





Abstract
Skin microbes play an important role in amphibian tissue regeneration. Xenopus spp. (African clawed frogs) are well-established model organisms, and standard husbandry protocols, including use of antibiotics, may affect experimental outcomes by altering bacterial assemblages. It is therefore essential to improve knowledge of Xenopus bacterial community characteristics and inheritance. We undertook bacterial 16S rRNA gene sequencing and source tracking of a captive Xenopus laevis colony, including various life stages and environmental samples across multiple aquarium systems. Tank environments supported the most complex bacterial communities, while egg jelly bacteria were the most diverse of frog life stages; tadpole bacterial communities were relatively simple. Rhizobium (Proteobacteria) and Chryseobacterium (Bacteroidota) were dominant in tadpoles, whereas Chryseobacterium, Vogesella (Proteobacteria), and Acinetobacter (Proteobacteria) were common in females. Tadpoles received approximately two-thirds of their bacteria via vertical transmission, though 23 genera were differentially abundant between females and tadpoles. Female frog skin appears to select for specific taxa, and while tadpoles inherit a proportion of their skin bacteria from females via the egg, they support a distinct and less diverse community. The outcomes of this study suggest the impacts of breaking the bacterial transmission chain with antibiotic treatment should be considered when raising tadpoles for experimental purposes.
Introduction
Symbiotic microbial communities are important to the function and health of their host. They instigate and mediate immune, metabolic, developmental, and other physiological pathways in a broad range of host taxa, from plants (Compant et al. 2019, Trivedi et al. 2020) to invertebrates (Mandel and Dunn 2016, Pita et al. 2018) and vertebrates (McFall-Ngai et al. 2013, Ross et al. 2019b), including humans (Cénit et al. 2014, Byrd et al. 2018). In amphibians, the skin is an organ of particular importance due to its roles in gas exchange, osmoregulation and immune defence (Heatwole and Barthalmus 1994, Varga et al. 2018). Microbes inhabiting the amphibian skin might influence their cohabiting community by producing antimicrobial compounds, stimulating the secretion of antimicrobial peptides from the host’s cutaneous mucous glands, or simply by competing for resources (Mangoni et al. 2001, Kommineni et al. 2015, Loudon et al. 2016). Due to conservation problems caused by the fungal pathogen Batrachochytrium spp., many amphibian microbiome studies have focused on the role of skin-dwelling bacterial communities in disease susceptibility and prevention [see for examples Longo et al. (2015), Rebollar et al. (2019), Jani et al. (2021), and Martins et al. (2022)]. Some have attempted to determine how and from where microbes colonize the amphibian integument, however conclusions vary. Several studies found that host species was the most important predictor of skin microbial composition among aquatic species, with cohabitation having little effect (McKenzie et al. 2012, Walke et al. 2014). However, others (Costa et al. 2016, Bird et al. 2018, Ellison et al. 2019) found that the environment was a significant, if not the primary, determinant of skin microbial assemblages [reviewed by Hernández-Gómez and Hua (2023)]. Differences in study design (e.g. use of high throughput sequencing versus culture-based or other methods) and host taxa/taxonomic levels (salamanders and newts versus frogs; species versus order) may account for some of these contrasting conclusions. While the microbiome may be seeded from various sources, including the habitat and vertical or horizontal transfer from conspecifics, the amphibian skin potentially selects for and provides a competitive advantage for certain microbes that may otherwise be encountered in low numbers (Walke et al. 2014).
Clawed frogs from the Xenopus genus are proven and widely used models for laboratory developmental and regeneration studies, yet their bacterial communities have only recently begun to receive attention. While Mashoof et al. (2013) were the first to characterize the Xenopus laevis gut bacterial community, and demonstrated that thymectomy did not affect its composition, Scalvenzi et al. (2021) found that the size and composition of such communities in the Xenopus tropicalis gut varied according to developmental stage. This can be at least partially explained by changes in diet and gut structure at different stages of ontogeny, however the potential role of these bacteria in influencing the developmental process is yet to be properly explored. Li et al. (2020) demonstrated that the gut bacteria of X. tropicalis froglets were distinct from that in their food and surrounding water, suggesting that frogs at this life stage may exhibit a degree of immune control over their microbiome composition. Piccinni et al. (2021) compared bacterial communities living on ‘clean’ (thimerosal/ethanol treated) and standard feeding stage X. laevis tadpoles and adults, finding distinct differences. Tadpoles hosted a relatively diverse bacterial assemblage which correlated with their environment (tank water), while those on adult frogs were less varied and showed little similarity to the surrounding environment, supporting the theory that amphibians acquire an ability to moderate their microbiome through immune responses or other means as they develop. Bacteria associated with adult frogs were not significantly affected by housing conditions (‘clean’ versus standard), however some differences in beta diversity and community composition were observed in tadpoles.
Bacterial communities play a critical role in activating pathways involved in development, limb regeneration, and wound healing in model organisms from other taxonomic groups, including planaria (Arnold et al. 2016, Williams et al. 2020), mice (Velasco et al. 2021, Wang et al. 2021), and axolotls (Debuque et al. 2022). Recently, it has become evident that bacteria are important in Xenopus tail regeneration (Bishop and Beck 2021, Chapman et al. 2022). Intriguingly, Bishop and Beck (2021) found that exposure to various antibiotics, commonly used by Xenopus breeding facilities during the embryo rearing process, resulted in a sharp decrease in tail regeneration success in X. laevis tadpoles. Meanwhile, the addition of either heat-killed Escherichia coli or commercially supplied lipopolysaccharide (LPS) from E. coli or Pseudomonas aeruginosa improved the likelihood of tail regeneration, including following antibiotic treatment. Further to this, Chapman et al. (2022) characterized skin bacterial communities from X. laevis tadpoles and demonstrated that LPS from other Gram-negative bacteria, including commensal taxa, can promote regeneration, with some taxa over-represented in successful regenerators.
While Piccinni et al. (2021) demonstrated the characteristics of X. laevis skin bacterial communities at two key life stages, their tadpoles were not descended from the adults studied and were routinely raised in antibiotics, so inferences on maternal transfer of skin bacteria (i.e. vertical transfer) were not possible. Chapman et al. (2022) compared bacteria between treated and untreated tadpole clutches (sibships) from different females and noted considerable variation between sibships. The relative roles of vertical familial transmission and the environment on associated skin bacteria at various stages of the lifecycle remain to be explored. As Xenopus spp. are widely used as models in regeneration and developmental studies, and intervention with their bacterial communities influences regenerative outcomes (Bishop and Beck 2021, Chapman et al. 2022), it is critical to understand how bacteria vary across individuals and colonies, and where they come from. To answer these questions, we characterized and compared the bacterial communities present on adults, eggs, and tadpoles from nonantibiotic treated X. laevis using 16S ribosomal RNA gene amplicon sequencing. We also sequenced samples taken from potential environmental sources to determine their contribution at various X. laevis life stages.
