





Circadian misalignment induces insulin resistance in both human and animal models, and skeletal muscle is the largest organ response to insulin. However, how circadian clock regulates muscle insulin sensitivity and the underlying molecular mechanisms are still largely unknown. Here we show circadian locomotor output cycles kaput (CLOCK) and brain and muscle aryl hydrocarbon receptor nuclear translocator-like protein (BMAL)-1, two core circadian transcription factors, are down-regulated in insulin-resistant C2C12 myotubes and mouse skeletal muscle. Furthermore, insulin signaling is attenuated in the skeletal muscle of ClockΔ19/Δ19 mice, and knockdown of CLOCK or BMAL1 by small interfering RNAs induces insulin resistance in C2C12 myotubes. Consistently, ectopic expression of CLOCK and BMAL1 improves insulin sensitivity in C2C12 myotubes. Moreover, CLOCK and BMAL1 regulate the expression of sirtuin 1 (SIRT1), an important regulator of insulin sensitivity, in C2C12 myotubes and mouse skeletal muscle, and two E-box elements in Sirt1 promoter are responsible for its CLOCK- and BMAL1-dependent transcription in muscle cells. Further studies show that CLOCK and BMAL1 regulate muscle insulin sensitivity through SIRT1. In addition, we find that BMAL1 and SIRT1 are decreased in the muscle of mice maintained in constant darkness, and resveratrol supplementation activates SIRT1 and improves insulin sensitivity. All these data demonstrate that CLOCK and BMAL1 regulate muscle insulin sensitivity via SIRT1, and activation of SIRT1 might be a potential valuable strategy to attenuate muscle insulin resistance related to circadian misalignment.
The circadian clock influences nearly all aspects of physiology and behavior, including sleep-wake cycles, cardiovascular activity, endocrine system, body temperature, blood pressure, renal activity, physiology of the gastrointestinal tract, memory formation, and metabolism (1, 2). Circadian misalignment has been described as a variety of circumstances, such as inappropriately timed sleep and wake, misalignment of sleep/wake with feeding rhythms, or misaligned central and peripheral rhythms (3). Epidemiological studies show that circadian misalignment increases the risk of developing a series of diseases, including obesity and type 2 diabetes (4, 5). Insulin resistance is the most powerful predictor of future development of type 2 diabetes, and it is also a therapeutic target once hyperglycemia is present (6). Overnutrition, low dietary fiber, sedentary lifestyle, and depression are associated with or can lead to insulin resistance (7–9). Circadian desynchrony in humans, a characteristic of shift work, jet lag, and/or sleep restriction, has also been reported to be correlated with or even directly induce insulin resistance (10–12). Similarly, housing mice in 20-hour light, 20-hour dark cycles to disrupt endogenous circadian clock results in accelerated weight gain and obesity, an increased insulin level, and an increased insulin to glucose ratio, suggesting the development of insulin resistance (13). Mice subjected to constant light exhibit a complete loss of circadian rhythms of insulin sensitivity (14). Circadian misalignment induced by constant darkness leads to insulin resistance in mice (15).
Central to the core clock mechanism is the heterodimeric transcription factor complex of circadian locomotor output cycles kaput (CLOCK) and brain and muscle aryl hydrocarbon receptor nuclear translocator-like protein (BMAL)-1, which induces the expression of the genes for two main oscillators, Per and Cry via E-box elements (CACGTG) in their promoters (1, 16). Mice with Clock mutation show normal insulin sensitivity at the age of 8 months, whereas 3-month-old ClockΔ19/Δ19 mutant mice display increased insulin sensitivity (17). Bmal1 knockout mice display a striking lean phenotype because of decreased sc adipose and muscle tissue mass (18) and show reduced insulin secretion and improved whole-body insulin sensitivity (17, 19). Circadian change of mouse hepatic insulin sensitivity is regulated by CLOCK/BMAL1 (15). Liver-specific deletion of Bmal1 results in a blunted sensitivity to insulin (20). Skeletal muscle is the predominant site of insulin-mediated glucose uptake in the postprandial state, and skeletal muscle insulin resistance is an early therapeutic target in young insulin-resistant individuals (21, 22). Both regular and inducible muscle-specific Bmal1 ablations cause decreased insulin-dependent glucose uptake in skeletal muscle (23). However, how circadian clock regulates muscle insulin sensitivity and the underlying molecular mechanisms need further investigation.
