





Abstract
For billions of years before electric light was invented, life on Earth evolved under the pattern of light during the day and darkness during the night. Through evolution, nearly all organisms internalized the temporal rhythm of Earth’s 24-hour rotation and evolved self-sustaining biological clocks with a ~24-hour rhythm. These internal rhythms are called circadian rhythms, and the molecular constituents that generate them are called molecular circadian clocks. Alignment of molecular clocks with the environmental light-dark rhythms optimizes physiology and behavior. This phenomenon is particularly true for reproductive function, in which seasonal breeders use day length information to time yearly changes in fertility. However, it is becoming increasingly clear that light-induced disruption of circadian rhythms can negatively impact fertility in nonseasonal breeders as well. In particular, the luteinizing hormone surge promoting ovulation is sensitive to circadian disruption. In this review, we will summarize our current understanding of the neuronal networks that underlie circadian rhythms and the luteinizing hormone surge.
The evolutionary advantage to time mating to the day of ovulation has allowed the development of numerous physiological and molecular mechanisms that together increase the chances of fertilizing the ovulated oocyte (Fig. 1). Some of these mechanisms include increased male-female attraction around ovulation, which is achieved through species-specific changes in pheromone secretion and behavior (1, 2). Together, these changes in the female increase the chances of attracting a male and mating behavior, resulting in fertilization of the oocyte during its 12- to 24-hour life span. To further increase chances of fertilization during the short window of oocyte receptivity, ovulation happens just around wake (activity onset) in most diurnal and nocturnal spontaneous ovulators (Fig. 1) (3, 4). To allow this precise timing of ovulation, the luteinizing hormone (LH) surge, which drives ovulation, is finely regulated by a complex set of precisely aligned neuroendocrine mechanisms. Specifically, the goal of this review is to summarize our current understanding of the role of circadian rhythms and peptides in the development and function of the neuronal network timing the LH surge that promotes ovulation. We will do this through the review of the neuronal network controlling the LH surge through 3 perspectives: (1) the development of the neurons in the hypothalamus regulating LH release; (2) the maturation of the circuit, including neuronal connectivity and a functioning molecular clock in the circuit; and (3) coordinated function and capacity to respond to sex steroids in the circuit. We will specifically focus on the role of homeodomain transcription factors that are expressed in the suprachiasmatic nucleus (SCN), gonadotropin-releasing hormone (GnRH) neurons, and kisspeptin neurons, in addition to molecular clock transcription factors in the regulation of neuronal circuit function, and discus the contribution of distinct SCN neuron subpopulations and neuropeptides in the functioning of the circuit driving the LH surge.
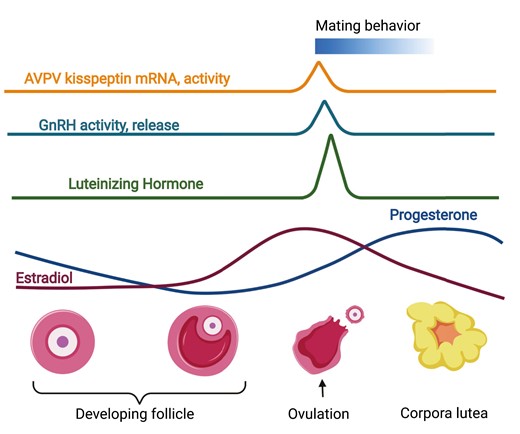
Ovulation is precisely timed to increase the chances of fertilization of the oocyte during its short life span. In most spontaneous ovulating mammals, the timing of ovulation happens shortly after activity onset/wake-up in both diurnal and nocturnal species. This timing of ovulation is thought to increase the chances of fertilization of the oocyte, which only lives 12 to 24 hours. Ovulating at activity onset increases the chances of mating behavior, which occurs during the wake hours. A sophisticated neuroendocrine-ovarian feedback mechanism generates increased estradiol levels, which are required for the LH (luteinizing hormone) surge, which promotes ovulation and female sexual behavior. Estradiol is produced by the maturing follicle, whereas the increase in progesterone is driven by the corpus luteum in the ovary that forms after ovulation. Progesterone is a primary hormone preparing the uterus for implantation and successful pregnancy maintenance. Figure made using BioRender.
The Case for Timing: Coordination Between Light, Neuronal, and Hormonal Signals Allowing the Timing of the LH Surge Makes it Vulnerable to Environmental Disruptors
The hypothalamic-pituitary-gonadal (HPG) axis regulates reproduction (Fig. 1). In the hypothalamus, kisspeptin neurons stimulate GnRH neurons, which release GnRH onto gonadotropes in the anterior pituitary. In response to GnRH, gonadotropes release LH and follicle-stimulating hormone (FSH) into the circulation, allowing these hormones to act on the gonads to stimulate gametogenesis and sex steroid production. In adult males, and throughout most of the adult female estrous cycle, this axis is characterized by sex steroid–mediated negative feedback through testosterone (in male) and estrogen (in female). The action of the sex steroids on the arcuate nucleus kisspeptin neurons reduces kisspeptin release onto GnRH neurons, resulting in reduced GnRH-promoted LH and FSH release. However, in females, the progressive development of ovarian follicle(s) results in increased estrogen production and release. When the follicle(s) are mature, the level of estrogen released reaches a threshold level that “switches” the action of estrogen, which then becomes an activator to kisspeptin neurons in the anteroventral periventricular nucleus (AVPV) (5). The increased activity and release of kisspeptin by AVPV kisspeptin neurons onto GnRH neurons results in a surge of GnRH, which prompts a surge of LH, and then ovulation. This LH surge is temporally confined to the period around activity onset, and neither time nor estrogen alone are sufficient to produce an LH surge in rodents. Specifically, ovariectomy with exogenous estrogen replacement produces a daily consecutive afternoon LH surge in rats (6), hamsters (7), and mice (8, 9). Phenobarbital treatment suppresses the central nervous system (and presumably the temporal signal) and delays the endogenous surge in rats for 1 to 3 days, depending on administration length (10), indicating neuronal activation is required to time the LH surge. Nonphotic circadian disruption, such as exposure to a running wheel on the day of proestrus, can similarly delay the LH surge by a day in hamsters (11). Thus, the sine qua non of the temporal signal imparts circadian sensitivity to the LH surge in females, but as males do not present a LH surge, male fertility is less disrupted by circadian disruption than females. In support of this observation, extensive work from the last 7 decades has identified and repeatedly confirmed that the hypothalamic SCN, the principal circadian timekeeping structure in the brain, regulates the timing of the LH surge (12, 13). As the SCN provides the “gate-keeping” signal that times the LH surge, this makes the LH surge sensitive to factors disrupting SCN function.