Materials and methods
Xenopus husbandry
The X. laevis colony at the University of Otago, New Zealand is comprised of several recirculating aquarium systems, each comprised of a number of smaller tanks and supplied by carbon filtered town tap water. The colony is closed, with no frogs introduced from outside sources since 2004, and current adults are captive-bred F1 or F2 generations. Frogs are fed with generic salmon farming pellets twice weekly. The aquarium systems use bioballs as biological filtration, which are plastic spheres with a network of channels and ridges designed to provide maximum surface area for nitrifying bacteria.
Embryo production and sample collection
All samples were collected in 2020. Samples represented major frog life stages, as well as potential sources of bacteria in their environment (Fig. 1A and B). Eggs and embryos from 16 female frogs were included in the study, with four females randomly selected from each of four different tanks split across two recirculating aquarium systems. While frogs from a tank were not necessarily related, they were of similar age, with frogs from Tank B being considerably older (13 years) than those from Tanks C—E (2–4 years). Induction of egg laying, in vitro fertilization, and embryo husbandry were as described by Chapman et al. (2022). Each female was placed in her own 1 l container of 1x Marc’s Modified Ringer’s (MMR) solution (pH 7.4: 100 mM NaCl, 2 mM KCl, 1 mM MgSO4.7H2O, 2 mM CaCl2, 5 mM HEPES, and 0.1 mM EDTA at pH 8.0) at 22°C the morning following induction injection, allowing her eggs to be kept separate from those of others. Each tadpole sibship (family group derived from a particular female) was split into two Petri dishes containing 24 tadpoles each (total 48 tadpoles per sibship) with 30 ml 0.1x MMR. Tadpole sibships did not have contact with other sibships at any stage, nor were they fed or in contact with any antibiotic or sterilizing agent.
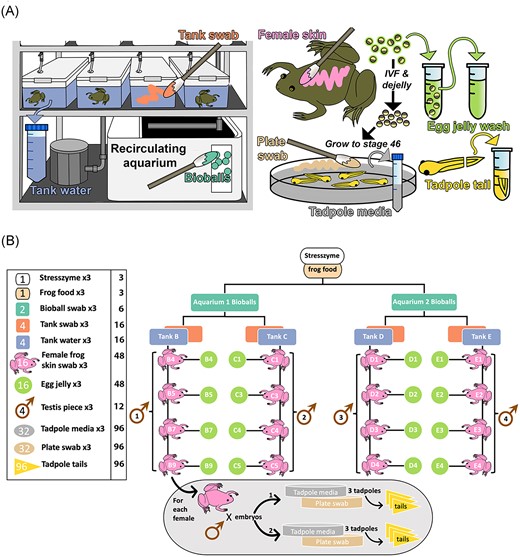
Sampling strategy for study. (A) Summary of sample types collected. Several sample types that were shown to contribute negligible microbes (STRESS ZYME/frog food/testis) were omitted, as preliminary findings suggested these do not contribute to tadpole microbial ecology. (B) Detailed sampling structure and sample numbers. Numbers within icons in the left panel denote numbers of biological replicates collected per sample type, with technical replicates indicated within accompanying text. For example, for bioballs, samples were collected from two biological replicates (i.e. two different aquarium systems), with three technical replicate swabs collected from each, giving a total of six samples. For each tadpole sibship, 48 tadpoles were split between two Petri dishes (plates), reared to stage 46 and sampled. All samples were collected for DNA extraction and amplification and sequencing of the 16S rRNA V4 hypervariable region.
During the egg production stage above, batches of 10 freshly laid, unfertilized eggs were collected from each female and placed in a 1.5-ml Eppendorf tube containing 200 µl filter-sterilized sodium chloride/Tween (0.15 M NaCl, 0.1% Tween20). Eggs were gently vortexed for 30 s to dislodge bacteria on the jelly coats, and the sample pulsed in a centrifuge to enable retrieval of the supernatant for DNA extraction. Samples from tadpoles consisted of tail tips (one-third of tail length) collected at developmental stage 46 (Nieuwkoop and Faber 1956) during a concurrent regeneration experiment. The removed tail portions were placed into 0.2 ml DNase/RNase-free Polymerase Chain Reaction strip tubes with 50 µl of filter-sterilized sodium chloride/Tween solution and vortexed for 1 min. A total of six tails were collected from each sibship. Unmoistened Puritan DNA-free cotton-tipped applicators were used to swab inner tank walls, the surfaces of bioballs, tadpole Petri dishes, and adult female frogs. For the latter, each frog was swabbed on the dorsal and ventral surface, thigh crease and foot web with one swab, then repeated twice more with new swabs; each swab covered approximately one quarter of the animal’s surface area. Male frogs were sacrificed as part of the standard fertilization protocol [refer Chapman et al. (2022)], with one testis used to fertilize eggs from a female tank group while the second was divided into three pieces for bacterial sequencing. Samples of tank water (50 ml), frog food (commercial salmon pellets), a bacterial aquarium conditioning additive (STRESS ZYME, API Fish Care) (200 µl), filtered Tween solution (200 µl), and tadpole raising media (0.1x MMR with residual 1/4000 v/v MS222 anaesthetic agent; 5 ml/sample), were also collected. Petri dish swabs and tadpole media were collected at the end of the regeneration experiment, 1 week after embryo fertilization. All tank associated samples were collected at the time of female swabbing, with swabbing/experiments being completed on a tank-by-tank basis over the course of ~2 months. In all cases except tadpole tails, three replicates were collected; final sample numbers can be found in Fig. 1(B). Samples were stored at −20°C until DNA extraction.
DNA extraction and 16S rRNA amplicon sequencing
DNA was extracted from all samples described in the previous section along with the following controls: blank Puritan swab, blank filter (Millex GV 0.22 µm PVDF membrane—used to filter Tween prior to use), Tween, mock community (ZymoBIOMICS® Microbial Community Standard), and blank (no sample) extractions.
A DNeasy PowerLyser PowerSoil DNA extraction kit (Qiagen) was used to obtain DNA from samples as per manufacturer’s instructions. DNA was eluted into a final volume of 30 µl and stored at −80°C. Amplification and sequencing of the 16S rRNA gene V4 hypervariable region was completed on the Illumina MiSeq platform by the Argonne National Laboratory (Illinois, USA). Sequencing was completed as per Caporaso et al. (2011) using primers 515F/806R (Apprill et al. 2015, Parada et al. 2016) and peptide nucleic acid PCR clamps (Lundberg et al. 2013) to inhibit amplification of host mitochondrial sequences, resulting in paired end reads of 2 × 250 bp.
Sequence processing
Sequences were demultiplexed using idemp (Wu 2014) and quality filtering and adaptor removal completed using fastp (v0.23.2) (Chen 2023). The DADA2 pipeline (v1.16) (Callahan et al. 2016) was used to further filter and trim sequence data, with paired reads merged and chimeras removed before taxonomic assignment using the naïve Bayesian classifier method with reference to the SILVA database (v138.1). Standard settings were used, with truncLen = c(175, 225) at the filterAndTrim() stage. The dataset was then passed to R (v4.2.2) (R Core Team 2022) for further processing and subsequent analysis. Data were formatted into a phyloseq (v1.42.0) (McMurdie and Holmes 2013) object, nonbacterial reads removed and decontam (v1.18.0) (Davis et al. 2018) used to identify and remove potential contaminant amplicon sequence variants (ASVs) with reference to negative controls. ASVs were then agglomerated at genus level to reduce noise and aid analysis, interpretation and comparability. A multiple sequence alignment was created using DECIPHER (v2.26.0) (Wright 2015) and a maximum-likelihood phylogenetic tree constructed with IQTree (v2.2.2.3) (Minh et al. 2020). The TVMe+R9 substitution model was selected based on a best-fit assessment using the inbuilt ModelFinder (Kalyaanamoorthy et al. 2017) utility; 1066 bootstrap replicates were required for convergence.