Sirtuin 1 (SIRT1), the mammalian homolog of yeast Sir2, is a NAD+-dependent deacetylase and an important regulator of insulin sensitivity (24–26). SIRT1 transgenic mice display reductions in blood insulin, fasted glucose, and more glucose tolerance, suggesting an increase in insulin sensitivity (27). Adenovirus-mediated overexpression of SIRT1 in the liver of ob/ob mice and diet-induced, insulin-resistant, low-density lipoprotein receptor-deficient mice ameliorates systemic insulin resistance (28). Consistently, liver-specific Sirt1 deficiency causes hepatic glucose overproduction and chronic hyperglycemia and also results in increased reactive oxygen species production and impaired mTorc2/Akt signaling, leading to insulin resistance (29). Overexpression of SIRT1 in C2C12 myotubes prevents fatty acid-induced insulin resistance (24). SIRT1 enhances skeletal muscle insulin sensitivity in mice during caloric restriction (30). Moreover, activation of SIRT1 by resveratrol, a natural polyphenolic compound, protects mice against diet-induced obesity and insulin resistance (31, 32) and improves insulin sensitivity in type 2 diabetic patients and obese subjects (33, 34). SIRT1 is expressed in a circadian manner, also binds to CLOCK-BMAL1 in a circadian manner, and regulates circadian clock gene expression through deacetylation of period 2 (PER2) (35). SIRT1 activity also has circadian change and can function as a molecular rheostat of CLOCK-mediated histone acetyltransferase function by deacetylation of histone H3 and BMAL1 (36). Our recent study showed that the hepatic circadian expression of SIRT1 is regulated by CLOCK/BMAL1 through binding to its promoter and is required for the circadian change of hepatic insulin sensitivity (15). However, whether SIRT1 is also involved in the regulation of muscle insulin sensitivity by the circadian clock is yet to be elucidated.
Here we show CLOCK and BMAL1 are down-regulated in insulin resistant myotubes and skeletal muscles and regulate insulin sensitivity via SIRT1 by affecting its transcription. The skeletal muscle of mice maintained in constant darkness shows decreased BMAL1 and SIRT1 protein levels and insulin resistance, and resveratrol supplementation to activate SIRT1 can efficiently reverse muscle insulin resistance induced by circadian misalignment.
Materials and Methods
Cell culture and treatments
Mouse myoblast cell line C2C12 was maintained in DMEM containing 10% fetal bovine serum (FBS; Gibco) and differentiated in DMEM supplemented with 2% horse serum (Gibco) after growing to 60%–80% confluent. After 4 days, the differentiated C2C12 myotubes were used for subsequent experiments.
C2C12 myotubes in 12-well plates were incubated in DMEM containing 10% FBS and 2% BSA in the absence or presence of 0.75 mM palmitate to induce insulin resistance for 16 hours.
To stimulate with insulin, cells were incubated in DMEM for 4 hours and were then incubated in DMEM containing 100 nM insulin (Sigma) for 15 minutes and harvested for Western blot.
For transfection of small interfering RNA (siRNA), C2C12 myotubes in 12-well plates were transfected with the indicated siRNAs at a final concentration of 80 nM in 1 mL culture medium per well using X-tremeGENE siRNA transfection reagent (Roche).
For adenovirus infection, C2C12 myotubes were infected with the indicated adenovirus at a multiplicity of infection of 200.
After transfection of siRNA for 96 hours or infection with adenovirus for 72 hours, the cells were harvested to extract total RNA or for Western blot or stimulated with insulin.
To combine the transfection of siRNA and infection of adenovirus, after transfection with siRNA for 24 hours, the cells were infected with adenovirus for 60 hours. Then the cells were incubated in culture medium or in DMEM containing 10% FBS, 1% BSA, and 0.5 mM palmitate to induce insulin resistance for 12 hours and were subsequently stimulated with insulin.