One of the primary roles of the SCN is to receive direct light input from the eye through the retino-hypothalamic tract and translate it into neuronal and hormonal signals, which coordinate tissue time-keeping throughout the body. The central role of the SCN in timing the LH surge is further enforced by studies highlighting that impaired SCN development (see “Disrupted Development and Maturation of the SCN and the Impact on the LH Surge”) (14-22). In addition, post-development SCN peptides (see “Role of Peptide Neurotransmission in the Neuronal Circuit Regulating the LH Surge”), and pace-making function (see “Role of Clock Genes in the Neuronal Network Regulating the LH Surge”) are equally important contributors to the LH surge and reproduction, where current research primarily has shown a key role of the SCN in female fertility.
The Neuronal Network Regulating the LH Surge in the Adult Rodent
The key role of SCN function in adulthood as a regulator of the LH surge has been established primarily using wild-type animals, where the SCN and neuroendocrine circuit regulating reproduction develops normally. Such work includes electrolytic lesion of the whole SCN, which abolished the LH surge and estrous cyclicity (23, 24). Further supporting that the function of the adult SCN is driving the timing of the LH surge comes from evidence in hamsters, who demonstrate “behavioral splitting” in low light which results in 2 12-hour rhythms, each governed by 1 hemisphere of the SCN (25). “Split” hamsters show 2 small LH surges 12 hours apart, with a corresponding increase in c-Fos in GnRH neurons in only one hemisphere (26), supporting GnRH neuron activation at the time of the LH surge. Similarly, unilateral SCN lesions in rats abolishes ipsilateral GnRH c-Fos and rise in kisspeptin mRNA at the time of the LH surge (27). This SCN signal requires direct SCN projections, as the transplant of a fetal SCN into an SCN-lesioned hamster did not restore the LH surge or estrous cyclicity (28), although it was sufficient to restore locomotor rhythms (29). Together, the fetal SCN transplant and hamster splitting experiments strongly support a direct neuronal projection from the SCN to the neurons that regulate the LH surge. The primary candidates are direct SCN neuron projections from vasoactive intestinal polypeptide (VIP) neurons to GnRH neurons, and arginine vasopressin (AVP) neurons to AVPV kisspeptin neurons.
Role of Peptide Neurotransmission in the Neuronal Circuit Regulating the LH Surge
Despite its small size of ~20 000 neurons in a mouse, the peptide expression in the SCN is highly heterogeneous. Four peptides expressed by the SCN have been implicated in fertility: VIP, AVP, neuromedin S (NMS), and prokineticin 2 (PK2).
VIP
VIP neurons in the SCN core send direct projections to GnRH neurons (30, 31) and GnRH neurons express the VIP receptor, VIP2R (32). In vitro studies have found conflicting results in which VIP increases GnRH neuronal firing at any time or estrous cycle stage (33), or only in a time- and estrogen-dependent manner (34). In vivo, intracranial VIP injection in ovariectomized, estradiol-replaced rats delays and attenuates the LH surge (35). However, VIP antagonism in the SCN also delays and attenuates the LH surge in rats (36), as does VIP antiserum (37). VIP knockout mice are subfertile, showing a reduced rate of ovulation (38), and mice with reduced VIP peptide in the SCN have reduced fertility (39). These findings indicate that VIP plays an important role in the LH surge, but the failure of VIP to stimulate LH suggests that it alone is not sufficient to promote GnRH release. However, the findings by Piet et al suggest that VIP can activate GnRH neurons at any time of day, indicating that a temporally confined VIP signal from the SCN may synergize with kisspeptin signaling to promote the surge at GnRH neurons (32, 33).
AVP
In addition to projecting onto GnRH neurons, VIP neurons also project onto AVP neurons, allowing AVP neurons to contribute to the light-sensitivity and finely tuned timing of the LH surge. AVP neurons in the SCN shell project to AVPV kisspeptin neurons in rodents (40-42), and kisspeptin neurons express the vasopressin receptor V1a (40). In brain slice preparations, AVP robustly stimulates kisspeptin neuron firing (43) and c-Fos induction (40), regardless of time of day or cycle stage. In SCN-lesioned animals, intracranial injection of AVP in the late afternoon rescues the LH surge through V1a (44, 45). Further supporting an important role of AVP in the LH surge is a recent study using optogenetic stimulation of AVP-containing projections in the AVPV (42), where high frequency light stimulation of AVP neurons increased firing in AVPV kisspeptin neurons specifically during proestrus. This effect of AVP neuron stimulation on kisspeptin neuron activity was blocked with the V1a antagonist Manning Compound, supporting that AVP neurons originating in the SCN promote AVPV kisspeptin neuron activity at proestrus, contributing to enhanced kisspeptin activity, which may in turn drive GnRH neuron–promoted LH release. Interestingly, stimulation of the SCN AVP projections inhibited AVPV kisspeptin neurons during estrus through the inhibitory neurotransmitter GABA, indicating that this circuit may be more dynamic than previously believed. However, while intracranial AVP stimulates kisspeptin neurons regardless of time of day, it only leads to downstream activation of GnRH neurons in the afternoon, suggesting time-dependent priming of GnRH neurons (43).
