





Abstract
Bisphenol A (BPA) is an estrogenic endocrine-disrupting chemical (EDC) that has a range of potential adverse health effects. Previously we showed that acute exposure to BPA promoted arrhythmias in female rat hearts through estrogen receptor rapid signaling. Progesterone (P4) and estrogen have antagonistic or complementary actions in a number of tissues and systems. In the current study, we examined the influence and possible protective effect of P4 on the rapid cardiac actions of BPA in female rat cardiac myocytes. Preincubation with physiological concentration (1 nM) of P4 abolished BPA-induced triggered activities in female cardiac myocytes. Further, P4 abrogated BPA-induced alterations in Ca2+ handling, including elevated sarcoplasmic reticulum Ca2+ leak and Ca2+ load. Key to the inhibitory effect of P4 is its blockade of BPA-induced increase in the phosphorylation of phospholamban. At myocyte and protein levels, these inhibitory actions of P4 were blocked by pretreatment with the nuclear P4 receptor (nPR) antagonist RU486. Analysis using membrane-impermeable bovine serum albumin–conjugated P4 suggested that the actions of P4 were mediated by membrane-initiated signaling. Inhibitory G (Gi) protein and phophoinositide-3 kinase (PI3K), but not tyrosine protein kinase activation, were involved in the observed effects of P4. In conclusion, P4 exerts an acute protective effect against BPA-induced arrhythmogenesis in female cardiac myocytes through nPR and the Gi/PI3K signaling pathway. Our findings highlight the importance of considering the impact of EDCs in the context of native hormonals and may provide potential therapeutic strategies for protection against the cardiac toxicities associated with BPA exposure.
Bisphenol A (BPA) is one of the most produced chemicals worldwide and is a common estrogenic endocrine-disrupting chemical (EDC). It is widely used in the production of epoxy resins and polycarbonate plastics and is pervasive in the environment. Humans are almost constantly exposed to BPA through diet, dermal contact, water, air, and dust (1). BPA has been detected in a number of human fluids and tissues, including urine, blood, maternal breast milk, fetal plasma, and placenta (2, 3).
BPA exposure has been linked to a wide range of adverse human health outcomes, such as cancer, obesity, diabetes, and reproductive and immune systems abnormalities (4, 5). In addition, growing evidence suggests that BPA exposure has potential toxicity on the cardiovascular (CV) system. Multiple epidemiological studies have shown that higher BPA exposure is associated with CV diagnoses or CV disease risk factors in humans (6–9). Experimental studies have shown that BPA exposure results in cardiac remodeling, alteration of cardiac function, oxidative stress, and altered autonomic regulation in a sex-specific manner in rodents (10–12). Studies in our laboratory have demonstrated that rapid BPA exposure has arrhythmogenic effects in female rat hearts, which are manifested as increased ventricular arrhythmias under pathophysiological conditions such as stress or ischemic injury (13, 14). The proarrhythmic actions of BPA are mediated by estrogen receptor signaling and alterations of myocyte Ca2+ handling through rapid phosphorylation of key Ca2+ handling proteins (15).
To better understand the biological and toxic impact of EDCs and the responsiveness of biological systems to such an impact, it is increasingly recognized that the effects of EDCs should be assessed not in isolation but in the context of native steroid hormones and other coexisting EDCs. Native steroid hormones can influence the actions of exogenous EDCs, including BPA. For instance, 17β-estradiol (E2) has been shown to either augment or alleviate the impact of BPA in a variety of cell types or systems (16–19). We previously showed that in rat female hearts the arrhythmogenic effect of BPA was particularly pronounced in the presence of physiological level of E2 (13).
Progesterone (P4) is a key ovarian hormone that has well-known interactions with estrogens. However, the possible influence of P4 on the actions of BPA, an estrogenic chemical, is unknown. P4 and native estrogens interact either synergistically or antagonistically to regulate diverse reproductive events such as embryo implantation, mammary gland development, ovulation, and epithelial cell proliferation/regression (20–22). Interactions between estrogens and P4 are also common in other physiological systems. For example, in the CV system, P4 attenuates E2-mediated vasoprotective effects on endothelial function and the protective effects of E2 against pathological conditions, such as inflammation and atherosclerosis (23–26). P4 and E2 also have counterbalancing effects on ventricular repolarization in postmenopausal women (27, 28). P4 has been shown to abolish E2-induced long QT–associated arrhythmias in a computational and genetic long QT rabbit model (29, 30). In addition, recent clinical and experimental evidence indicates that the neuroprotective actions of E2 can be either attenuated or augmented by coadministration with P4 (31–34). Based on the interactions between P4 and estrogens, we hypothesized that P4 may influence, and even counterbalance, the proarrhythmic effects of BPA in cardiac myocytes.
P4 is known to exert its biological effects through binding to the nuclear P4 receptors (nPRs), which function as ligand-regulated transcriptional factors to regulate gene expression. In addition, a nongenomic mechanism is well recognized. Through this mechanism, P4 can elicit rapid effects by the activation of membrane-initiated intracellular signaling pathways. There is growing evidence that nongenomic actions contribute to a range of CV effects of P4. For example, acute P4 administration results in rapid declines in blood pressure and vascular tone in animal models (35, 36). P4 exerts rapid cardioprotective effects against ischemia/reperfusion injury in female, but not male, rats, reducing both infarction size and incidence of ventricular arrhythmias (37). Moreover, physiological levels of P4 have been shown to protect hearts against isoproterenol-induced cardiac arrhythmias in long-QT models (models of a type of congenital arrhythmia disease associated with abnormal ventricular repolarization) via a nongenomic pathway (38).
In the current study, we examined the acute influence of P4 on BPA-induced arrhythmogenesis in female rat cardiac myocytes and elucidated their underlying mechanisms.