Immunofluorescence
C2C12 myoblasts or myotubes were fixed with 4% paraformaldehyde, blocked with 5% BSA in PBS containing 0.1% Triton X-100 and then incubated overnight at 4°C with antimyosin antibody (Developmental Studies Hybridoma Bank) as listed in Table 1 and then detected with Cy3-conjugated goat antimouse IgG antibody (Jackson ImmunoResearch). Immunofluorescence images were obtained on an Olympus IX51 fluorescence microscope.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised (Monoclonal or Polyclonal) | Dilution Used |
|---|---|---|---|---|---|
| BMAL1 | C-DMIDNDQGSSSPS | Anti-BMAL1 antibody-ChIP grade | Abcam, ab3350 | Rabbit, polyclonal | 1:1000 |
| PER2 | S (11)SIVDRDDSSIFDGLVEESS (30)C | Anti-PER2 antibody | Abcam, ab180655 | Rabbit, polyclonal | 1:1000 |
| CLOCK | Anti-KAT13D/CLOCK antibody-ChIP grade | Abcam, ab3517 | Rabbit, polyclonal | 1:1000 | |
| Acetyl-RelA | Anti-NFκB p65 (acetyl K310) antibody-ChIP grade | Abcam, ab19870 | Rabbit, polyclonal | 1:1000 | |
| p-Gsk3β | Phospho-GSK-3β (Ser9) antibody | Cell Signaling Technology, number 9336 | Rabbit, polyclonal | 1:2000 | |
| SIRT1 | SirT1 (1F3) mouse mAb | Cell Signaling Technology, number 8469 | Mouse, monoclonal | 1:1000 | |
| p53 | p53 (1C12) mouse mAb | Cell Signaling Technology, number 2524 | Mouse, monoclonal | 1:1000 | |
| Acetyl-p53 | Acetyl-p53 (Lys379) antibody | Cell Signaling Technology, number 2570 | Rabbit, polyclonal | 1:1000 | |
| GAPDH | GAPDH (14C11) rabbit mAb | Cell Signaling Technology, number 2119 | Rabbit, monoclonal | 1:10 000 | |
| p-InsR | Phospho-IGF-1 receptorβ (Tyr1131)/insulin receptorβ (Tyr1146) antibody | Cell Signaling Technology, number 3021 | Rabbit, monoclonal | 1:1000 | |
| p-Akt | Phospho-Akt (Ser473) (193H12) rabbit mAb | Cell Signaling Technology, number 4058 | Rabbit, monoclonal | 1:1000 | |
| RelA | NFκB p65 antibody (C-20) | Santa Cruz Biotechnology, sc-372 | Rabbit, polyclonal | 1:1000 | |
| Myosin | MF20 | Developmental Studies Hybridoma Bank | Mouse, monoclonal | 1:100 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised (Monoclonal or Polyclonal) | Dilution Used |
|---|---|---|---|---|---|
| BMAL1 | C-DMIDNDQGSSSPS | Anti-BMAL1 antibody-ChIP grade | Abcam, ab3350 | Rabbit, polyclonal | 1:1000 |
| PER2 | S (11)SIVDRDDSSIFDGLVEESS (30)C | Anti-PER2 antibody | Abcam, ab180655 | Rabbit, polyclonal | 1:1000 |
| CLOCK | Anti-KAT13D/CLOCK antibody-ChIP grade | Abcam, ab3517 | Rabbit, polyclonal | 1:1000 | |
| Acetyl-RelA | Anti-NFκB p65 (acetyl K310) antibody-ChIP grade | Abcam, ab19870 | Rabbit, polyclonal | 1:1000 | |
| p-Gsk3β | Phospho-GSK-3β (Ser9) antibody | Cell Signaling Technology, number 9336 | Rabbit, polyclonal | 1:2000 | |
| SIRT1 | SirT1 (1F3) mouse mAb | Cell Signaling Technology, number 8469 | Mouse, monoclonal | 1:1000 | |
| p53 | p53 (1C12) mouse mAb | Cell Signaling Technology, number 2524 | Mouse, monoclonal | 1:1000 | |
| Acetyl-p53 | Acetyl-p53 (Lys379) antibody | Cell Signaling Technology, number 2570 | Rabbit, polyclonal | 1:1000 | |
| GAPDH | GAPDH (14C11) rabbit mAb | Cell Signaling Technology, number 2119 | Rabbit, monoclonal | 1:10 000 | |
| p-InsR | Phospho-IGF-1 receptorβ (Tyr1131)/insulin receptorβ (Tyr1146) antibody | Cell Signaling Technology, number 3021 | Rabbit, monoclonal | 1:1000 | |
| p-Akt | Phospho-Akt (Ser473) (193H12) rabbit mAb | Cell Signaling Technology, number 4058 | Rabbit, monoclonal | 1:1000 | |