Neuromedin S
NMS expression in adults is relatively confined to the SCN (46). NMS is expressed in approximately half of all SCN neurons, including most VIP and AVP neurons as well as other populations (47, 48). Intracerebroventricular injection of NMS induces LH release in an estrogen-dependent manner in rats (49). Transcripts for the central NMS receptor, Nmur2, is expressed in immortalized GnRH neurons (Mellon lab, unpublished data) as well as at low levels in AVPV kisspeptin neurons in ovariectomized adult female mice (personal communication with Alexander Kauffman (50)). However, NMS knockout mice are reported to be fertile. More work is needed to determine the contribution of NMS as a timing signal for the LH surge.
Prokineticin-2
PK2 is another SCN peptide linked to female reproduction. Recent single-cell RNA sequencing analysis of SCN neurons revealed that approximately half of PK2 neurons also contain AVP (51). Knockout mouse models of PK2 (52) and its receptor, PK2R (53), cause infertility due to mismigration of GnRH neurons. Heterozygous loss of PK2 and PK2R, or just PK2R alone, have no effect on GnRH neuron migration but are sufficient to disrupt estrous cycle length, and administration of a PK2R antagonist on the morning of proestrus blocks the LH surge (54); however, the site of action of this effect is unknown. GnRH neurons do not express PK2R (52, 55, 56), and there is little overlap with PK2R- and estrogen receptor alpha-expressing cells in the AVPV (eg, kisspeptin neurons; (56)). Disruption to PK2/PK2R could be exerting a reproductive effect through the SCN onto other SCN outputs, and more work is needed to determine the role of SCN-derived PK2 in the LH surge.
Disrupted Development and Maturation of the SCN and the Impact on the LH Surge
Correct ventral forebrain and hypothalamic development as well as neuronal circuit maturation are required to establish a functional neuronal network that can drive the LH surge around activity onset. To establish this neuronal circuit, it is important to consider when the SCN develops in comparison to GnRH and kisspeptin neurons.
The SCN
The SCN arises from a rapid cell proliferation of the neuroepithelium caudal to the optic recess ~embryonic day (e)14 in the mouse (57-60). The emergence of the neuropeptides VIP, AVP, and NMS, along with the emergence of strong circadian rhythms, are indicators of SCN neuron maturity (61-64).
GnRH neurons
GnRH neurons arise in the olfactory placode ~e11 in the mouse, from where they migrate through the cribriform plate and ventral forebrain, before they arrive in the preoptic area of the hypothalamus ~e15 to postnatal day (P)0. GnRH neurons can be identified from ~e11 onwards due to their expression of GnRH. Mature GnRH neurons have a functional molecular clock, which is thought to contribute to daily changes in GnRH neuron sensitivity to VIP and AVP (40, 65).
AVPV and arcuate nucleus kisspeptin neurons
Kisspeptin neurons are sexually dimorphic, and develop at different times in different brain areas, depending on the sex. AVPV kisspeptin neurons arise around P25, with adult levels of kisspeptin neurons in the AVPV at P31 in female, and P45 in male mice. Although AVPV kisspeptin neurons are present in both sexes, females have ~10 times more kisspeptin neurons in the AVPV than males (66). Arcuate nucleus kisspeptin neurons are not thought to be sexually dimorphic. The arcuate nucleus starts to express key neuropeptide markers as early as e10, where kisspeptin expressing neurons are detectable ~e13 (66-68).
After SCN formation, VIP neurons project directly onto GnRH neurons, whereas AVP neurons project onto kisspeptin neurons. When during development these projects are established has not yet been determined, but as reviewed below, diminished SCN development impairs its control of the LH surge. In contrast to VIP and AVP SCN neurons, which are distinct neuronal populations, NMS is co-expressed in in most VIP neurons, as well as most AVP neurons.
Role of Homeodomain Transcription Factors in SCN Development and Fertility
Despite our relatively advanced understanding of the role of the SCN as the brain’s primary circadian pacemaker, the transcriptional program directing specification of the SCN remains poorly defined (69). Recent reviews have summarized our understanding of SCN development (70-73), where 4 homeodomain transcription factors have been identified as required for SCN development, Lhx1 (LIM homeobox) (12, 74), Six3 (SIX homeobox, transcription factor 3) (72), Six6, and most recently, Vax1 (Ventral Anterior Homeobox 1) (75). Deletion of Lhx1, Six3, Six6, or Vax1 before e14 causes loss of SCN morphology, and in the mouse models that are viable, disrupts SCN output (summarized in Table 1). None of these studies directly investigated whether impaired SCN function impacted the LH surge. However, Vax1 heterozygous and Six3 heterozygous mice are subfertile (76, 77), whereas Six6 knockout mice were almost completely infertile (16). However, these changes in fertility appear to be primarily associated with loss of GnRH neurons or abnormal GnRH neuron migration (78, 79).
Impact of homeodomain transcription factor KO on the SCN, GnRH neurons, and kisspeptin neurons
| Model | SCN | GnRH | Kisspeptin (diestrus female) | Female fertility | Reference |
|---|---|---|---|---|---|
| Six3 HET | Normal in LD12:12 (male) | 55% | N/A | Subfertile | (76) |
| Six6 HET | N/A | 100% | N/A | Normal | (16) |
| Six6 KO | Loss of morphology, no AVP, no VIP, impaired SCN output | 10% | N/A | Subfertile | (15) |
| Vax1 HET | Normal morphology reduced VIP, modest | 40% | Arcuate: normal | Subfertile | (77) |
| AVPV: ~20-fold increase | |||||
| Vax1 KO | Loss of morphology, no AVP | 0% (at P0) | N/A | N/A | (75) |
| Model | SCN | GnRH | Kisspeptin (diestrus female) | Female fertility | Reference |
|---|---|---|---|---|---|
| Six3 HET | Normal in LD12:12 (male) | 55% | N/A | Subfertile | (76) |
| Six6 HET | N/A | 100% | N/A | Normal | (16) |
| Six6 KO | Loss of morphology, no AVP, no VIP, impaired SCN output | 10% | N/A | Subfertile | (15) |
| Vax1 HET | Normal morphology reduced VIP, modest | 40% | Arcuate: normal | Subfertile | (77) |
| AVPV: ~20-fold increase | |||||
| Vax1 KO | Loss of morphology, no AVP | 0% (at P0) | N/A | N/A | (75) |
Abbreviations: AVP, arginine vasopressin; AVPV, anteroventral periventricular nucleus; GnRH, gonadotropin-releasing hormone; N/A, not assessed; SCN, suprachiasmatic nucleus; Six, SIX homeobox; VAX1, ventral anterior homeobox 1; VIP, vasoactive intestinal peptide.