For downstream analysis, the three replicates from each sample (i.e. frog, tank, etc) were merged, with read counts for each bacterial genus averaged (speedyseq v0.5.3; McLaren 2023). For tadpole tails, the three individual tadpoles taken from each plate were treated as sample replicates.
After sequence processing and replicate merging, we identified 466 bacterial genera across 138 samples. Read counts in all control samples (excluding mock community samples) were negligible. Mock community profiles reflected those expected. A summary of read counts for each sample type is provided in Supplementary Table S1. Following an initial analysis of read counts and composition, samples for further analysis were narrowed down to a ‘core’ group of eight sample types comprised of bioballs, tank water, tank wall swabs, female frog skin swab, egg jelly wash, tadpole tails, tadpole media, and tadpole plate swabs. Sample numbers for each sample type are detailed in Fig. 1(B).
Data analyses
All analyses were completed using R v4.2.2, with statistics calculated using the base Rstats package unless otherwise specified. A summary decision tree for statistical analyses is provided as Supplementary Fig. S1.
Alpha diversity
Rarefaction curves were prepared prior to analysis. Samples were subsequently rarefied to 1000 reads based on examination of rarefaction curves and read count distribution histograms (Supplementary Fig. S2A and B). Observed, Shannon and Faith’s phylogenetic diversity were calculated for each sample type using phyloseq (Observed/Shannon) and picante v1.8.2 (Kembel et al. 2010) (Faith’s). Shapiro–Wilk test of normality was used to determine distribution of data; Kruskal–Wallis tests and post hoc Dunn’s test with Bonferroni multiple-comparison correction (the latter with FSA v0.9.4; Ogle et al. 2023) were used to find significant pairwise differences between sample types where Shapiro–Wilk resulted in P < .05; alternatively, ANOVA and Tukey’s post hoc test were used where P > .05. Diversity metrics were visualized as boxplots created using ggplot2 (v3.4.0) (Wickham 2016).
Beta diversity
Bray–Curtis distances were calculated between unrarefied samples using the phyloseq::distance function. Samples with less than 500 reads were excluded. Hopkins Test (clustertend v1.6) (Wright et al. 2022) and a silhouette analysis (factoextra v1.0.7) (Kassambara and Mundt 2020) were calculated for the Bray–Curtis distance dataset to assess the degree of clustering, and nonmetric multidimensional scaling (NMDS) ordinations were prepared using phyloseq and ggplot2. Permutational analysis of multivariate dispersions (PERMDISP—vegan v2.6–4 betadisper function) (Oksanen et al. 2022), Permutational multivariate analysis of variance (PERMANOVA—vegan adonis2 function), and Wilcoxon rank-sum test (rstatix v0.7.2) (Kassambara and Mundt 2023) or Kruskal–Wallis test were used to compare Bray–Curtis distances between sample types. Female ID, tank, and aquarium system were designated as fixed effects with interactions for PERMANOVA’s dependent on the comparison being made. The following analyses/comparisons were undertaken:
PERMDISP and PERMANOVA were used to assess Bray–Curtis distances between samples, with sample type, aquarium and tank included as fixed effects for PERMANOVA.
Bray–Curtis distances between samples of the same type were compared to assess relative variability of bacterial communities associated with different environments. Means were compared using Kruskal–Wallis test and Dunn’s post hoc test. Pearson’s correlation coefficients were also computed to assess correlations between samples of different types.
Bray–Curtis distances between (a) female skin swab samples and (b) tadpole tail clippings and other sample types were assessed to determine those sample types most similar or different. Median Bray–Curtis distances were compared using Kruskal–Wallis test and Dunn’s post hoc test.
To assess the effect of the female’s tank or aquarium system on her skin bacteria and that of her tadpoles, Bray–Curtis distances between female skin swab samples from the same tank or aquarium and those from different tanks/aquaria were compared. The same comparison was made for tadpoles. Median Bray–Curtis distances were compared using Wilcoxon’s rank-sum test. PERMDISP and PERMANOVAs were also performed for female and tadpole samples, with aquarium and tank as fixed effects for the latter test. For tadpoles, an additional PERMANOVA with tank and female as fixed effects was performed.
Community composition and differential abundance
Rarefied relative abundance of bacterial taxa in sample types was visualized using the microshades (v1.0) (Dahl et al. 2022) package.
The ANCOMBC2 (v2.0.1) (Lin et al. 2022) and MaAsLin2 (v1.12.0) (Mallick et al. 2021) packages were used to calculate differential abundance of genera between key sample types, using unrarefied data. Taxa not observed at least three times in 5% of samples were filtered out before analysis. Taxa were determined to be differentially abundant if their q-value (false discovery rate adjusted P-value) was below 0.25. Bacterial taxa found to be differentially abundant by both packages were extracted and mean relative abundance visualized across all sample types with ggplot2.
Source tracking of bacterial communities
The FEAST package (v0.1.0) (Shenhav et al. 2019) was used to determine the likely origin of bacterial taxa found in different sample types. Analyses were completed on rarefied data in four broad groups (Table 1) selected based on likely direct interaction between samples. Where relevant, analyses were run individually for subgroups (for e.g. Tank B females with relevant Tank B environmental samples) and averaged for visualization in a Sankey plot using networkD3 (v0.4) (Gandrud et al. 2017). After initial analysis we determined that tadpoles were most likely to be seeding their environment rather than vice versa as tadpoles shared a number of taxa with female skin and egg jelly. Additionally, the MMR media, while not sterile after opening, was autoclaved prior to use.
Groupings of sources and sinks for FEAST analyses.
| Group | Source/s | Sink |
|---|---|---|
| 1 | Tank water, tank swab | Female skin swab |
| 2 | Female skin swab | Egg jelly wash |
| 3 | Egg jelly wash | Tadpole tail |
| 4 | Tadpole tail, egg jelly wash | Tadpole media |
| Group | Source/s | Sink |
|---|---|---|
| 1 | Tank water, tank swab | Female skin swab |
| 2 | Female skin swab | Egg jelly wash |
| 3 | Egg jelly wash | Tadpole tail |
| 4 | Tadpole tail, egg jelly wash | Tadpole media |
Groupings of sources and sinks for FEAST analyses.