Materials and Methods
Reagents
All reagents and solvents used were of the highest purity available. All aqueous solutions were prepared using BPA-free water (18 MΩ, <6 parts per billion total oxidizable organics) (Millipore A10 System; Millipore Corp., Milford, MA). BPA (CAS 80-05-7, lot 111909; TCI America, Portland, OR) (ground by Battelle, Columbus, OH) was provided by the Division of the National Toxicology Program at the National Institute of Environmental Health Sciences. P4-bovine serum albumin (BSA) conjugate (catalog no. 7720-0750, batch number 101014) was from AbD Serotec (Raleigh, NC). Mifepristone (RU486) (CAS 84371-65-3, batch number 1B/157396) and pertussis toxin (PTX) (CAS 70323-44-3, batch number 12A) were from Tocris Bioscience (Ellisville, MO). P4 (CAS 57-83-0, lot SLBC2633V), 4-amino-3-(4-chlorophenyl)-1-(t-butyl)-1H-pyrazolo [3,4-d] pyrimidine (PP2) (high-performance liquid chromatography ≥98%; catalog no. P0042, lot 112M4626V), and wortmannin (high-performance liquid chromatography ≥98%; CAS 19545-26-7) were from Sigma-Aldrich (St. Louis, MO). Other chemicals were from Sigma-Aldrich unless otherwise stated (Table 1).
List of Antibodies
| Peptide/Protein Target | Name of Antibody | Manufacturer, Catalog #, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|
| Phosphorylated serine 2808 of ryanodine receptor | RYR2 phospho serine 2808 antiserum | Badrilla, A010-30 | Rabbit; polyclonal | 1:5000 |
| Ryanodine receptor | Anti-RYR antibody | Thermo Fisher Scientific, MA3-916 | Mouse; nonoclonal | 1:1000 |
| Phosphorylated threonine 17 of phospholamban | Phospholamban phospho threonine-17 antiserum | Badrilla, A010-13 | Rabbit; polyclonal | 1:5000 |
| Phosphorylated serine 16 of phospholamban | Phospholamban phospho serine-16 antiserum | Badrilla, A010-12 | Rabbit; polyclonal | 1:5000 |
| Phospholamban | Anti-phospholamban A1 antibody | Badrilla, A010-14 | Mouse; monoclonal | 1:1000 |
| Phosphorylated serine 473 of Akt | Phospho-Akt (Ser473) antibody | Cell Signaling, 9271 | Rabbit; polyclonal | 1:1000 |
| Akt | Anti-Akt antibody | Cell Signaling, 9272 | Rabbit; polyclonal | 1:1000 |
| Rabbit IgG | IRDye 800CW goat anti-rabbit antibody | LI-COR, 926-32211 | Goat; polyclonal | 1:5000 |
| Mouse IgG | IRDye 680RD goat anti-mouse antibody | LI-COR, 926-68070 | Goat; polyclonal | 1:5000 |
| Peptide/Protein Target | Name of Antibody | Manufacturer, Catalog #, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|
| Phosphorylated serine 2808 of ryanodine receptor | RYR2 phospho serine 2808 antiserum | Badrilla, A010-30 | Rabbit; polyclonal | 1:5000 |
| Ryanodine receptor | Anti-RYR antibody | Thermo Fisher Scientific, MA3-916 | Mouse; nonoclonal | 1:1000 |
| Phosphorylated threonine 17 of phospholamban | Phospholamban phospho threonine-17 antiserum | Badrilla, A010-13 | Rabbit; polyclonal | 1:5000 |
| Phosphorylated serine 16 of phospholamban | Phospholamban phospho serine-16 antiserum | Badrilla, A010-12 | Rabbit; polyclonal | 1:5000 |
| Phospholamban | Anti-phospholamban A1 antibody | Badrilla, A010-14 | Mouse; monoclonal | 1:1000 |
| Phosphorylated serine 473 of Akt | Phospho-Akt (Ser473) antibody | Cell Signaling, 9271 | Rabbit; polyclonal | 1:1000 |
| Akt | Anti-Akt antibody | Cell Signaling, 9272 | Rabbit; polyclonal | 1:1000 |
| Rabbit IgG | IRDye 800CW goat anti-rabbit antibody | LI-COR, 926-32211 | Goat; polyclonal | 1:5000 |
| Mouse IgG | IRDye 680RD goat anti-mouse antibody | LI-COR, 926-68070 | Goat; polyclonal | 1:5000 |
List of Antibodies
| Peptide/Protein Target | Name of Antibody | Manufacturer, Catalog #, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|
| Phosphorylated serine 2808 of ryanodine receptor | RYR2 phospho serine 2808 antiserum | Badrilla, A010-30 | Rabbit; polyclonal | 1:5000 |
| Ryanodine receptor | Anti-RYR antibody | Thermo Fisher Scientific, MA3-916 | Mouse; nonoclonal | 1:1000 |
| Phosphorylated threonine 17 of phospholamban | Phospholamban phospho threonine-17 antiserum | Badrilla, A010-13 | Rabbit; polyclonal | 1:5000 |
| Phosphorylated serine 16 of phospholamban | Phospholamban phospho serine-16 antiserum | Badrilla, A010-12 | Rabbit; polyclonal | 1:5000 |
| Phospholamban | Anti-phospholamban A1 antibody | Badrilla, A010-14 | Mouse; monoclonal | 1:1000 |
| Phosphorylated serine 473 of Akt | Phospho-Akt (Ser473) antibody | Cell Signaling, 9271 | Rabbit; polyclonal | 1:1000 |
| Akt | Anti-Akt antibody | Cell Signaling, 9272 | Rabbit; polyclonal | 1:1000 |
| Rabbit IgG | IRDye 800CW goat anti-rabbit antibody | LI-COR, 926-32211 | Goat; polyclonal | 1:5000 |
| Mouse IgG | IRDye 680RD goat anti-mouse antibody | LI-COR, 926-68070 | Goat; polyclonal | 1:5000 |
| Peptide/Protein Target | Name of Antibody | Manufacturer, Catalog #, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|
| Phosphorylated serine 2808 of ryanodine receptor | RYR2 phospho serine 2808 antiserum | Badrilla, A010-30 | Rabbit; polyclonal | 1:5000 |