Impact of homeodomain transcription factor KO on the SCN, GnRH neurons, and kisspeptin neurons
| Model | SCN | GnRH | Kisspeptin (diestrus female) | Female fertility | Reference |
|---|---|---|---|---|---|
| Six3 HET | Normal in LD12:12 (male) | 55% | N/A | Subfertile | (76) |
| Six6 HET | N/A | 100% | N/A | Normal | (16) |
| Six6 KO | Loss of morphology, no AVP, no VIP, impaired SCN output | 10% | N/A | Subfertile | (15) |
| Vax1 HET | Normal morphology reduced VIP, modest | 40% | Arcuate: normal | Subfertile | (77) |
| AVPV: ~20-fold increase | |||||
| Vax1 KO | Loss of morphology, no AVP | 0% (at P0) | N/A | N/A | (75) |
| Model | SCN | GnRH | Kisspeptin (diestrus female) | Female fertility | Reference |
|---|---|---|---|---|---|
| Six3 HET | Normal in LD12:12 (male) | 55% | N/A | Subfertile | (76) |
| Six6 HET | N/A | 100% | N/A | Normal | (16) |
| Six6 KO | Loss of morphology, no AVP, no VIP, impaired SCN output | 10% | N/A | Subfertile | (15) |
| Vax1 HET | Normal morphology reduced VIP, modest | 40% | Arcuate: normal | Subfertile | (77) |
| AVPV: ~20-fold increase | |||||
| Vax1 KO | Loss of morphology, no AVP | 0% (at P0) | N/A | N/A | (75) |
Abbreviations: AVP, arginine vasopressin; AVPV, anteroventral periventricular nucleus; GnRH, gonadotropin-releasing hormone; N/A, not assessed; SCN, suprachiasmatic nucleus; Six, SIX homeobox; VAX1, ventral anterior homeobox 1; VIP, vasoactive intestinal peptide.
In summary, Lhx1, Six3, Six6, and Vax1 are required for SCN development; however, the severe brain development defects and impact on GnRH neuron development in most of these full body knockout mouse models make it challenging to determine how impaired embryonic development of the SCN impacts the neuronal network regulating the LH surge and fertility.
The use of conditional knockout mouse models has proven a valuable tool to start dissecting the role of homeodomain transcription factors in late SCN maturation as well as SCN control of the LH surge and fertility. The summarized findings from the discussed studies can be found in Table 2.
Impact of conditional deletion of Six3, Six6, Vax1, or Lhx1 on the neuronal circuit regulating the LH surge
| Model | Cre expression | SCN | GnRH neurons | Female fertility | Reference |
|---|---|---|---|---|---|
| Six3fl/fl:Syncre | Synapsincre expression starts ~e16-P10. Synapsincre targets most mature neurons, but a few GnRH neurons | Impaired SCN output (shorter or longer Tau). Intact SCN morphology, weak circadian function, normal AVP and VIP expression | 100% (mRNA) | Subfertile-infertile. | (39, 80) |
| Loss of OVX + E induced LH surge | |||||
| Six3fl/fl:Nestincre | Nestincre expression targets nervous tissue from e11 | Loss of SCN morphology, loss of AVP, and Rorα (at e18). | N/A | N/A | (72) |
| Vax1fl/fl:Syncre | Synapsincre expression starts ~e16-P10. Synapsincre targets most mature neurons, but a few GnRH neurons | Impaired SCN output. Intact SCN morphology, weak circadian function, reduced AVP and VIP expression. | 2.5-fold increase | Subfertile-infertile | (39) |
| Loss of OVX + E induced LH surge | |||||
| Vax1fl/fl:Vipcre | Vipcre expression starts ~P0 | SCN morphology intact, reduced VIP, shorter free- running period, shorter SCN period (Per2::LUC). | N/A | Fertile | (83) |
| Lhx1fl/fl:Six3cre | Six3cre expression starts ~e9 | Intact SCN morphology. | N/A | N/A | (12) |
| Targets retina, optic nerve, optic stalk, and ventral forebrain | Reduced Vip, Grp, Avp, and Avpr1a, normal clock gene expression. Weakened SCN output. | ||||
| Lhx1fl/fl:Rorα cre | Rorα-cre onset ~e14.5 onward, primarily in eye and SCN | Intact SCN morphology. Reduced Vip and Avpr1a, normal clock gene expression. Weakened SCN output. | N/A | N/A | (72, 74) |
| Model | Cre expression | SCN | GnRH neurons | Female fertility | Reference |
|---|---|---|---|---|---|
| Six3fl/fl:Syncre | Synapsincre expression starts ~e16-P10. Synapsincre targets most mature neurons, but a few GnRH neurons | Impaired SCN output (shorter or longer Tau). Intact SCN morphology, weak circadian function, normal AVP and VIP expression | 100% (mRNA) | Subfertile-infertile. | (39, 80) |
| Loss of OVX + E induced LH surge | |||||
| Six3fl/fl:Nestincre | Nestincre expression targets nervous tissue from e11 | Loss of SCN morphology, loss of AVP, and Rorα (at e18). | N/A | N/A | (72) |
| Vax1fl/fl:Syncre | Synapsincre expression starts ~e16-P10. Synapsincre targets most mature neurons, but a few GnRH neurons | Impaired SCN output. Intact SCN morphology, weak circadian function, reduced AVP and VIP expression. | 2.5-fold increase | Subfertile-infertile | (39) |
| Loss of OVX + E induced LH surge | |||||
| Vax1fl/fl:Vipcre | Vipcre expression starts ~P0 | SCN morphology intact, reduced VIP, shorter free- running period, shorter SCN period (Per2::LUC). | N/A | Fertile | (83) |
| Lhx1fl/fl:Six3cre | Six3cre expression starts ~e9 | Intact SCN morphology. | N/A | N/A | (12) |
| Targets retina, optic nerve, optic stalk, and ventral forebrain | Reduced Vip, Grp, Avp, and Avpr1a, normal clock gene expression. Weakened SCN output. | ||||
| Lhx1fl/fl:Rorα cre | Rorα-cre onset ~e14.5 onward, primarily in eye and SCN | Intact SCN morphology. Reduced Vip and Avpr1a, normal clock gene expression. Weakened SCN output. | N/A | N/A | (72, 74) |
Abbreviations: GnRH, gonadotropin-releasing hormone; N/A, not assessed; OVX+E, ovariectomy + estrogen supplementation (a model to detect the LH surge at activity onset); SCN, suprachiasmatic nucleus.