| Group | Source/s | Sink |
|---|---|---|
| 1 | Tank water, tank swab | Female skin swab |
| 2 | Female skin swab | Egg jelly wash |
| 3 | Egg jelly wash | Tadpole tail |
| 4 | Tadpole tail, egg jelly wash | Tadpole media |
| Group | Source/s | Sink |
|---|---|---|
| 1 | Tank water, tank swab | Female skin swab |
| 2 | Female skin swab | Egg jelly wash |
| 3 | Egg jelly wash | Tadpole tail |
| 4 | Tadpole tail, egg jelly wash | Tadpole media |
Results
Different Xenopus life stages and environments support distinct bacterial communities
Sample types, or groups of sample types, were broadly distinguishable based on their bacterial communities. Samples associated with the adult frog tank environment (i.e. tank water, tank wall swabs, and bioballs) were the most complex, with tadpole environment samples (media and plate swabs) being least complex (Fig. 2A; Supplementary Fig. S3A and B; Supplementary Table 2A–C). Frog samples (female skin swabs, egg jelly, and tadpole tail clips) fell in between the environmental samples, with tadpole samples being consistently the least complex. This pattern remained generally consistent across all alpha diversity metrics calculated. For beta diversity, PERMDISP indicated that dispersion of Bray–Curtis distances was significantly different between sample types (P = .002; Supplementary Table S3A). Sample type accounted for 30% of Bray–Curtis distance variation (PERMANOVA (vegan:: adonis2), P = .001, R2 = 0.296; Supplementary Table S3B) while the aquarium system accounted for 3.2% (P = .001, R2 = 0.032) and the interaction between system and tank 9.2% (P = .001, R2 = 0.092). Inverse Hopkins statistic calculated on Bray–Curtis distances (0.157) indicated clustering within the data, with cluster (silhouette) analysis identifying two clusters (Supplementary Fig. S4). NMDS ordination (Fig. 2B) showed that aquarium surface samples (i.e. bioballs and tank swabs) overlapped substantially, forming a cluster distinct from tank water samples and the various Xenopus samples and tadpole environmental samples. Female skin swabs and egg jelly samples clustered together, as did tadpole samples, tadpole plate swabs and media.
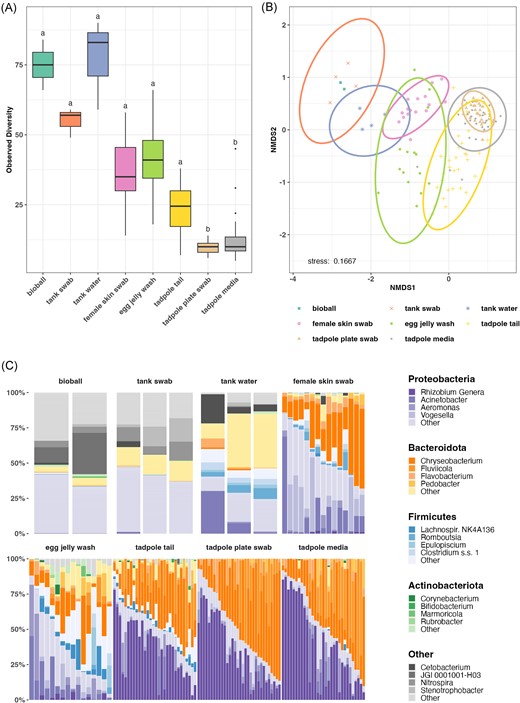
Comparison of microbial communities in core sample types based on 16S amplicon sequencing. Each analysis is based on three merged replicates for each sample. Total sample numbers for each sample type are detailed in Fig. 1(B) and Table S1. (A) Observed alpha diversity (rarefied at 1000 reads). Letters above each sample type denote statistical significance in compact letter display format; sample types with the same letter designation have no statistically significant difference (P < .05). (B) NMDS ordination of Bray–Curtis distance matrix of microbial communities from core sample types. (C) Relative abundance of microbial phyla and genera in core sample types (rarefied at 1000 reads).
Overall, Proteobacteria and Bacteroidota were the most dominant bacterial phyla (Fig. 2C, Table 2). In most cases, relative abundances of genera differed markedly based on the sample type. Chryseobacterium (Bacteroidota) was common across all frog-associated samples, being either the most or second most abundant taxon in female skin, egg jelly, and tadpole tail samples (Table 2), and was also common in the tadpole environment. Vogesella (Proteobacteria) was the second most abundant taxon on female skin, while Rhizobium spp. (assigned as Allorhizobium–Neorhizobium–Pararhizobium–Rhizobium by the SILVA (v138.1) database, summarized here as Rhizobium genera for brevity) (Proteobacteria) was dominant in tadpole samples, followed by Chryseobacterium and Fluviicola. These latter three taxa also dominated tadpole environment samples. The adult frog environment shared relatively few taxa with tadpole tails or their associated environment, although Acinetobacter (Proteobacteria) was common in adult tank water, as well as in female skin and egg samples. The top five taxa by relative abundance for frog samples are provided in Table 2. Top five genera for all core sample types are provided in Supplementary Table S4.
The top five most abundant genera for each of the three Xenopus sample types (based on mean relative abundance).
| Sample type | Genus | Phylum | Abundance |
|---|---|---|---|
| Female skin swab | Chryseobacterium | Bacteroidota | 0.27 |
| Vogesella | Proteobacteria | 0.17 | |
| Acinetobacter | Proteobacteria | 0.13 | |
| Pseudomonas | Proteobacteria | 0.06 | |
| Rheinheimera | Proteobacteria | 0.04 | |
| Egg jelly wash | Chryseobacterium | Bacteroidota | 0.13 |
| Acinetobacter | Proteobacteria | 0.10 | |
| Aeromonas | Proteobacteria | 0.07 | |
| Lachnospiraceae NK4A136 | Firmicutes | 0.06 | |
| Bacteroides | Bacteroidota | 0.03 | |
| Tadpole tail | Rhizobium genera | Proteobacteria | 0.36 |
| Chryseobacterium | Bacteroidota | 0.22 | |
| Fluviicola | Bacteroidota | 0.10 | |
| Aeromonas | Proteobacteria | 0.04 | |
| Bosea | Proteobacteria | 0.03 |
| Sample type | Genus | Phylum | Abundance |
|---|---|---|---|
| Female skin swab | Chryseobacterium | Bacteroidota | 0.27 |
| Vogesella | Proteobacteria | 0.17 | |
| Acinetobacter | Proteobacteria | 0.13 | |
| Pseudomonas | Proteobacteria | 0.06 | |
| Rheinheimera | Proteobacteria | 0.04 | |
| Egg jelly wash | Chryseobacterium | Bacteroidota | 0.13 |
| Acinetobacter | Proteobacteria | 0.10 | |
| Aeromonas | Proteobacteria | 0.07 | |
| Lachnospiraceae NK4A136 | Firmicutes | 0.06 | |
| Bacteroides | Bacteroidota | 0.03 | |
| Tadpole tail | Rhizobium genera | Proteobacteria | 0.36 |
| Chryseobacterium | Bacteroidota | 0.22 | |
| Fluviicola | Bacteroidota | 0.10 | |
| Aeromonas | Proteobacteria | 0.04 | |
| Bosea | Proteobacteria | 0.03 |
The top five most abundant genera for each of the three Xenopus sample types (based on mean relative abundance).