| Ryanodine receptor | Anti-RYR antibody | Thermo Fisher Scientific, MA3-916 | Mouse; nonoclonal | 1:1000 |
| Phosphorylated threonine 17 of phospholamban | Phospholamban phospho threonine-17 antiserum | Badrilla, A010-13 | Rabbit; polyclonal | 1:5000 |
| Phosphorylated serine 16 of phospholamban | Phospholamban phospho serine-16 antiserum | Badrilla, A010-12 | Rabbit; polyclonal | 1:5000 |
| Phospholamban | Anti-phospholamban A1 antibody | Badrilla, A010-14 | Mouse; monoclonal | 1:1000 |
| Phosphorylated serine 473 of Akt | Phospho-Akt (Ser473) antibody | Cell Signaling, 9271 | Rabbit; polyclonal | 1:1000 |
| Akt | Anti-Akt antibody | Cell Signaling, 9272 | Rabbit; polyclonal | 1:1000 |
| Rabbit IgG | IRDye 800CW goat anti-rabbit antibody | LI-COR, 926-32211 | Goat; polyclonal | 1:5000 |
| Mouse IgG | IRDye 680RD goat anti-mouse antibody | LI-COR, 926-68070 | Goat; polyclonal | 1:5000 |
Animals
All animal procedures were performed as previously described (13) and in accordance with protocols approved by the University of Cincinnati Institutional Animal Care and Use Committee. Adult female Sprague-Dawley rats (200 to 250 g) (Charles River, Wilmington, MA) were used as nonsurviving sources of cardiac tissue. Animals were maintained on a 14-hour light, 10-hour dark cycle in standard polycarbonate caging with Sani-chip bedding (Irradiated Aspen Sani-chip; P. J. Murphy Forest Products Corp., Montville, NJ) to eliminate possible corn-based mycoestrogen exposure. All animals were fed Teklad diet 2020 (Harlan Laboratories Inc., Indianapolis, IN) ad libitum; this feed lacks soybeans meal, alfalfa, or animal products that may introduce uncontrolled levels of estrogenic compounds. Animals were provided with BPA-free drinking water, which was generated by a dedicated water purification system (Millipore Rios 16 with ELIX ultraviolet/Progard 2) that decreases oxidizable organics to <1% of source levels. Drinking water was dispensed from glass water bottles.
Isolation of ventricular myocytes
Ventricular myocytes from female rat hearts were enzymatically dissociated using Langendorff perfusion as previously described (13). Briefly, rats were anesthetized by pentobarbital (80 mg/kg, intraperitoneally), and hearts were quickly removed for myocyte dissociation. The excised hearts were cannulated and perfused with a Tyrode’s solution containing 0.7 mg/mL collagenase type II (Worthington Biochemical, Lakewood, NJ), 0.2 mg/mL hyaluronidase, 0.1% BSA, and 25 μm Ca2+. After digestion, the ventricles were minced, and myocytes were collected and kept in Tyrode’s solution. For immunoblotting, myocytes were plated on laminin-coated (CAS 114956-81-9, various lots; Sigma-Aldrich) dishes and counted.
Myocyte Ca2+ transient and spark analysis
Myocyte Ca2+ transient and Ca2+ spark were analyzed as previously described (13). Briefly, isolated ventricular myocytes were loaded with fluo-4 acetoxymethyl ester (5 μM) (Molecular Probes, Eugene, OR). For Ca2+ after-transients recording, myocytes were paced with field stimulation (Grass S48 stimulator; Grass Instruments, Quincy, MA) with 2-ms 1.5× threshold pulses at a rate of 2 Hz. For Ca2+ sparks measurement, fluorescence signals were measured with line-scan imaging at 3.07-ms intervals, with each line comprising 512 pixels spaced at 0.056 mm. Fluorescence signals from fluo-4–loaded myocytes were imaged for Ca2+ after-transients and Ca2+ sparks using an inverted confocal microscope (Zeiss LSM 710; Carl Zeiss Microscopy, LLC, Thornwood, NY) at an excitation wavelength of 488 nm. Image processing and data analysis were performed using IDL 6.3 software (Exelis Visual Information Solutions, Boulder, CO). Ca2+ spark frequency was calculated as the count of sparks divided by the length of scanning line divided by time of scanning and expressed as number of sparks per 100 μm/s. Ca2+ spark amplitude was calculated as the peak signal intensity of the spark and expressed as peak fluorescence intensity of the spark divided by baseline intensity. For caffeine-induced Ca2+ transient measurement, fluo-4 loaded myocytes were field stimulated at 0.5 Hz for 20 seconds and exposed to solution containing 10 mM caffeine immediately after cessation of pacing. Fluorescence signals were recorded by a microscope (TE 2000; Nikon, Melville, NY) and an InCyt Standard PM photometry system (Intracellular Imaging, Cincinnati, OH). All experiments were performed at room temperature (24°C).
Myocyte after-contraction analysis
Myocyte after-contractions were analyzed as previously described (13). Myocytes were placed in a plexiglass cell chamber filled with Tyrode solution containing 1.0 mM CaCl2 and were paced with field stimulation (Grass S48 stimulator; Grass Instruments, Quincy, MA) at 2 Hz. Myocyte shortening was imaged with a charge-coupled device camera and examined using a video-edge detector (Crescent Electronics, Sandy, UT). Data were sampled through an Axon Digidata 1322A board using the PCLAMP 9 software (both from Molecular Devices, Sunnyvale, CA).