Impact of conditional deletion of Six3, Six6, Vax1, or Lhx1 on the neuronal circuit regulating the LH surge
| Model | Cre expression | SCN | GnRH neurons | Female fertility | Reference |
|---|---|---|---|---|---|
| Six3fl/fl:Syncre | Synapsincre expression starts ~e16-P10. Synapsincre targets most mature neurons, but a few GnRH neurons | Impaired SCN output (shorter or longer Tau). Intact SCN morphology, weak circadian function, normal AVP and VIP expression | 100% (mRNA) | Subfertile-infertile. | (39, 80) |
| Loss of OVX + E induced LH surge | |||||
| Six3fl/fl:Nestincre | Nestincre expression targets nervous tissue from e11 | Loss of SCN morphology, loss of AVP, and Rorα (at e18). | N/A | N/A | (72) |
| Vax1fl/fl:Syncre | Synapsincre expression starts ~e16-P10. Synapsincre targets most mature neurons, but a few GnRH neurons | Impaired SCN output. Intact SCN morphology, weak circadian function, reduced AVP and VIP expression. | 2.5-fold increase | Subfertile-infertile | (39) |
| Loss of OVX + E induced LH surge | |||||
| Vax1fl/fl:Vipcre | Vipcre expression starts ~P0 | SCN morphology intact, reduced VIP, shorter free- running period, shorter SCN period (Per2::LUC). | N/A | Fertile | (83) |
| Lhx1fl/fl:Six3cre | Six3cre expression starts ~e9 | Intact SCN morphology. | N/A | N/A | (12) |
| Targets retina, optic nerve, optic stalk, and ventral forebrain | Reduced Vip, Grp, Avp, and Avpr1a, normal clock gene expression. Weakened SCN output. | ||||
| Lhx1fl/fl:Rorα cre | Rorα-cre onset ~e14.5 onward, primarily in eye and SCN | Intact SCN morphology. Reduced Vip and Avpr1a, normal clock gene expression. Weakened SCN output. | N/A | N/A | (72, 74) |
| Model | Cre expression | SCN | GnRH neurons | Female fertility | Reference |
|---|---|---|---|---|---|
| Six3fl/fl:Syncre | Synapsincre expression starts ~e16-P10. Synapsincre targets most mature neurons, but a few GnRH neurons | Impaired SCN output (shorter or longer Tau). Intact SCN morphology, weak circadian function, normal AVP and VIP expression | 100% (mRNA) | Subfertile-infertile. | (39, 80) |
| Loss of OVX + E induced LH surge | |||||
| Six3fl/fl:Nestincre | Nestincre expression targets nervous tissue from e11 | Loss of SCN morphology, loss of AVP, and Rorα (at e18). | N/A | N/A | (72) |
| Vax1fl/fl:Syncre | Synapsincre expression starts ~e16-P10. Synapsincre targets most mature neurons, but a few GnRH neurons | Impaired SCN output. Intact SCN morphology, weak circadian function, reduced AVP and VIP expression. | 2.5-fold increase | Subfertile-infertile | (39) |
| Loss of OVX + E induced LH surge | |||||
| Vax1fl/fl:Vipcre | Vipcre expression starts ~P0 | SCN morphology intact, reduced VIP, shorter free- running period, shorter SCN period (Per2::LUC). | N/A | Fertile | (83) |
| Lhx1fl/fl:Six3cre | Six3cre expression starts ~e9 | Intact SCN morphology. | N/A | N/A | (12) |
| Targets retina, optic nerve, optic stalk, and ventral forebrain | Reduced Vip, Grp, Avp, and Avpr1a, normal clock gene expression. Weakened SCN output. | ||||
| Lhx1fl/fl:Rorα cre | Rorα-cre onset ~e14.5 onward, primarily in eye and SCN | Intact SCN morphology. Reduced Vip and Avpr1a, normal clock gene expression. Weakened SCN output. | N/A | N/A | (72, 74) |
Abbreviations: GnRH, gonadotropin-releasing hormone; N/A, not assessed; OVX+E, ovariectomy + estrogen supplementation (a model to detect the LH surge at activity onset); SCN, suprachiasmatic nucleus.