| Sample type | Genus | Phylum | Abundance |
|---|---|---|---|
| Female skin swab | Chryseobacterium | Bacteroidota | 0.27 |
| Vogesella | Proteobacteria | 0.17 | |
| Acinetobacter | Proteobacteria | 0.13 | |
| Pseudomonas | Proteobacteria | 0.06 | |
| Rheinheimera | Proteobacteria | 0.04 | |
| Egg jelly wash | Chryseobacterium | Bacteroidota | 0.13 |
| Acinetobacter | Proteobacteria | 0.10 | |
| Aeromonas | Proteobacteria | 0.07 | |
| Lachnospiraceae NK4A136 | Firmicutes | 0.06 | |
| Bacteroides | Bacteroidota | 0.03 | |
| Tadpole tail | Rhizobium genera | Proteobacteria | 0.36 |
| Chryseobacterium | Bacteroidota | 0.22 | |
| Fluviicola | Bacteroidota | 0.10 | |
| Aeromonas | Proteobacteria | 0.04 | |
| Bosea | Proteobacteria | 0.03 |
| Sample type | Genus | Phylum | Abundance |
|---|---|---|---|
| Female skin swab | Chryseobacterium | Bacteroidota | 0.27 |
| Vogesella | Proteobacteria | 0.17 | |
| Acinetobacter | Proteobacteria | 0.13 | |
| Pseudomonas | Proteobacteria | 0.06 | |
| Rheinheimera | Proteobacteria | 0.04 | |
| Egg jelly wash | Chryseobacterium | Bacteroidota | 0.13 |
| Acinetobacter | Proteobacteria | 0.10 | |
| Aeromonas | Proteobacteria | 0.07 | |
| Lachnospiraceae NK4A136 | Firmicutes | 0.06 | |
| Bacteroides | Bacteroidota | 0.03 | |
| Tadpole tail | Rhizobium genera | Proteobacteria | 0.36 |
| Chryseobacterium | Bacteroidota | 0.22 | |
| Fluviicola | Bacteroidota | 0.10 | |
| Aeromonas | Proteobacteria | 0.04 | |
| Bosea | Proteobacteria | 0.03 |
The influence of the environment on Xenopus skin bacteria is dependent on life stage
To assess the variability of bacterial communities within the various sample types, Bray–Curtis distances were compared between samples of the same type (‘within-type variation’) and between samples of different types (‘between-type variation’). An overall high degree of within-type variation was observed for all sample types, with frog samples (female skin swabs, egg jelly wash, and tadpole tail clippings) generally being more variable than environmental samples (Fig. 3A, Supplementary Table S5A–C).
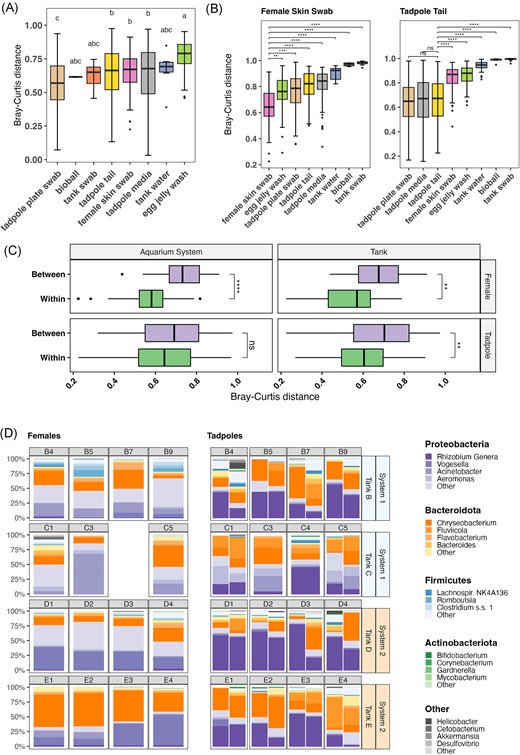
Variability of microbial communities in core sample types based on 16S amplicon sequencing. (A) Box plot of Bray–Curtis distances between samples of the same type. Each box shows the distribution of distances between samples of that sample type. Bars within boxes indicate median value. Statistical significance is shown using compact letter display, sample types within the same letter group have no statistically significant difference from each other based on Dunn’s post hoc test (P < .05). (B) Box plot showing relative distances between mother skin swab samples and tadpole tail samples and other sample types. Asterisks indicate significance levels determined by Dunn’s post hoc test; **** = < 0.0001, *** = < 0.001, ** = < 0.01, * = < 0.05, and ns = not significant. (C) Comparison of Bray–Curtis distance variation in mother skin swabs and tadpole tail samples. On the y-axis, ‘Within’ refers to distances between samples from the same tank or from within the same aquarium system according to the panel. ‘Between’ refers to distances between samples taken from different tanks or systems. Asterisks indicate significance levels determined by Wilcoxon rank-sum; **** = < 0.0001, *** = < 0.001, ** = < 0.01, * = < 0.05, and ns = not significant. (D) Comparison of relative abundance of microbial phyla and genera on individual mother skin and tadpole tail samples within sibships. Each bar within the tadpole panel represents tadpoles from an individual plate (i.e. two plates per mother). Samples are rarefied at 1000 reads. Horizontal labels indicate identity of mother, while vertical labels on right of plot indicate mother’s tank of residence and the aquarium system the tank is within (A or B). Mother samples represent multiple skin swabs, while tadpole samples are tail clips from multiple tadpoles per mother.
Between-type Bray–Curtis distances were frequently greater than within-type distances. We focussed on the distances between female skin swab or tadpole tail samples and other sample types (Fig. 3B). For female skin swabs, the greatest median distances were observed to tank samples, indicating little relationship between female skin bacteria and their environment; distances within female samples were significantly shorter than between females and other sample types (Supplementary Table S6A–C). For tadpole tail samples, Bray–Curtis distances to corresponding environmental samples (tadpole media/plate swabs) were similar to tadpole tail within-type distances (Supplementary Table S6A–C), suggesting that tail bacterial communities were more similar to their environment.
We also calculated and compared mean pairwise Pearson’s correlation coefficients within and between all sample types (Supplementary Fig. S5). Samples were strongly correlated with other samples of the same type and less so with samples of different types, although tadpole tail samples were also strongly correlated with tadpole environmental samples.
To determine whether the female’s tank or aquarium system influenced the female or tadpole skin bacterial community, Bray–Curtis distances between samples within the same tank or aquarium system were compared with distances between samples from different tanks or aquaria (Fig. 3C) (refer to Fig. 1 and 'Embryo Production and Sample Collection' for detail on tank/aquarium system structure). For female skin samples, there was significantly less distance between samples from the same tank or aquarium than between those from different tanks/aquaria (Wilcoxon rank-sum; Supplementary Table S7B), indicating that both variables affect adult skin bacteria. Dispersion of female samples based on aquarium system and tank were significantly different (PERMDISP, P = .04/.01; Supplementary Table 7C), while PERMANOVAs confirmed a significant effect of aquarium system, which accounted for 27% of variation observed (Supplementary Table S7D). The interaction between aquarium system and tank accounted for 22%. For tadpoles, there was a significant difference observed between within- and between-group Bray–Curtis distances for tanks, but not for aquarium system (Wilcoxon rank-sum; Supplementary Table S7B). There was no difference in dispersion between samples based on aquarium system or tank (PERMDISP; Supplementary Table 7C). The aquarium system accounted for 10% of variation, whereas the combined effect of aquarium system and tank accounted for 22%. (PERMANOVA; Supplementary Table S7D). A second PERMANOVA to assess the effect of tank and female found that the tank accounted for 32% of variation, while the combined effect of tank and female ID accounted for 35% (Supplementary Table S7D).