Western blotting analysis
Western blotting experiments were performed as previously described (15). Ventricular myocytes were subjected to different treatments for the indicated length of time, centrifuged, and snap frozen in liquid nitrogen. Each treatment group typically contained about 3 × 105 myocytes. Proteins were extracted with 1x Cell Lysis Buffer (Cell Signaling Technology, Danvers, MA) supplemented with protease inhibitor and phosphatase inhibitors. Equal amounts of samples were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA). The membrane was blocked by Odyssey Blocking Buffer (LI-COR, Lincoln, NE) followed by incubation with primary antibodies and secondary antibodies. The following primary and secondary antibodies were used: phosphorylated ryanodine receptor (pSer2808-RYR), phosphorylated phospholamban (pSer16-PLN and pThr17-PLN), phospholamban (PLN), ryanodine receptor (RyR), and IRDye goat anti-mouse and anti-rabbit secondary antibodies. The Odyssey CLx Imaging System (LI-COR) was used to detect the bands on the membrane. Densitometric analysis was performed using Image J software (National Institutes of Health, Bethesda, MD). Net values for all bands were generated by deducting the inverted background from the inverted band value. To quantify phosphorylation levels, densitometry values for phosphorylated bands were normalized to their corresponding total proteins.
Treatments of myocytes
Hormones and reagents used in various treatments in the study were prepared as stock solutions in dimethyl sulfoxide. Vehicle control solution contained the maximum concentration of dimethyl sulfoxide (<0.1%) contained in any treatment groups. To examine the influence of P4 or P4-BSA on BPA-induced effects, female rat ventricular myocytes were preincubated with 1 nM of P4 or P4-BSA for 10 minutes before exposure to 1 nM BPA for 5 minutes. To define the signaling pathways of the effects of P4, myocytes were preincubated with the nPR antagonist RU486 (1 μM), the inhibitory G (Gi) protein inhibitor PTX (200 ng/mL), the phosphatidylinositol-3 kinase (PI3K) inhibitor wortmannin (1 μM), or the tyrosine protein kinase (cSrc) inhibitor PP2 (1 μM) for 30 minutes before treatment with 1 nM P4 and exposure to BPA.
Statistical analysis
All experiments were independently repeated using myocytes isolated from at least three rat hearts. Statistical analysis was conducted using one-way analysis of variance, with differences between treatment groups assessed using a multiple comparison post-test. The χ2 test was used to analyze the frequency of events (e.g., percentage of myocytes with triggered activities). Minimal level of statistical significance for differences in values is considered to be P < 0.05. Data were analyzed with SigmaPlot 11.0 or Excel and expressed as average ± standard error of the mean.
Results
P4 inhibits BPA-induced triggered activities in female rat cardiac myocytes
Previously we showed that acute exposure to low-dose BPA promoted aberrant spontaneous excitation known as “triggered activities” in female rat myocytes (13). The effect of P4 on BPA-induced triggered activities, measured as spontaneous contractions after repeated pacing, was examined in female rat ventricular myocytes. BPA (1 nM) rapidly increased the percentage of myocytes with triggered activities (5.3% in control versus 27.1% in BPA) [Fig. 1(a)]. Interestingly, preincubation with 1 nM P4 for 10 minutes abolished the effect of BPA, reducing the percentage of myocytes with triggered activities by 1.2%. At a concentration of 1 μM, RU486, an nPR antagonist, blocked the inhibitory action of P4 on BPA-induced triggered activities, whereas either P4 or RU486 alone had no significant effect on triggered activities [Fig. 1(a)]. Similar results were obtained when triggered activities were measured as spontaneous Ca2+ after-transients [Fig. 1(b)]. These results suggest that, when mediated by nPR activation, P4 elicits a rapid inhibition of the proarrhythmic effect of BPA in female rat ventricular myocytes.
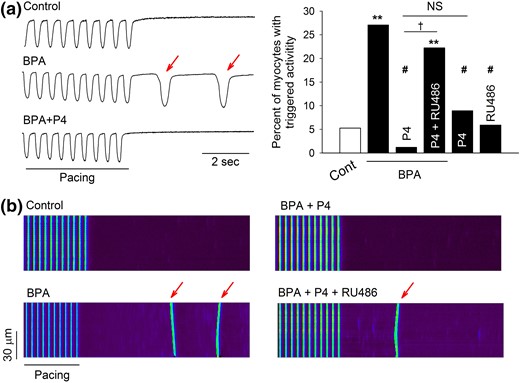
P4 inhibits BPA-induced triggered activities in isolated female rat ventricular myocytes. (a) Left: Representative contraction traces under control (Cont), BPA, and BPA + P4. Red arrows indicate spontaneous after-contraction (i.e., triggered activity) after pacing. Right: Percentages of myocytes with triggered activities under indicated treatments (n = 30 to 50 myocytes for each group from four hearts). (b) Representative confocal images of Ca2+ transients in myocytes elicited by repeated pacing under various treatments. Red arrows indicate spontaneous Ca2+ after-transients (i.e., triggered activity) after pacing. For all experiments, BPA and P4 = 1 nM; RU486 = 1 μM. **P < 0.01; #P > 0.1 versus control; †P < 0.01. NS, not significant (χ2 test).
P4 inhibits BPA-induced alteration of myocyte Ca2+ handling
We previously showed that BPA-induced triggered activities in cardiac cells were mediated by alterations of myocyte Ca2+ handling, including elevated sarcoplasmic reticulum (SR) Ca2+ release and Ca2+ load (13). To investigate the cellular mechanism underlying the rapid inhibitory action of P4, the effects of P4 on BPA-induced alterations in SR Ca2+ handling were assessed in female rat ventricular myocytes. Consistent with our previous study, 1 nM BPA increased spontaneous Ca2+ release, or “leak,” from the SR, as indicated by an increased frequency of Ca2+ sparks in quiescent myocytes [Fig. 2(a) and 2(c)]. This effect of BPA was blocked by P4 [Fig. 2(a) and 2(c)]. BPA markedly increased the amplitude of caffeine-induced Ca2+ transients, an indication of SR Ca2+ load. This stimulatory effect of BPA on SR load was also blocked by 1 nM P4 [Fig. 2(b) and 2(d)]. Consistent with the results in triggered activity, RU486 eliminated the inhibitory effects of P4 in BPA-induced alterations of Ca2+ spark frequency and SR Ca2+ load [Fig. 2(a), 2(c), and 2(d)].