| RelA | NFκB p65 antibody (C-20) | Santa Cruz Biotechnology, sc-372 | Rabbit, polyclonal | 1:1000 | |
| Myosin | MF20 | Developmental Studies Hybridoma Bank | Mouse, monoclonal | 1:100 |
Abbreviations: ChIP, chromatin immunoprecipitation; mAb, monoclonal antibody.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised (Monoclonal or Polyclonal) | Dilution Used |
|---|---|---|---|---|---|
| BMAL1 | C-DMIDNDQGSSSPS | Anti-BMAL1 antibody-ChIP grade | Abcam, ab3350 | Rabbit, polyclonal | 1:1000 |
| PER2 | S (11)SIVDRDDSSIFDGLVEESS (30)C | Anti-PER2 antibody | Abcam, ab180655 | Rabbit, polyclonal | 1:1000 |
| CLOCK | Anti-KAT13D/CLOCK antibody-ChIP grade | Abcam, ab3517 | Rabbit, polyclonal | 1:1000 | |
| Acetyl-RelA | Anti-NFκB p65 (acetyl K310) antibody-ChIP grade | Abcam, ab19870 | Rabbit, polyclonal | 1:1000 | |
| p-Gsk3β | Phospho-GSK-3β (Ser9) antibody | Cell Signaling Technology, number 9336 | Rabbit, polyclonal | 1:2000 | |
| SIRT1 | SirT1 (1F3) mouse mAb | Cell Signaling Technology, number 8469 | Mouse, monoclonal | 1:1000 | |
| p53 | p53 (1C12) mouse mAb | Cell Signaling Technology, number 2524 | Mouse, monoclonal | 1:1000 | |
| Acetyl-p53 | Acetyl-p53 (Lys379) antibody | Cell Signaling Technology, number 2570 | Rabbit, polyclonal | 1:1000 | |
| GAPDH | GAPDH (14C11) rabbit mAb | Cell Signaling Technology, number 2119 | Rabbit, monoclonal | 1:10 000 | |
| p-InsR | Phospho-IGF-1 receptorβ (Tyr1131)/insulin receptorβ (Tyr1146) antibody | Cell Signaling Technology, number 3021 | Rabbit, monoclonal | 1:1000 | |
| p-Akt | Phospho-Akt (Ser473) (193H12) rabbit mAb | Cell Signaling Technology, number 4058 | Rabbit, monoclonal | 1:1000 | |
| RelA | NFκB p65 antibody (C-20) | Santa Cruz Biotechnology, sc-372 | Rabbit, polyclonal | 1:1000 | |
| Myosin | MF20 | Developmental Studies Hybridoma Bank | Mouse, monoclonal | 1:100 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised (Monoclonal or Polyclonal) | Dilution Used |
|---|---|---|---|---|---|
| BMAL1 | C-DMIDNDQGSSSPS | Anti-BMAL1 antibody-ChIP grade | Abcam, ab3350 | Rabbit, polyclonal | 1:1000 |
| PER2 | S (11)SIVDRDDSSIFDGLVEESS (30)C | Anti-PER2 antibody | Abcam, ab180655 | Rabbit, polyclonal | 1:1000 |
| CLOCK | Anti-KAT13D/CLOCK antibody-ChIP grade | Abcam, ab3517 | Rabbit, polyclonal | 1:1000 | |
| Acetyl-RelA | Anti-NFκB p65 (acetyl K310) antibody-ChIP grade | Abcam, ab19870 | Rabbit, polyclonal | 1:1000 | |
| p-Gsk3β | Phospho-GSK-3β (Ser9) antibody | Cell Signaling Technology, number 9336 | Rabbit, polyclonal | 1:2000 | |
| SIRT1 | SirT1 (1F3) mouse mAb | Cell Signaling Technology, number 8469 | Mouse, monoclonal | 1:1000 | |
| p53 | p53 (1C12) mouse mAb | Cell Signaling Technology, number 2524 | Mouse, monoclonal | 1:1000 | |
| Acetyl-p53 | Acetyl-p53 (Lys379) antibody | Cell Signaling Technology, number 2570 | Rabbit, polyclonal | 1:1000 | |
| GAPDH | GAPDH (14C11) rabbit mAb | Cell Signaling Technology, number 2119 | Rabbit, monoclonal | 1:10 000 | |
| p-InsR | Phospho-IGF-1 receptorβ (Tyr1131)/insulin receptorβ (Tyr1146) antibody | Cell Signaling Technology, number 3021 | Rabbit, monoclonal | 1:1000 | |
| p-Akt | Phospho-Akt (Ser473) (193H12) rabbit mAb | Cell Signaling Technology, number 4058 | Rabbit, monoclonal | 1:1000 | |
| RelA | NFκB p65 antibody (C-20) | Santa Cruz Biotechnology, sc-372 | Rabbit, polyclonal | 1:1000 | |
| Myosin | MF20 | Developmental Studies Hybridoma Bank | Mouse, monoclonal | 1:100 |
Abbreviations: ChIP, chromatin immunoprecipitation; mAb, monoclonal antibody.