SIX3
It appears that SIX3 plays distinct roles during SCN development vs in post-proliferating SCN neurons. During development, SIX3 is required for SCN formation, whereas after birth, SIX3 is a regulator of molecular clock function, SCN output, and required for the correct timing of the LH surge. Six3 was first shown to be involved in SCN maturation, where early developmental loss of Six3 (~e11) was associated with lack of Avp and Rorα gene expression (72), whereas deletion of Six3 in post-proliferative SCN neurons using the Synapsincre allele (Cre expression varies with neuronal subtype but happens around ~e17) did not impact VIP or AVP expression in the SCN (39, 80). Six3fl/fl:Synapsincre did not present with an LH surge at activity onset, causing substantially decreased fertility in females. The mechanism by which SIX3 impacted SCN control of the LH surge in post-proliferative neurons was found to potentially be through weakened SCN timekeeping mechanism, by the loss of SIX3 regulation of the molecular clock gene Per2 and Bmal1 (39, 80). To further support that the loss of the LH surge was caused by a deregulation of circadian timekeeping within the SCN, and not due to a disruption of the neuroendocrine circuit regulating the LH surge, we showed that these mice presented with normal Gnrh expression levels in the hypothalamus and intact sensitivity of GnRH neurons to kisspeptin, as well as gonadotrope sensitivity to GnRH, along with relatively normal basal levels in LH, FSH, and sex steroids (39).
SIX6
During development, SIX6 is required for SCN formation and fertility (15), although the infertility of SIX6KO most likely is caused by a loss of GnRH neurons (16). It is less clear if SIX6 is required for SCN maturation and the control of the LH surge. The best evidence we currently have as to a potential role of SIX6 in late SCN maturation is a study showing that a Cre-allele, which partially targets the late developing SCN, disrupts SCN morphology and weakens SCN output. However, as the Cre-allele used also caused loss of GnRH neurons and hypogonadism, this study was unable to determine if SIX6 in the maturing SCN regulates the LH surge (Table 2) (75). Thus, it remains unknown if SIX6 in one or more mature SCN neuron populations is involved in regulating the LH surge.
VAX1
The most recent homeodomain transcription factor identified to be required for SCN development is VAX1 (75). In contrast to its broad expression in the eye and ventral forebrain during development (81), in the adult brain, Vax1 expression is highly enriched in the SCN, and only expressed in a small additional number of neurons in the adult brain outside of the SCN (39, 75, 82). After neuronal proliferation, VAX1 maintains a role in defining SCN peptide expression, where deletion of VAX1 in post-proliferative SCN neurons, using the Synapsincre allele, was associated with reduced SCN VIP expression, and reduced AVP neuron numbers (39). Although both AVP and VIP regulate circadian timekeeping in the SCN and their downregulation likely contributes to the shorter SCN period in Vax1fl/fl:Synapsincre mice, the additional role of VAX1 as a regulator of the clock gene Per2 makes it difficult to identify the specific origin of the shorter period of the SCN in this mouse model (Table 2). That said, the weakened SCN function in Vax1fl/fl:Synapsincre females impaired the LH surge at activity onset (using the ovariectomy + high estradiol model). The loss of the LH surge was not caused by loss of GnRH neurons or impaired pituitary function, and the HPG axis was functioning as evidenced by normal basal hormone levels and intact gonads. The requirement of VAX1 in post-proliferative SCN neurons in circadian timekeeping function is further supported by a recent preliminary study, showing that conditional deletion of Vax1 in Vipcre neurons shortens SCN period (39, 83). However, in contrast to the Vax1fl/fl:Synapsincre, a Cre-allele that targets both VIP and AVP neurons and which caused severe female subfertility and loss of the LH surge (39), the Vax1fl/fl:Vipcre females did not have impaired fertility. This suggest that loss of VAX1 in both AVP and VIP (and potentially other SCN neuron populations), is required to disrupt SCN function sufficiently to disrupt the LH surge and female fertility.
LIM homeodomain factor 1
Developmental loss of LIM homeodomain factor 1 (Lhx1) does not disrupt SCN neuron specification or SCN morphology but is associated with reduced Vip and Avpr1a (AVP receptor 1a) expression (12). Although no studies have deleted Lhx1 after SCN formation, SCN morphology is intact in mice with conditional knockout of Lhx1 in the ventral forebrain and neuroepithelium giving rise to the SCN at both ~e9 and ~e14.5 (Table 2). This indicates that LHX1 is not required for SCN neuron proliferation but is important in peptide expression and SCN neuron communication. In contrast to both Six3 and Vax1, Lhx1 primarily impacts neuropeptide and receptor expression in the SCN and does not appear to directly regulate clock gene expression. The reproductive phenotype of this mouse strain was not reported, but would be of interest to study, to understand how the LH surge might be impacted in this mouse model.
Role of Clock Genes in the Neuronal Network Regulating the LH Surge
Circadian rhythms are regulated by a molecular transcription-translation feedback loop that takes approximately 24 hours to complete. Bmal1 and Clock dimerize and promote transcription through binding to E-boxes. In addition to regulating transcription of several clock-controlled genes, BMAL1 and CLOCK promote Cryptochrome (CRY) 1 and 2, and Period (PER) 1, 2, and 3 transcription. Per and Cry are transcribed and dimerize in the cytoplasm, accumulating and eventually returning to the nucleus to bind the BMAL1/CLOCK dimer and inhibit their own transcription. PER and CRY eventually degrade, and BMAL1/CLOCK resumes transcription. Many more genes are involved in the molecular clock, which is reviewed in more detail (84). Only 2 mutated alleles are sufficient alone to cause circadian arrhythmicity: Clock mutant and Bmal1 knockout (Table 3).
