kobayashi, 2002). Furthermore, higher bacterial biomass was observed in microbial mats. These results corroborate geochemical studies (Masazawa et al., 1992; Olu et al., 1997), which show that microbial mats, rather than the Calyptogena communities, are more closely associated with the fluid conduits. Thus, these biological communities in Sagami Bay are sustained not only by locally produced reduced species (e.g. sulphide and methane, see below), but also by nutrients of allochthonous origin.
The bacterial community was dominated by Gram-negative bacteria, as suggested by the high concentrations of monounsaturated fatty acids in PLFA (Ratledge & Wilkinson, 1988). 16S rRNA gene analyses confirmed their presence. The PLFA profiles suggested the presence of abundant sulphate-reducing bacteria. The high levels of cis-vaccenic acid (18:1Δ11), which is formed by the anaerobic fatty acid desaturase pathway, point to the presence of anaerobic bacteria (White et al., 1996). The branched C15 and C17 fatty acids can be attributed to Desulfococcus multivorans, whereas 10Me16:0 is characteristic of Desulfobacter (Londry & Des Marais, 2004). The detection of 16:1Δ11t also suggests the presence of Desulfobacter as well as Desulfomonas acetoxidans (Londry & Des Marais, 2004). The presence of cy17:0 suggests the presence of Desulfobacter and Desulfobacterium (White et al., 1996). The C17 monounsaturated fatty acids (e.g. 17:1Δ9) are commonly associated with sulphate reducers, such as Desulfobulbus and Desulfovibrio (Londry & Des Marais, 2004). The lipid results are thus congruent with molecular analysis. The Deltaproteobacteria clones detected in the Sagami Bay samples are closely related to psychrophilic sulphate reducers in marine sediments of the Arctic Ocean: Desulfuromonas, Desulfovibrio, Desulfotalea, Desulfobulbus and Desulfobacterium sp. (Rovenschla et al., 1999). Thus, molecular and lipid evidence suggests the presence of a possible syntrophic relationship between sulphate-reducing bacteria and ANME-2, because ANME-2 lineages have been found in sites with active anaerobic oxidation of methane by sulphate-reducing bacteria and ANME-2 (Hinrichs et al., 1999; Boetius et al., 2000; Orphan et al., 2002). It is interesting that we detected two ANME-2 groups (a and c) in the same sample site, in contrast with previous studies where only one group was found in a particular habitat (Teske et al., 2003).
The PLFA profiles suggest the presence of sulphur-oxidizing bacteria, consistent with the 16S rRNA gene sequence. The exceptionally large amounts of monounsaturated fatty acids 16:1Δ9 (17–27%) and 18:1Δ11 (9–15%) suggest a possible contribution by sulphur oxidizers (Larkin, 1980; McCaffrey et al., 1989; Guezennec & Fiala-Medioni, 1996; Zhang et al., 2005). The high concentration of 18:1Δ11c is indicative of the presence of Beggiatoa and Thioploca spp., which also contain significant amounts of 16:1Δ9 (Jacq et al., 1989; McCaffrey et al., 1989; Zhang et al., 2005). This PLFA characteristic has been observed in chemoautotrophic sulphur-oxidizing bacteria in hydrothermal vents (Jannasch, 1985), in deep-sea sediments of the Barbados Trench (Bidle et al., 1999), and in a microbial mat associated with gas hydrates in the Gulf of Mexico (Zhang et al., 2005).
The detection of caldarchaeol and crenarchaeol in Sagami Bay cold-seep sediments and microbial mats suggests the presence of Crenarchaeota and, possibly, Euryarchaeota. Crenarchaeol is a specific biomarker of nonthermophilic planktonic Crenarchaeota (Schouten et al., 2000; Sinninghe Damsté, 2002), and is one of the most abundant GDGTs in the ocean (Sinninghe Damsté, 2002). This lipid is widely distributed in marine sediments, the water column and particulate matter (Schouten et al., 2000; Pancost et al., 2001; Sinninghe Damsté, 2002; Wakeham et al., 2003), and has recently also been found in some terrestrial hot springs (Pearson et al., 2004). The detection of this biomarker suggests the presence of marine planktonic group I archaea. Caldarchaeol is produced by nonthermophilic Crenarchaeota, but is also present in certain Euryarchaeota, including those in the ANME-1 group in the marine environment (Blumenberg et al., 2004). However, in the GDGT distribution, no significant enhancement of GDGTs with one or two cyclopentane rings was observed, typical of GDGTs from the ANME-1 group (Blumenberg et al., 2004), suggesting that the ANME-1 group was not present in substantial amounts. This agrees with the 16S rRNA gene data.
In conclusion, using combined 16S rRNA gene sequencing and lipid biomarker analysis, we studied the microbial diversity of the Sagami Bay cold-seep sediment and microbial mat. Both the sediment and microbial mat contained abundant proteobacterial phylotypes and archaeal phylotypes that included novel and unique lineages, indicating as yet uncultured and undescribed groups of bacteria and archaea. PLFA and GDGT analyses provided complementary insights into microbial biomass and community structure in the cold-seep site. Our results demonstrated the advantage of combining 16S rRNA gene sequencing and lipid analysis in determining and better understanding microbial communities in the marine environment.
Acknowledgements
We thank Olivia Chan (Iowa State University, Ames, IA) and Joost Brandsma (Royal Netherlands Institute for Sea Research, Texel, the Netherlands) for their skilful laboratory assistance in GC-MS and HPLC-APCI-MS analyses of lipids. We are very grateful to the science group of the cruises in NT01-11, the Shinkai 2000 operation team and the crew of the R/V Natsushima for helping us to collect the cold-seep samples. This work was supported by the National Science Foundation (to J.F.), the INOUE ENRYO Memorial Foundation for Promoting Sciences (to S.A.) and, in part, by a Grant-in-Aid for Exploratory Research (No. 15651008) provided by the Ministry of Education, Culture, Sports, Science and Technology of Japan (to C.K.). We thank Gary M. King and an anonymous reviewer for their constructive comments which have greatly assisted us in improving the manuscript.
References

{kind=link}
{kind=link}
{kind=link}