Dimethylthiazol-diphenyltetrazolium-bromide (MTT) assay
C2C12 myoblasts were seeded in 96-well plates. After differentiation, the cells were treated with the indicated concentrations of palmitate for 16 hours and then were used for the MTT assay as described previously (37). In brief, 20 μL 5 mg/ml MTT dissolved in PBS was added to each well containing 200 μL culture medium. After incubation at 37°C for 4 hours, the culture medium containing MTT in each well was removed. The MTT-formazan crystals formed by metabolically viable cells were dissolved in 150 μL dimethylsulfoxide, and the absorbance at 490 nm was measured by using a 96-well plate reader.
Western blot
Protein samples were analyzed with antibodies against CLOCK, BMAL1, PER2 (Abcam), Tyr1131/1146-phosphorylated (p) insulin receptor (p-InsR), Ser473-p-Akt, Ser9-phosphorylated glycogen synthase kinase 3β (p-Gsk3β), SIRT1, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), p53, acetyl-p53 (K379) (Cell Signaling), RelA, and acetyl-RelA (Santa Cruz Biotechnology) as listed in Table 1. Each blot shown in the figures is representative of at least three experiments. Protein quantification was analyzed by ImageJ (http://rsb.info.nih.gov/ij/index.html), and the intensity values were normalized to GAPDH.
Glucose uptake assay
Glucose uptake was determined using a method described previously (38). Briefly, cells were incubated in Krebs-Ringer phosphate buffer (128 mM NaCl; 4.7 mM KCl; 5 mM NaH2PO4; 5 mM Na2HPO4; 1.25 mM MgSO4; 1.25 mM CaCl2, pH 7.4) with or without 100 nM insulin for 30 minutes at 37°C. [3H]2-deoxyglucose (Amersham) and 2-deoxyglucose were then added to a final concentration of 0.5 μCi/mL and 0.05 mM, respectively. After incubation for 5 minutes, the cells were washed four times with ice-cold PBS and then were lysed with 0.2 M NaOH for 30 minutes. Radiolabeled glucose in lysates was measured in a high-flashpoint scintillation cocktail (PerkinElmer) using a liquid scintillation counter.
Animal experiments
All animals were maintained and used in accordance with the guidelines of the Institutional Animal Care and Use Committee of the Institute for Nutritional Sciences. The db/db mice, ClockΔ19/Δ19 mice with a deletion of exon 19 in the Clock gene and their wild-type littermates were obtained from the Jackson Laboratory. C57BL/6 mice were obtained from Slac. Mice were housed in a 12-hour light, 12-hour dark cycle (LD) or constant darkness (DD) as indicated. Zeitgeber time (ZT) 0 corresponds to the time of light onset, and circadian time (CT) 0 corresponds to ZT0 of the preceding LD cycle. Except where indicated, male mice at the age of 10 weeks were used for the following experiments.
Insulin tolerance tests were performed at ZT7 on db/db mice fasted for 4 hours. Glucose concentrations were measured from tail blood at the indicated times after an ip injection of 1 U/kg body weight of human insulin (Novo Nordisk).