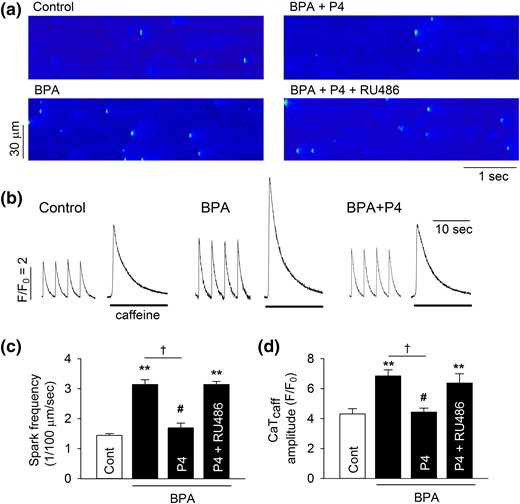
P4 inhibits BPA-induced alterations of Ca2+ handling in female rat ventricular myocytes. (a) Representative images of Ca2+ sparks in quiescent myocytes under control, BPA, BPA + P4, or BPA + P4 + RU486 (1 μM). (b) Representative Ca2+ transients elicited by caffeine after field stimulation under various treatments. The amplitudes of caffeine-induced Ca2+ transients are indications of SR Ca2+ load. (c) Mean Ca2+ sparks frequency in myocytes under various treatments (n = 7 to 9 myocytes from four hearts). (d) Caffeine-induced Ca2+ transients amplitude (CaTcaff) in myocytes under various treatments (n = 8 to 10 myocytes from four hearts). BPA and P4 at 1 nM for all experiments. **P < 0.01; #P > 0.1 versus control; †P < 0.01 (one-way analysis of variance). Error bars are standard error of the mean.
P4 inhibits BPA-induced increase in the phosphorylation of PLN17
We previously showed that BPA altered myocyte Ca2+ handling by increasing the phosphorylation levels of RyR and PLN, two key Ca2+ handling proteins of SR (15). Thus, BPA rapidly increased RyR phosphorylation at the serine 2808 protein kinase A (PKA) site (RyR2808) and increased PLN phosphorylation at the threonine 17 Ca2+/calmodulin-dependent protein kinase II (CAMKII) site (PLN17) (15). P4 treatment completely blocked the BPA-induced increase in PLN17 phosphorylation in female rat myocytes [Fig. 3(a) and 3(b)]. By contrast, P4 did not affect the BPA-induced increase in RyR2808 phosphorylation or alter the phosphorylation level of PLN16 (PKA site) [Fig. 3(a) and 3(b)]. RU486 abolished P4’s effect on BPA-induced PLN17 phosphorylation [Fig. 3(c) and 3(d)]. These results suggest that P4 selectively abolishes BPA-induced increase in PLN17 but not RyR2808 phosphorylation through an nPR-dependent mechanism and thus partially normalizes myocyte Ca2+ handling.
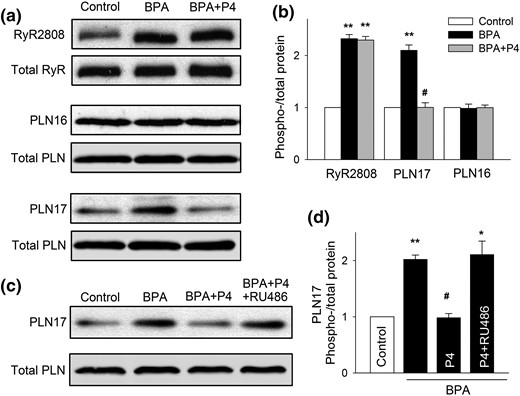
P4 abolishes BPA-induced increase in the phosphorylation of PLN17 in female rat ventricular myocytes. (a and b) Representative immunoblot and quantification of phosphorylated RyR at serine 2808 (RyR2808) and phosphorylated PLN at serine 16 (PLN16) or threonine 17 (PLN17) and total RyR and PLN under control, BPA, and BPA + P4 (n = 3 hearts). (c and d) Representative immunoblot and quantification of PLN17 phosphorylation and total PLN under control and indicated treatments (n = 4 hearts). BPA and P4 = 1 nM; RU486 = 1 μM for all experiments. All values were normalized to control. *P < 0.05; **P < 0.01; #P > 0.1 versus control (one-way analysis of variance). Error bars are standard error of the mean.
Inhibitory effect of P4 is mediated by membrane-initiated signaling
P4-BSA, a membrane-impermeable protein conjugated P4, was used to examine whether the rapid inhibitory effects of P4 in female rat myocytes were initiated at the cell surface. Similar to P4, 1 nM P4-BSA abolished BPA-induced increases in Ca2+ park frequency [Fig. 4(a) and 4(b)], reduced the percentage of myocytes with triggered activities [Fig. 4(c)], and abolished PLN17 phosphorylation [Fig. 4(d) and 4(e)]. In fact, the inhibitory effects of P4-BSA were indistinguishable from those of P4. Again, nPR blockade with RU486 abolished all the observed inhibitory effects of P4-BSA (Fig. 4).