Impact of circadian gene deletion on the LH surge and female fertility
| Model | Female fertility | LH surge | Reference |
|---|---|---|---|
| Clock mutant | Subfertile | Not detected (OVX + E or endogenous) | (85, 86) |
| Bmal1 KO | Infertile | Not detected (OVX + E or endogenous) | (17, 87, 88) |
| Bmal1flox/flox:GnRHcre | Fertile | Normal/mistimed (OVX + E) | (88, 89) |
| Bmal1flox/flox:Kisscre | Fertile | Normal/mistimed (OVX + E) | (88, 89) |
| Bmal1flox/flox:Avpcre | Fertile | Normal/mistimed (OVX + E) | (88, 89) |
| Bmal1flox/flox:Vipcre | Fertile | Normal (OVX + E) | (88) |
| Bmal1flox/flox:Vipcre + Avpcre | Fertile | Normal (OVX + E) | (88) |
| Model | Female fertility | LH surge | Reference |
|---|---|---|---|
| Clock mutant | Subfertile | Not detected (OVX + E or endogenous) | (85, 86) |
| Bmal1 KO | Infertile | Not detected (OVX + E or endogenous) | (17, 87, 88) |
| Bmal1flox/flox:GnRHcre | Fertile | Normal/mistimed (OVX + E) | (88, 89) |
| Bmal1flox/flox:Kisscre | Fertile | Normal/mistimed (OVX + E) | (88, 89) |
| Bmal1flox/flox:Avpcre | Fertile | Normal/mistimed (OVX + E) | (88, 89) |
| Bmal1flox/flox:Vipcre | Fertile | Normal (OVX + E) | (88) |
| Bmal1flox/flox:Vipcre + Avpcre | Fertile | Normal (OVX + E) | (88) |
Impact of circadian gene deletion on the LH surge and female fertility
| Model | Female fertility | LH surge | Reference |
|---|---|---|---|
| Clock mutant | Subfertile | Not detected (OVX + E or endogenous) | (85, 86) |
| Bmal1 KO | Infertile | Not detected (OVX + E or endogenous) | (17, 87, 88) |
| Bmal1flox/flox:GnRHcre | Fertile | Normal/mistimed (OVX + E) | (88, 89) |
| Bmal1flox/flox:Kisscre | Fertile | Normal/mistimed (OVX + E) | (88, 89) |
| Bmal1flox/flox:Avpcre | Fertile | Normal/mistimed (OVX + E) | (88, 89) |
| Bmal1flox/flox:Vipcre | Fertile | Normal (OVX + E) | (88) |
| Bmal1flox/flox:Vipcre + Avpcre | Fertile | Normal (OVX + E) | (88) |
| Model | Female fertility | LH surge | Reference |
|---|---|---|---|
| Clock mutant | Subfertile | Not detected (OVX + E or endogenous) | (85, 86) |
| Bmal1 KO | Infertile | Not detected (OVX + E or endogenous) | (17, 87, 88) |
| Bmal1flox/flox:GnRHcre | Fertile | Normal/mistimed (OVX + E) | (88, 89) |
| Bmal1flox/flox:Kisscre | Fertile | Normal/mistimed (OVX + E) | (88, 89) |
| Bmal1flox/flox:Avpcre | Fertile | Normal/mistimed (OVX + E) | (88, 89) |
| Bmal1flox/flox:Vipcre | Fertile | Normal (OVX + E) | (88) |
| Bmal1flox/flox:Vipcre + Avpcre | Fertile | Normal (OVX + E) | (88) |
Clock mutants
Clock mutants (also referred to as ClockΔ19) have a 51-bp deletion in the transcriptional activation domain of CLOCK, causing CLOCK to act as a dominant-negative transcription factor (90). Female Clock mutants are subfertile and have disrupted estrous cyclicity (85, 86, 91). Serial bleeds of Clock female mice do not show an LH surge on the day of proestrus when measured hourly for 16 hours (86), but they also show low estradiol levels, which can impair the LH surge on its own. When the level of estradiol is ameliorated in an ovariectomy + estradiol benzoate treatment paradigm, Clock mutants are still unable to produce an LH surge around lights off, supporting the loss of the LH surge in this model is likely driven by changes within the hypothalamic circuit timing the surge. Light conditions may also play a role in the reduced fertility of Clock mutants, as the behavioral phenotype is more pronounced in constant dark (DD); however, Clock females did not show a difference from WT animals in the amount of time pregnant in constant darkness (85). Avp mRNA is low and nonrhythmic in Clock mutants, and intracerebroventricular administration of AVP in the morning of proestrus rescues the LH surge in approximately half of these mice (45).
Bmal1
The Bmal1 knockout mouse is infertile (17, 19). Male mice have mild HPG axis impairments but are infertile because they do not engage in mating behavior; this is likely due to neural circuitry defects in transmitting the olfactory cues to mating behavior (92). In female Bmal1 knockout mice, endogenous or induced LH surges have not been detected (87, 88); however, like the Clock mutants, they still ovulate. The infertility is proposed to be due to insufficient progesterone synthesis by the ovary during early embryonic development (18), although ovaries transplanted from infertile steroidogenic cell-specific Bmal1 knockout mice are able to produce pups when transplanted into a wild-type female (93). Bmal1 knockout mice have additional neuroendocrine disruptions, including prolonged estrous cycles and elevated LH response to exogenous kisspeptin administration. In a tamoxifen-inducible Bmal1 knockout model, in which mice are allowed to develop normally with Bmal1, the induced Bmal1 knockout mice are subfertile but able to produce pups (~30%); the interpretation of this study is confounded by the tamoxifen treatment, and the LH surge was not examined (94). Conditional knockout of Bmal1 in the myometrium (18) or steroidogenic cells of the ovary (93), but not pituitary gonadotropes (87), results in mistimed birth and impaired fertility, respectively, demonstrating the power of these endogenous clocks in female fertility.
The molecular clock in GnRH and kisspeptin neurons
The importance of the endogenous molecular clock in generating the preovulatory LH surge is demonstrated by the absence of an appropriately timed surge in the Clock mutants and Bmal1 knockouts. However, the cell population(s) in which the clock is acting is unknown. The endogenous circadian clock has been shown to regulate GnRH pulses, Kiss1R expression, and activation by VIP in vitro in the immortalized GnRH neurons, GT1-7 cells (65, 91, 95). AVPV kisspeptin neurons demonstrate a molecular circadian clock, and temporal regulation of Kiss1 and the vasopressin receptor V1a (58, 96). However, loss of Bmal1 in either GnRH or Kisspeptin neurons does not alter fertility (88, 89). We found that Bmal1flox/floxGnRHcre and Bmal1flox/flox:Kisscre mice had normal estrous cycles, ovarian morphology, kisspeptin responsiveness, and demonstrated an LH surge at the time of lights off (88). Using a similar model with serial blood sampling, Bittman observed that both Bmal1flox/flox:GnRHcre and Bmal1flox/flox:Kisscre failed to show a united LH surge over a 5-hour time window (89). The sources of this variation may be due to differences in mouse strains (C57BL6 vs C57BL6/CBA), LH assay methods, and/or sampling strategy. If there are changes to the timing of the LH surge in these mice, they are insufficient to alter overall reproductive success.