The composition of communities from each female and each plate of tadpoles within a sibship are shown in Fig. 3(D). Differences in female skin bacteria can be seen according to tank as well as aquarium system. Vogesella was more abundant in females from System 2 (Tanks D and E), while Acinetobacter was proportionately more common in System 1 (Tanks B and C). Chryseobacterium (followed by Vogesella) was the overall most abundant genus in Tank E, although Vogesella was more common on one frog. Firmicutes did not comprise a majority of bacteria from any frog, but they were more common in frogs from Tank B (mean 16.5%, c.f. 1.0%–6.7%), which comprised frogs considerably older than those from Tanks C–E (Supplementary Table S8). Tadpole tail bacterial composition also appeared to vary according to the female’s tank; for example, tadpoles from Tank D females were most often dominated by Rhizobium genera, which were in relatively low abundance in Tank C sibships. Firmicutes were in highest relative abundance in Tank B tadpoles, which corresponds to a higher profusion of this phylum in Tank B female frogs.
Tadpole skin bacterial communities were similar to their environment, while those of adult frogs were more distinct
Differential abundance of taxa between female skin swab and tadpole tail samples was assessed using ANCOMBC2 and MaAsLin2. Twenty-three taxa were found to be significantly differentially abundant (FDR-adjusted P-value < .25) by both analyses, with a further 17 taxa identified by one analysis but not the other. The mean relative abundances of the 23 common taxa are shown for each of the sample types in Fig. 4(A); full results of the analyses can be found in Supplementary Tables S9 and S10. Further, the mean relative abundance across sample types of a selection of genera that were notably abundant in Supplementary Table S4 are shown in Fig. 4(B).
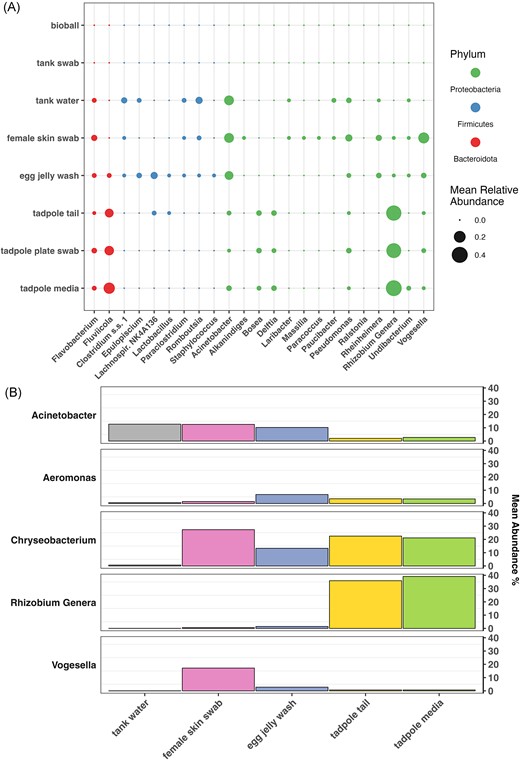
(A) Mean relative abundance of significantly differentially abundant taxa across sample types. Taxa shown here are those found to be differentially abundant between mother skin swabs and tadpole tails by both ANCOMBC2 and MaAsLin2 (q < 0.25). (B) Mean relative abundance of selected taxa across sample types, demonstrating changes in abundance through the progression of the X. laevis life cycle (left to right). Taxa shown were noted as being particularly abundant across one or more sample types of interest in Supplementary Table S4.
Rhizobium were markedly more abundant in tadpole tails and their immediate environment than any other sample type, while taxa such as Romboutsia, Acinetobacter, and Clostridium were more abundant in the tank water and/or on females and eggs (Fig. 4A and B). Meanwhile, Vogesella was notably abundant on female frogs and to a lesser extent, eggs, while being relatively uncommon in the corresponding environment. Female and tank water samples seemed to be less favourable to taxa such as Fluviicola and Rhizobium. Chryseobacterium was abundant in all three frog life stages, as well as in tadpole media.
A microbial source tracking analysis was completed using FEAST v0.1.0 (Fig. 5; Supplementary Table S11). While the source of a proportion of bacterial taxa was ultimately unknown for each sink (female skin, egg jelly, tadpole tails, and tadpole environmental samples) the analysis indicates the likely most significant sources of bacteria. The skin bacteria of female adult frogs were largely sourced from their tank water, while the bacteria adhering to the tank wall made a comparatively negligible contribution. The majority of the bacteria associated with egg jelly was sourced from the female’s skin, and tadpoles subsequently received most of theirs from the eggs before seeding their environment (MMR media).
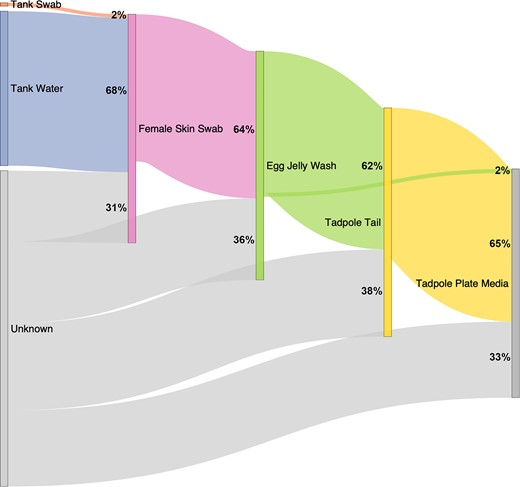
Sankey diagram conceptualizing flow of microbial taxa through the environment and frog life stages, based on FEAST analysis. Percentages indicate the proportion of bacteria attributed to that particular source for each sample type (‘sink’)—e.g. on average, 68% of the bacterial communities found on female skin were determined to be contributed by tank water. Further details of calculations are provided in Supplementary Table S11. Bioballs are not included in this analysis as they are not in direct contact with frogs and are similar in composition to tank wall swabs. Tadpole plate swabs were not analysed as a sink due to their similar composition to tadpole plate media.
Discussion
Frogs of the Xenopus genus, most notably X. laevis and X. tropicalis, are widely used model organisms, finding particular utility in tissue regeneration, developmental, genetic, and neurobiological research. Many frogs are supplied to research laboratories by central breeding facilities which routinely treat tadpoles with antibiotics or preservatives (e.g. gentamicin and thimerosal) (Wlizla et al. 2018, Shaidani et al. 2021), which inherently alters microbiome community structure and transmission (Chapman et al. 2022). However, frogs are sometimes also raised in smaller, in-house facilities which may employ different rearing protocols, which, in combination with local environmental differences, is likely to affect microbiome composition. It is becoming evident that bacteria play an important role in Xenopus tail regeneration (Bishop and Beck 2021, Chapman et al. 2022) and are undoubtedly critical in the development of immune and other physiological systems. For this reason, it is imperative to characterize microbiomes associated with Xenopus and consider how they may be influencing experimental results.