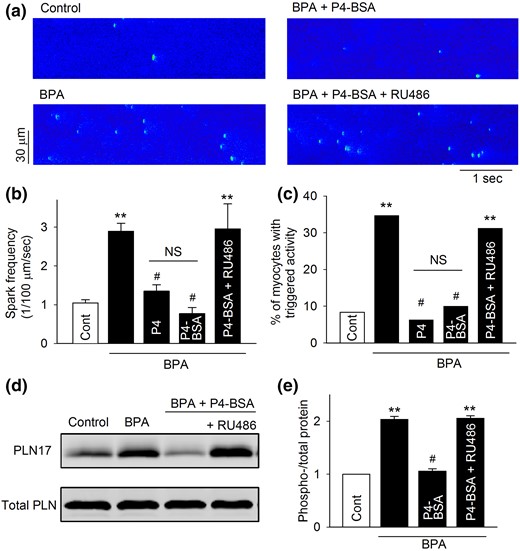
Effect of P4-BSA on BPA-induced alterations of Ca2+ handling and PLN17 phosphorylation in female rat ventricular myocytes. (a) Representative recordings of Ca2+ sparks in quiescent myocytes under control and various treatments as indicated. (b) Mean Ca2+ sparks frequency under various treatments (n = 8 to 9 myocytes from four hearts). (c) Percentages of myocytes with triggered activities under various treatments (n = 35 to 45 myocytes from four hearts). (d and e) Representative immunoblot and quantification of PLN17 phosphorylation and total PLN under control and indicated treatments. All values were normalized to control (n = 3 hearts). BPA, P4, and P4-BSA = 1 nM; RU486 = 1 μM for all experiments. **P < 0.01; #P > 0.1 versus control (one-way analysis of variance or χ2 test). NS, not significant. Error bars are standard error of the mean.
P4 signaling involves activation of Gi protein
We next examined the downstream signaling pathway underlying the inhibitory effects of P4 against BPA’s proarrhythmic actions using PLN17 phosphorylation and triggered activities as endpoints. Rapid signaling of P4 has been shown to involve direct interaction between nPR and cSrc (39, 40). The cSrc inhibitor PP2 did not alter the inhibitory effects of P4 on either BPA-induced triggered activities or PLN17 phosphorylation [Fig. 5(a) and 5(b)]. PP2 did not affect PLN17 phosphorylation or the incidence of triggered activities under baseline conditions or in the presence of BPA. These results show that cSrc does not play a role in mediating P4’s rapid inhibitory actions.
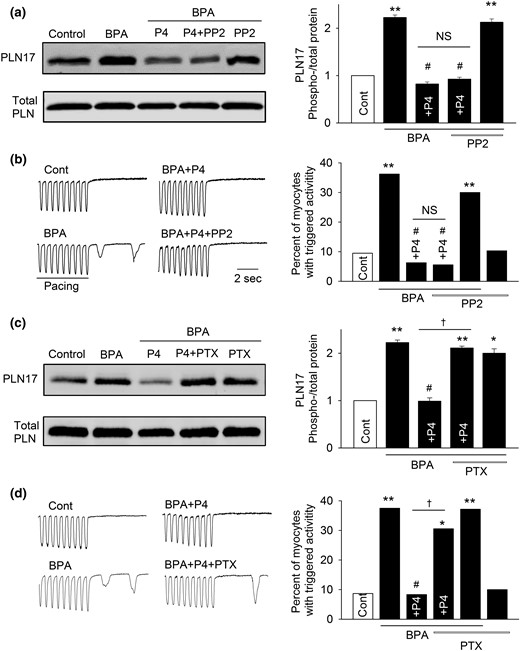
Gi, but not cSrc, mediates P4’s inhibition of the rapid actions of BPA in female rat ventricular myocytes. (a) Representative immunoblot and quantification of PLN17 phosphorylation and total PLN under control, BPA, and BPA in the presence of P4, P4 + 1 μM PP2, or PP2. (b) Representative recordings and percentages of myocytes with triggered activities under various treatments (n = 32 to 47 myocytes from four hearts). (c) Representative immunoblot and quantification of PLN17 phosphorylation and total PLN under control, BPA, and BPA in presence of P4, P4 + 200 ng/mL PTX, or PTX. (d) Representative recordings and percentages of myocytes with triggered activities under various treatments (n = 30 to 42 myocytes from four hearts). BPA and P4 = 1 nM for all experiments. *P < 0.05; **P < 0.01; #P > 0.1 versus control; †P < 0.01 (χ2 test). NS, not significant.
Activation of Gi protein is a key downstream event of nPR signaling mediating the rapid effects of P4 in breast cancer cells and rat cortical neurons (41, 42). Consistent with these reports, inhibition of Gi with PTX (200 ng/mL) abolished, or significantly suppressed, the inhibitory effects of P4 on BPA-induced PLN17 phosphorylation and triggered activities [Fig. 5(c) and 5(d)]. PTX did not affect PLN17 phosphorylation or the incidence of triggered activities under baseline conditions or in the presence of BPA, suggesting that the observed effects of PTX were not due to its lack of specific effect on PLN or triggered activities. These results indicate that Gi, but not cSrc, contributes to P4’s rapid inhibitory effect in cardiac myocytes.
Rapid inhibitory effect of P4 is mediated by nPR-Gi-PI3K signaling cascade
PI3K activation has been implicated in the rapid signaling of P4 in various experimental models (38, 43–45). Further, PI3K can be activated by the Gi protein (46). Thus, we examined the role of PI3K in mediating the rapid inhibitory effect of P4. Blockade of PI3K with wortmannin (1 μM) abolished the inhibitory effects of P4 on BPA-induced increases in PLN17 phosphorylation and in the incidence of triggered activities [Fig. 6(a) and 6(b)]. Wortmannin did not affect PLN17 phosphorylation or the incidence of triggered activities under baseline conditions or in the presence of BPA. We further used the phosphorylation status of Akt, a downstream effector of PI3K, to confirm the activation of PI3K by P4. Indeed, 1 nM P4 rapidly increased phosphorylation of Akt, and the effect was abolished by both PTX and RU486 [Fig. 6(c)], supporting the activation of the nPR-Gi-PI3K signaling cascade by P4.