AVP and VIP neurons and the LH surge
Bmal1 may also be exerting its effects on the LH surge in the SCN, either through transcriptional regulation of signaling peptides or the timing of an activating signal. Loss of Bmal1 in AVP neurons (Bmal1flox/flox:Avpcre) is sufficient to alter locomotor rhythms (97). We have found that these mice are fertile and have an appropriately timed LH surge (98), findings which were partially confirmed by Bittman, who, although using a different Avpcre allele to delete Bmal1flox/flox, confirmed Bmal1flox/flox:Avpcre have an LH surge; however, Bittman found it to be mistimed (89). Similarly, loss of Bmal1 in VIP neurons (Bmal1flox/flox:Vipcre), or mice with both the Avpcre and Vipcre alleles (Bmal1flox/flox:Avpcre + Vipcre:) had no effect on the timing of the LH surge (using the ovariectomy + estradiol paradigm) or female fertility (98). While Bmal1 knockout mice fail to show a surge in normal light-dark cycles, it may be that these conditional knockouts are more functional in normal light/dark conditions and would require a circadian stressor to show a more robust phenotype. Alternative, there may be compensation in different parts of the circuit, or incomplete knockdown of Bmal1flox/flox using these Cre alleles, which could allow sufficient BMAL1 to maintain the timing cues driving the LH surge and fertility.
Relevance to Human Reproduction: Evidence Supporting the Conservation of the Neuronal Network Relaying Light Information to the Reproductive Axis and Its Role in Light-Induced Disruption of Fertility
Because the LH surge is gated by the SCN, the impact of circadian disruption, which reduces SCN timekeeping function, markedly impacts female, but not male fertility. Of importance in today’s society is the great sensitivity of the neuroendocrine system regulating the LH surge to light-induced circadian disruption, which occurs in response to repeated light at night, and irregular sleep-wake cycles, as previously reviewed (99, 100). Today, it is estimated that 50 to 70 million Americans suffer from disrupted circadian rhythms. Circadian disruption not only impacts female fertility, but it is also associated with a large number of serious medical illnesses in men and women, including increased risk of cardiovascular disease, obesity, and decreased cognitive capacities. Further supporting an important modulatory role of properly timed light in fertility are smaller human studies using light therapy, where therapeutical light treatments were primarily used to modulate mood and counteract seasonal depression. Specifically, light therapy impacts fertility through increased LH and FSH (101, 102). Appropriately timed light therapy-induced changes in LH and FSH can also improve ovulation and menstrual cycles in women (103-105). Furthermore, a case study of a menopausal woman experiencing seasonal depression found that her seasonal depression was accompanied by changes in FSH and estradiol, causing seasonal hot flashes. These studies support that the reduced fertility of women having frequent shiftwork schedules is, at least partially, driven by a direct regulation by light of LH and FSH, and is not exclusively the results of weakened circadian rhythms, and desynchrony of tissue function to the time of day.
Conclusions
Using primarily rodent models, it has become clear that proper SCN function is required to gate the LH surge. Recent and ongoing work is focused on identifying the neuronal subpopulations within the SCN that regulate the LH surge, where VIP and AVP neurons are currently the strongest candidates. Due to the strong buffer effect and capacity to compensate for loss of function of subpopulations of neurons of the SCN, the true contribution of each neuronal population is likely obfuscated. Despite this, it is clear that the SCN does provide a timing signal to promote the LH surge, and that this signal is transmitted through neuronal projections originating in the SCN onto neurons of the reproductive axis. The neuronal circuit relaying light information to the reproductive axis appears to be conserved in humans, where light therapy can promote LH and FSH release. This effect of light on LH and FSH release provides a mechanism to explain how shiftwork and extensive light at night negatively impact menstrual cycles and women’s fertility. Future research focused on how light can be used to improve fertility would be of interest. Indeed, there is evidence that light (day length) impacts artificial reproductive success rates (106, 107). Understanding how day length and shiftwork impact the success of in vitro fertilization would be of great interest, as in vitro fertilization rates currently have a modest success rate.
Abbreviations
- AVP
arginine vasopressin
- AVPV
anteroventral periventricular nucleus
- e#
embryonic day
- FSH
follicle-stimulating hormone
- GnRH
gonadotropin-releasing hormone
- KO
knockout
- LH
luteinizing hormone
- NMS
neuromedin S
- P#
postnatal day
- PK2
prokineticin 2
- SCN
suprachiasmatic nucleus
- SIX
SIX homeobox
- VAX1
ventral anterior homeobox 1
- VIP
vasoactive intestinal peptide
Financial Support
This work was supported by NIH grants R01 HD082567, R01 HD100580, R01 HD072754, and P50 HD012303 (to P.L.M.). P.L.M. was partially supported by P30 DK063491, P42 ES010337, and P30 CA023100. K.J.T. was partially supported by T32 HD007203, F32 HD090837, and P42 ES010337. H.M.H. was partially supported by R00 HD084759.
Disclosures
The authors have nothing to disclose.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
Morris EL, Patton AP, Chesham JE, Crisp A, Adamson A, Hastings MH. Single-cell transcriptomics of supraciasmatic nuclei reveal a Prokineticin-driven circadian network. The EMBO Journal. 2021;40:e108614.
Meadows JD, Breuer JA, Lavalle SN, et al. Deletion of Six3 in post-proliferative neurons produces weakened SCN circadian output, improved metabolic function, and dwarfism in male mice. Mol Metab. 2021;101431.

{kind=link}