To study the mRNA and protein levels in skeletal muscle, db/db mice, ClockΔ19/Δ19 mice and their wild-type littermates were killed at ZT7 except as indicated to collect quadriceps muscle samples. To detect the insulin signaling in vivo, the ClockΔ19/Δ19 mice and their wild-type littermates were fasted for 16 hours and then injected ip with PBS or human insulin at a dose of 5 U/kg at ZT7 for 15 minutes and killed to collect quadriceps muscle samples.
To study the effect of constant darkness, 10-week-old mice were housed in a 12-hour light, 12-hour dark cycle or constant darkness. After 22 days, the mice were killed at the indicated time points to collect quadriceps muscle samples.
To investigate the effect of resveratrol in vivo, the 10-week-old mice were fed with or without 250 mg/L transresveratrol (Sigma) for 14 days as described previously (24) and then housed on a 12-hour light, 12-hour dark cycle or constant darkness with the same treatment of resveratrol for 22 days. The dose of resveratrol is about 25 mg/kg · d, which is similar as previous studies (24, 39). Then the mice were killed to collect quadriceps muscle samples or fasted for 16 hours and then injected ip with PBS or insulin (Novo Nordisk) at a dose of 5 U/kg at the indicated time points. Fifteen minutes later, the mice were killed to collect quadriceps muscle samples.
Small interfering RNAs
The siRNA for Clock is a mixture of GACGAGACUGUGAGAGGAA (dTdT), CAGAGUGCUACAGUCACUA(dTdT) and AGUAUGCCACAGAACAGUA(dTdT) (where dT is deoxythymidine) synthesized by Bioneer at a ratio of 1:1:1. The siRNA for Bmal1 is a mixture of GUGCAACAGGCCUUCAGUA(dTdT), GUAUCUACCACAGGAACUU(dTdT) and UGAAUGCUUUUGGAACCAA(dTdT) by Bioneer at a ratio of 1:1:1. The siRNA for Sirt1 is a mixture of GUGGCAGAUUGUUAUUAAU(dTdT) and CCGUCUCUGUGUCACAAAU(dTdT) by Genepharma at a ratio of 1:1.
Adenoviruses
Adenoviruses (Ads) for overexpressing green fluorescent protein, CLOCK (Ad-CLOCK), BMAL1 (Ad-BMAL1), and SIRT1 (Ad-SIRT1) were prepared as described previously (15).
RNA isolation and quantitative PCR
Total RNA was extracted with TRIzol reagent (Invitrogen) and then reverse transcribed to cDNA using Moloney murine leukemia virus reverse transcriptase (Promega) with random hexamer primers. Real-time quantitative PCR was performed with ABI Prism 7900 sequence detection system using FastStart Universal SYBR Green Master (Roche). The results were normalized to 36b4. Primers were as follows: GGGCATCACCACGAAAATCTC and CTGCCGTTGTCAAACACCT for mouse 36b4, ATGACGCTGTGGCAGATTGTT and CCGCAAGGCGAGCATAGAT for mouse Sirt1, CCAAGAAAGTATGGACACAGACAAA and GCATTCTTGATCCTTCCTTGGT for mouse Bmal1, and AGAACTTGGCATTGAAGAGTCTC and GTCAGACCCAGAATCTTGGCT for mouse Clock.
Plasmids
Plasmids for expressing CLOCK (pcDNA3-CLOCK) or BMAL1 (pcDNA3-BMAL1) were kind gifts from K. Yagita (40). pGL3-91, pGL3-629, pGL3-1487 luciferase reporter plasmids containing 91, 629, and 1487 bp mouse Sirt1 promoter were constructed as described previously (41). pGL3-629 mutant luciferase reporter plasmid with mutation in the two E-boxes of Sirt1 promoter was constructed as described previously (15).
Luciferase assay
C2C12 myoblasts in 24-well plates were transfected with a luciferase reporter plasmid containing the indicated Sirt1 promoter (0.05 μg/well), pSV40-β-gal (0.05 μg/well), pcDNA3-CLOCK and pcDNA3-BMAL1, or pcDNA3 (0.3 μg/well) using Lipofectamine 2000 (Invitrogen). After transfection for 24 hours, cells were harvested for the luciferase assay as described previously (42), and normalized to β-galactosidase activity, which was determined as described previously (43).