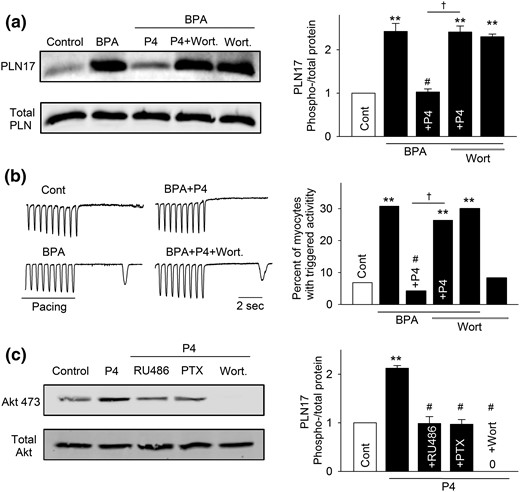
PI3K activation is involved in P4 and nPR signaling in female rat ventricular myocytes. (a) Representative immunoblot and quantification of PLN17 phosphorylation and total PLN under control (Cont), BPA, and BPA in the presence of P4, P4 + 1 μm wortmannin (Wort), or Wort. (b) Representative recordings and percentages of myocytes with triggered activities under various treatments (n = 32 to 46 myocytes from four hearts). (c) Representative immunoblot and quantification of Akt473 phosphorylation and total Akt under control, P4, and P4 in the presence of RU486, PTX, or Wort. For all experiments, the concentrations of treatments are as follows: BPA and P4 = 1 nM; RU486 and Wort = 1 μM; PTX = 200 ng/mL. **P < 0.01; #P > 0.1 versus control; †P < 0.01 (χ2 test).
Discussion
The native hormonal background should be taken into consideration when evaluating the biological and toxic effects of EDCs. P4 has well-known counterbalancing effects on estrogens, yet its influence on the actions of the estrogenic BPA is unknown. In this study, we show that the physiological level of P4 has a protective effect against BPA’s arrhythmogenic actions in female rat ventricular myocytes. P4 abolishes BPA-induced aberrant spontaneous excitation and alterations in Ca2+ handling through normalization of BPA-induced CAMKII phosphorylation of PLN. The protective effect of P4 is mediated by a membrane nPR-initiated signaling pathway involving Gi protein and PI3K. Although BPA exposure has been shown to affect P4 receptor expression and signaling in mice and is associated with altered P4 levels in humans (47, 48), our study demonstrates the influence of P4 on the actions of BPA.
BPA rapidly increased aberrant spontaneous excitation in female rat ventricular myocytes and promoted the development of arrhythmias in female rat hearts (13). The proarrhythmic effect of BPA is mediated by alteration of myocyte Ca2+ handling (13). In cardiac myocytes, Ca2+ handling is a coordinated process that involves several key Ca2+ handling proteins, such as l-type Ca2+ channels, RyR, and PLN (49). RyR is the primary channel for SR Ca2+ release, and PLN, an SR transmembrane inhibitor of sarco/endoplasmic reticulum Ca2+ ATPase, plays a central role in regulating SR Ca2+ reuptake (50). BPA rapidly activates two parallel signaling pathways, the cAMP/PKA pathway and the phospholipase C (PLC)/ inositol 1,4,5-trisphophate (IP3)/Ca2+/CAMKII pathway, which phosphorylate RyR and PLN, respectively (15). These actions of BPA result in increased SR Ca2+ load and SR Ca2+ leak and in aberrant myocyte excitation. P4 completely abolishes BPA-induced CAMKII phosphorylation of PLN17, which abrogates BPA’s stimulatory effect on SR Ca2+ reuptake and normalizes SR Ca2+ load. Further, it is known that SR Ca2+ release/leak is determined by both RyR open probability and SR Ca2+ load (49). Although P4 does not affect PKA phosphorylation of RyR, it indirectly inhibits BPA-induced SR Ca2+ leak likely through normalization of SR Ca2+ load. P4’s inhibition of PLN17 phosphorylation can be mediated either by direct inhibition of the CAMKII pathway or indirectly through activation of phosphatases that target PLN17. The inhibition of PLN17 phosphorylation likely accounts for P4’s blockade of BPA-induced triggered activities. This notion is consistent with our previous findings that pharmacological blockade of either the CAMKII pathway or the PKA pathway is sufficient to abolish BPA-induced triggered activities in female rat myocytes (15).
P4 evokes its biological effects on cell function through activation of nPR or nPR-like receptors. Those receptors have been shown to be expressed in cardiac myocytes and tissues in several species, including humans (38, 51, 52). Our results suggest that nPR signaling plays a key role in mediating P4 inhibition of BPA-induced arrhythmogenesis in female rat myocytes. We found that the nPR antagonist RU486 abolished all aspects of P4’s inhibitory actions on BPA-induced rapid effects, including triggered activity, SR Ca2+ cycling, and PLN17 phosphorylation. Further, the effects of P4 were mimicked by the membrane-impermeable P4-BSA, suggesting that P4 elicited rapid effects via a membrane nPR-initiated mechanism. Similarly, a central role of nPR in mediating the rapid effects of P4 has been described in various CV cell types, including vascular endothelial cells, coronary smooth muscle cells, and cardiomyocytes (38, 45, 53).
We show that the downstream elements of P4’s signaling include Gi protein and PI3K. We show that P4 increased Akt phosphorylation, an indicator of PI3K activation, and that blockade of Gi or PI3K abolished P4’s inhibition of BPA-induced triggered activities and PLN17 phosphorylation. Further, we show that blockade of Gi abrogated P4-induced Akt phosphorylation, suggesting that Gi is an upstream mediator of PI3K activation. Our findings agree with previous reports that Gi is one of the mechanisms mediating nPR-dependent rapid actions of P4. Gi plays a key role in nPR signaling and rapid P4 induction of actin remodeling and neuronal spine formation in rat cortical neurons (41). In breast cancer cells, nPR-dependent recruitment of Gi mediates the rapid P4 facilitation of cell movement and invasion (42). Further, it is well recognized that PI3K can be activated by G protein–coupled receptors through G proteins, including Gi. It has been shown that PI3K, activated by Gi, inhibits interleukin 12 production in human peripheral monocytes (54). In COS-7 cells, Gi activates Akt and promotes cell survival through a pathway involving PI3K (55). A previous report found that β2-adrenergic receptor, a Gi coupled receptor, protected rat neonatal cardiomyocytes from hypoxia- and reactive oxygen species–induced apoptosis through a PI3K-dependent mechanism (56).