A large proportion of studies into amphibian skin microbiota to date have been undertaken with reference to chytridiomycosis [summarized by Jiménez and Sommer (2017)]. Chytrid infection was shown to drive changes in skin bacterial communities (Jani and Briggs 2014, Rebollar et al. 2016, Jani et al. 2017, 2021, Bates et al. 2018); indeed, some bacterial taxa are shown to have antichytrid properties (Woodhams et al. 2015, 2016). These taxa are conceivably selected for in epi/enzootic scenarios, and have been investigated as potential probiotic therapies (Bletz et al. 2013, Woodhams et al. 2016, 2020). The X. laevis colony involved in this study previously tested negative to chytrid infection, and is assumed to remain so due to the closed nature of the colony, biosecurity protocols and lack of symptoms. However, we note the presence of potentially antifungal genera identified by Woodhams et al. (2016), such as Chryseobacterium and Pseudomonas, on our frogs and tadpoles.
Female Xenopus skin bacterial communities are affected by the environment, but the frogs have ultimate control
Female frogs had less variable bacterial communities than tadpoles, and Bray–Curtis distances to their corresponding environmental samples were relatively high, suggesting they may select for microbes occurring at low abundance in the environment (e.g. Vogesella). We note that although females were moved into clean filtered water before swabbing, they were not rinsed to remove transient environmental DNA, which may have resulted in some tank water microbiota remaining on the skin. Regardless, tank and female sample analyses suggest that the adult X. laevis skin favours certain skin bacteria adapted to withstand and exploit the unique physical conditions. This is a similar conclusion to Piccinni et al. (2021)’s laboratory X. laevis study, and was also suggested by Li et al. (2020)’s research on X. tropicalis froglet gut bacteria. Studies of wild amphibians (McKenzie et al. 2012, Kueneman et al. 2014, 2016, Walke et al. 2014) yield comparable results. Host control of the skin microbiome could be accomplished through a variety of mechanisms, including skin shedding (Meyer et al. 2012, Cramp et al. 2014, Colombo et al. 2015) and mucous secretion (Walke et al. 2014). Importantly, amphibian skin mucous contains unique assemblages of antimicrobial peptides (Woodhams et al. 2007, 2016, Tennessen et al. 2009, Myers et al. 2012, Walke et al. 2014) that probably dictate the suite of microbes able to successfully colonize. The microbes themselves produce metabolites that may inhibit growth of other microbes (Mangoni et al. 2001, Kommineni et al. 2015, Loudon et al. 2016). While female frogs are likely to select for certain skin microbiota through the above means, it was clear from the analysis of Bray–Curtis distances that bacteria still vary between individual frogs, and are also affected by the tank and aquarium system. Individual differences in physiology and immune development may account for some variation between frogs. Frogs from Tank B were born in 2007, while those from other tanks were considerably younger (2016–2018), so age-related physiological factors may account for some variation observed in this study. Differences between tanks may also be attributed to small differences in environmental factors due to the tank’s location in the aquarium room, or may be related to the cohort of tank mates and the assemblage of microbiota each potentially shares with others when introduced.
Female skin seeds the egg jelly, which in turn provides some bacteria to the tadpoles
A significant portion of the tadpole skin bacteria appear to be received vertically from the female via the egg jelly. A large portion (mean 64% ± 19%) of egg jelly bacteria was derived from the female’s skin. Scalvenzi et al. (2021) reported that, while in many cases a large portion of X. tropicalis egg bacteria could not be traced to the parents, the parent’s skin contributed significantly more than did their faeces/gut. The relative unimportance of the adult gut bacteria in seeding egg bacterial communities is surprising given the close contact between eggs and the female digestive tract in the amphibian cloaca. We did not sample the parent gut bacteria in this study as this was difficult to achieve effectively without sacrificing female frogs, however the skin appears to be a major contributor to egg bacterial flora. Scalvenzi et al. (2021)’s study differs from the present experiment in that parents were allowed to mate naturally, whereas our embryos were produced using in vitro fertilization. Eggs therefore had no direct contact with male frogs. While testis samples were sequenced and returned variable read counts, a large proportion had low bacterial biomass, and it was deemed likely that contamination during the necropsy and collection process was responsible for most reads. Testes are therefore considered unlikely to be a primary contributor to colonization of eggs and subsequent life stages under the study conditions. Given that the majority of laboratories produce Xenopus embryos using in vitro fertilization, the female is logically a more significant source of microbes in these cases.
Kueneman et al. (2016) found that larval stages of boreal toads (Anaxyrus boreas) and Cascades frogs (Rana cascadae) (Kueneman et al. 2014) had the lowest skin bacterial alpha diversity of all life stages, and we demonstrated the same for X. laevis. We also found that tadpole bacterial communities were more variable than adult females; this may be at least partially due to tadpole replicates representing different individuals rather than true technical replicates. However, newly developed embryos and tadpoles are also a prospect for colonization by opportunistic microbiota, and their early communities may reflect those taxa, which either happen to encounter them first or are most suited to rapid exploitation of that environment. McGrath-Blaser et al. (2021) determined that tadpole skin bacteria established via both vertical transmission and environmental exposure in Bornean foam-nesting frogs (Polypedates spp.). While it is common practice to raise laboratory X. laevis eggs/embryos in ‘clean’ environments separate to the adult colony, had tadpoles in our study been raised in a more complex environment, it is possible that we would have observed a lower contribution of vertical transmission due to increased competition by environmental microbes. However, while the majority of our tadpole’s bacteria were contributed by the egg jelly (and indirectly, the female), and despite the relatively ‘clean’ environment, a proportion were derived from unknown, presumably environmental sources. It was not considered likely that the plates (Petri dishes) or media (0.1x MMR) were significant contributors. Plates used were new and provided in sterile packaging, and media was autoclaved prior to use. While this should have minimized the number of microbes present, it is possible that some bacteria may have entered media bottles after opening. Media samples sequenced here (as well as plate swabs) were collected after exposure to tadpoles for a period of 1 week, and were quite similar in bacterial composition to tadpole samples. We determined that, given the high calculated contribution of the egg jelly to tadpole samples and previous records of dominant taxa in frogs/tadpoles, it was likely that tadpoles were seeding their environment rather than vice versa. However, in future it would be preferable to sequence the media prior to tadpole exposure.
Similarly to the tadpole stage, a notable proportion of bacteria were attributed to unknown sources at the egg and adult female life stages. We used a variety of controls to detect potential contaminants introduced throughout the sample collection and processing pipeline. The ‘kit microbiome’ or ‘kitome’ is a recognized source of contaminants during DNA extraction (Glassing et al. 2016) and while we cannot eliminate this as a contributor to ‘unknown’ sources, our blank controls revealed negligible reads and we have taken measures to computationally account for and remove contaminants prior to analysis. Other potential ‘unknown’ sources include airborne bacteria in the lab and those potentially inadvertently introduced by the sample collector.
Proteobacteria and Bacteroidota dominate X. laevis skin bacterial communities
Proteobacteria and Bacteroidota were the dominant phyla across frog, egg, and tadpole samples. This is similar to Piccinni et al. (2021)’s study which found that Bacteroidota (in particular Flavobacteriia) followed by Proteobacteria were the most common bacterial phyla on adult X. laevis. Proteobacteria were dominant in their feeding stage tadpoles, with Bacteroidota making up a relatively minor portion of the skin bacteria.
In our study, tadpole skin bacteria were dominated by a genus of Proteobacterium, Rhizobium (designated as Allorhizobium–Neorhizobium–Pararhizobium–Rhizobium by the SILVA v. 138.1 database). Most Rhizobium are Gram-negative, nitrogen-fixing plant symbionts and would not be immediately associated with the aquatic environment; however some species are free-living (Sullivan et al. 1996). While Rhizobium were not among the top 50 taxa in Piccinni et al. (2021)’s dataset, bacteria from the Rhizobiales order were reported from Scalvenzi et al. (2021)’s X. tropicalis metamorph/froglet guts, and Rhizobiaceae were detected in postmetamorphic X. laevis by Mashoof et al. (2013). A Rhizobium genus was also associated with faeces and gut samples in laboratory raised Asiatic toad tadpoles (Bufo gargarizans) (Song et al. 2018). Interestingly, a previous study of tadpoles from the same colony as this study (Chapman et al. 2022) did not detect this Rhizobium genotype, nor did it identify any Rhizobium as a dominant genus despite an absence of major changes in husbandry. These tadpoles were instead dominated by Shinella or Delftia (for antibiotic treated tadpoles) or Shinella, Acinetobacter, Escherichia–Shigella, or Chryseobacterium (untreated). The differences between the two studies may be attributed to different sampling techniques (Chapman et al. (2022) extracted DNA from the entire tadpole, while the current study used only tail clippings), antibiotic effects in the earlier study, fluctuations in microbe populations in the various inputs (food, water, and so on) or natural community drift/succession. Differences in computational processing and analysis of data may also be a factor.
The other notably dominant taxa on both females and tadpoles was a genus of Bacteroidota, Chryseobacterium (once classified as Flaviobacterium; Vandamme et al. 1994). In some tadpoles, this taxa was more abundant than Rhizobium, and was also relatively common in tadpoles from the Chapman et al. (2022) study. Neither Scalvenzi et al. (2021) nor Piccinni et al. (2021) reported Chryseobacterium from their tadpoles, however several epibiotic strains of ultrasmall Chryseobacterium have previously been reported from adult X. laevis (Ross et al. 2019a). Piccinni et al. (2021) did report that Bergeyella, a related species (Vandamme et al. 1994, Bernardet et al. 1996) was dominant on adult X. laevis skin, although very low or undetectable in tadpole and water samples. Chryseobacterium is also well documented from newts (Kirk et al. 2013) and fish (Loch and Faisal 2015) and the closely related Elizabethkingi genus (once considered to be a Chryseobacterium) has been associated with disease in frogs (Mauel et al. 2002, Xie et al. 2009, Trimpert et al. 2021) including X. laevis (Green et al. 1999). We have successfully cultured Chryseobacterium from X. laevis adults and tadpoles and found that it correlated with improved tadpole tail regeneration outcomes (Chapman et al. 2022).
Other notable taxa on frog samples included Acinetobacter, Aeromonas, Fluviicola, and Vogesella. A strain of the latter genus (Vogesella sp. strain XCS3) was successfully cultured and sequenced from our frogs by Hudson et al. (2021). Tank/aquarium samples were dominated by both Proteobacteria and Bacteroidota as well as Acidobacteriota. Aeromonas, a taxon commonly found in aquatic environments and among the most abundant in our egg jelly samples, was only reported by Piccinni et al. (2021) on ‘clean’ (thimerosal/ethanol treated) tadpoles and in standard ‘unclean’ water, while the remaining taxa listed above were not within their top 50 abundant taxa. Aeromonas was common in Scalvenzi et al. (2021)’s tadpole gut samples, but the others were once again absent.
Captive X. laevis skin bacterial communities depend on laboratory environment and husbandry
As is clear from the above, bacterial communities appear to vary between species of the same host genus, colonies, tanks/aquaria and probably over time. The marked differences are likely due to differences in skin microenvironment (skin structure, secretions, and so on) between species, variations in environment (i.e. microbial inputs from water and surrounding environment) and husbandry (e.g. food source, aquarium additives, and antibiotic use). We note that Piccinni et al. (2021)’s tadpoles were ~1 month old, premetamorphic, and being fed on algae. We sampled our tadpoles at a stage where they were not sufficiently developed to require formal feeding (stage 46; Nieuwkoop and Faber 1956), as food input may have confounded attempts to detect vertical transmission of bacteria. Any potential food intake for our tadpoles would have been comprised of material derived from other tadpoles (tissue or waste product) or other incidental microscopic items scavenged from the media, with waste material derived from these sources or from yolk digestion/absorption. The difference in feeding status is likely to impact opportunities for colonization of the gut and the skin through different/additional microbial inputs into the tadpole environment, and through concurrent changes to the gut and immune system associated with development stage. In our colony, we determined that frog food (generic salmon pellets) was unlikely to contribute substantively to the skin bacteria of adult frogs due to a low number of bacterial reads; we also determined that the tank water conditioner, comprised almost entirely of Firmicutes, was not a major contributor.
Conclusions
This study has explored the bacterial communities associated with nonantibiotic treated, captive X. laevis and the dynamics of transmission between different frog life stages and their environment. Female frog skin bacterial communities were less diverse than those in their environment, and were dominated by different taxa, indicating that adult frogs have some ability to select for certain taxa that are present at low abundances in their environment. Tadpoles inherit a proportion of their bacteria from females via the egg, however have a less diverse and distinct bacterial community, which may reflect opportunistic colonization of an available niche. After comparison with previous studies, these results indicate that although adult frogs retain some control over their bacteria, the ultimate composition is dependent on environmental and experimental conditions. Skin bacterial communities and the impact of husbandry procedures should be an important consideration in Xenopus laboratory experiments. In lieu of microbial community profiling, detailed reporting of husbandry practices and environmental conditions should accompany all experimental results.
Acknowledgements
We would like to acknowledge Joanna Ward and Nikita Woodhead for their skilled assistance in the laboratory and the aquarium, respectively. Xenopus laevis egg and embryo production methodology was approved by the University of Otago’s Animal Ethics Committee (permit number AUP19-01).
Author contributions
Phoebe A. Chapman (Data curation, Formal analysis, Methodology, Project administration, Visualization, Writing – original draft, Writing – review & editing), Daniel Hudson (Data curation, Investigation, Methodology, Writing – review & editing), Xochitl C. Morgan (Conceptualization, Formal analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – review & editing), and Caroline W. Beck (Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Visualization, Writing – review & editing)
Conflict of interest
None declared.
Funding
This work was supported by the Royal Society of New Zealand through the Marsden Fund program (grant number MFP-UOO1910).
Data Availability
R code for all analyses is provided at https://gitlab.com/pachapman/xenopus_microbiomes. Sequence data has been deposited in Sequence Read Archive (SRA) under BioProject ID PRJNA823390 (BioSamples SAMN33958523–SAMN33958729).
References
Author notes
Equal contribution

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}