In addition to Gi, it is well recognized that nPR can directly interact with the SH3 domain of cSrc, thereby activating cSrc and its downstream signaling (39). A key role of cSrc in the nPR-mediated rapid signaling has been shown in various cell types, including vascular endothelial cells, arterial smooth muscle cells, cortical neurons, breast cancer cells, and cardiomyocytes (38, 40, 41, 45, 57). By contrast, we found that in female rat myocytes, the rapid inhibitory effect of P4 was independent of cSrc activation. Blockade of cSrc with PP2 did not affect P4’s inhibitory effects on BPA’s rapid actions. Our results contrast with a previous report in guinea pig ventricular myocytes, where activation of cSrc is essential for P4’s acute modulation of cardiac ion channels (38). This discrepancy may reflect the complexity and target specificity in rapid P4 signaling in cardiac myocytes.
Our results demonstrate that the nPR/Gi/PI3K signaling pathway plays a key role in P4’s inhibition of BPA-induced PLN17 phosphorylation. A plausible mechanism underlying this effect is depletion of membrane phosphatidylinositol (3,4)-bisphosphate (PIP2). PIP2 is a low-abundance phospholipid in the plasma membrane, accounting for less than 1% of total plasma membrane phospholipids (58). Activation of PI3K phosphorylates PIP2 and converts it into phosphatidylinositol (3,5)-trisphosphate, thereby reducing, or even depleting, membrane PIP2. This PIP2 depletion occurs rapidly (i.e., within minutes) and lasts several minutes or longer depending on the cell type (59–62). It is well established that consumption of the low-abundance membrane PIP2 inhibits PIP2-dependent physiological processes. For example, PIP2 is a key modulator of a variety of ion channels, including the inwardly rectifying K+ channel, the voltage-gated K+ channel, the voltage-gated Ca2+ channel, and the transient receptor potential channel; sequestration or depletion of PIP2 by events such as stimulation of G protein–coupled receptors and/or PI3K activation affect the activity of these ion channels (58, 63–65). Previously, we showed that BPA increased PLN17 phosphorylation by CAMKII through activation of PLC and generation of IP3 through hydrolysis of PIP2. This in turn resulted in Ca2+ release through the IP3 receptors and in activation of CAMKII (15). We speculate that P4 inhibits this pathway by PI3K activation and PIP2 depletion. This reduces the substrate of PLC, blocks IP3 production and downstream CAMKII activation, and inhibits BPA-induced PLN17 phosphorylation. We also recognize that P4’s action could be mediated through indirect mechanisms such as activation of phosphatases that target PLN17. Further studies are needed to determine the direct relationship between membrane PIP2 level and PLN17 phosphorylation and to further define the role of PI3K-activated PIP2 depletion in the rapid effect of P4.
Previously we showed that the acute proarrhythmic action of BPA was particularly pronounced in the presence of E2 (13, 14). Here, we show that P4 counterbalances the proarrhythmic action of BPA. The balance of the two major ovarian sex hormones, E2 and P4, varies during different life stages in women, and this balance may influence the response of the female hearts to the cardiac impact of BPA. For instance, P4 declines to the postmenopausal level during the early phase of menopausal transition, whereas the E2 level is usually well preserved until late menopausal transition (66). The perimenopause period is marked by unique susceptibility to certain CV diseases, including arrhythmias. The mortality rate after myocardial infarction (i.e., heart attack) is particularly high in perimenopausal women and is more than double that of men of the same age (67, 68), and a significant contributing factor to the worse prognosis after myocardial infarction in women is arrhythmia (69, 70). Due to the change in hormone levels, female hearts may also have a higher susceptibility to the proarrhythmic toxicity of BPA.
In the current study, we used myocyte-level triggered activities as the endpoint to assess the influence of P4 on the cardiac actions of BPA. Myocyte-level arrhythmia events are one of the major arrhythmogenic mechanisms in the heart (49). They, however, do not necessarily translate to arrhythmia in vivo. Additional animal studies are needed to further investigate the interactions between BPA and native sex hormones and the resulting cardiac outcome.
In summary, we show that the physiological level of P4 has a protective effect against BPA-induced arrhythmogenesis in female rat cardiac myocytes. The underlying mechanism involves membrane-initiated signaling and activation of an nPR/Gi/PI3K pathway, which leads to the inhibition of BPA-induced alterations of myocyte Ca2+ handling. Our findings may help to better understand the response and susceptibility of female hearts to the cardiac toxicity of BPA and may provide mechanistic insights that could lead to the development of potential protective strategies against such toxicity.
Abbreviations:
- BPA
bisphenol A
- BSA
bovine serum albumin
- CAMKII
CaM-dependent protein kinase II
- CV
cardiovascular
- E2
17β-estradiol
- EDC
endocrine-disrupting chemical
- Gi
inhibitory G protein
- IP3
inositol 1,4,5-trisphophate
- nPR
nuclear porgesterone receptor
- P4
progesterone
- PI3K
phophoinositide-3 kinase
- PKA
protein kinase A
- PLC
phospholipase C
- PLN
phospholamban
- PP2
4-amino-3-(4-chlorophenyl)-1-(t-butyl)-1H-pyrazolo [3,4-d] pyrimidine
- PTX
pertussis toxin
- RyR
ryanodine receptor
- SR
sarcoplasmic reticulum.
Acknowledgments
This work was supported by National Institutes of Health Grant R01-ES017263 (to H.S.W.).
Disclosure Summary: The authors have nothing to disclose.
References
Shankar A, Teppala S. Urinary bisphenol A and hypertension in a multiethnic sample of US adults. J Environ Public Health. 2012; 2012:481641.
Author notes
Address all correspondence and requests for reprints to: Hong-Sheng Wang, PhD, Department of Pharmacology, University of Cincinnati College of Medicine, Cincinnati, Ohio 45267-0575. E-mail: [email protected]; or Kui Hong, MD, Department of Cardiology, Second Affiliated Hospital of Nanchang University, Nanchang, Jiangxi 330006, China.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}