Statistical analysis
Data are expressed as mean ± SD of at least three independent experiments. Statistical significance was assessed by the Student's t test or a one-way ANOVA. Differences were considered statistically significant at P < .05.
Results
Down-regulation of CLOCK and BMAL1 protein levels is correlated with muscle insulin resistance
To investigate the correlation between CLOCK, BMAL1, and insulin sensitivity in skeletal muscle, we treated C2C12 myotubes with palmitate to induce insulin resistance as described previously (24). The differentiation of C2C12 myoblasts into myotubes was confirmed by immunofluorescence to detect the expression of myosin (Supplemental Figure 1A). Treatment with palmitate for 16 hours had no obvious effect on the morphological appearance of C2C12 myotubes at the concentration up to 0.75 mM (Supplemental Figure 1B) and had slight but significant effect on cell viability only at the high concentration (Supplemental Figure 1C). Palmitate-induced insulin resistance was confirmed by impaired insulin-induced phosphorylation of InsR, Akt, and Gsk3β and glucose uptake (Supplemental Figure 1, D and E). As shown in Figure 1, A–C, palmitate induced insulin resistance and also led to a significant decrease in protein levels of CLOCK and BMAL1. These data show that the decrease of CLOCK and BMAL1 is correlated with insulin resistance in cultured myotubes. Furthermore, as shown in Figure 1, D–F, CLOCK and BMAL1 protein levels were significantly decreased in the skeletal muscle of db/db mice, an insulin-resistant mouse model confirmed by an insulin tolerance test (Supplemental Figure 2). These data show that the down-regulation of CLOCK and BMAL1 protein levels is correlated with muscle insulin resistance.
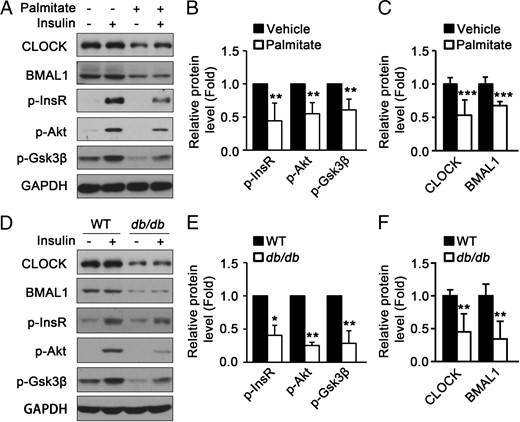
The protein levels of CLOCK and BMAL1 in mouse skeletal muscle and cultured myotubes are down-regulated under insulin resistant conditions. A, The protein levels of CLOCK, BMAL1, and insulin-induced phosphorylation of InsR, Akt, and Gsk3β were down-regulated in insulin-resistant C2C12 myotubes induced by palmitate. B, Quantification of p-InsR, p-Akt, and p-Gsk3β protein levels in panel A. C, Quantification of CLOCK and BMAL1 protein levels in panel A. D, The protein levels of CLOCK, BMAL1, insulin-induced phosphorylation of InsR, Akt, and Gsk3β were decreased in the muscle of db/db mice compared with those in wild type (WT). E and F, Quantification of the protein levels in panel D (n = 4 for each group). Protein levels were analyzed by Western blot. *, P < .05; **, P < .01; ***, P < .001.
CLOCK and BMAL1 regulate muscle insulin sensitivity
To investigate whether the down-regulation of CLOCK or BMAL1 leads to muscle insulin resistance, we detected the insulin signaling in the skeletal muscle of mice with Clock mutation. As shown in Figure 2, A and B, insulin-induced phosphorylation of InsR, Akt, and Gsk3β in wild-type mice was significantly attenuated in the skeletal muscle of ClockΔ19/Δ19 mice. Moreover, knockdown of CLOCK or BMAL1 by siRNAs induced insulin resistance in C2C12 myotubes, as shown by the down-regulated phosphorylation levels of InsR, Akt, and Gsk3β (Figure 2, C–F). Consistently, ectopic expression of CLOCK and BMAL1 by adenovirus attenuated insulin resistance in C2C12 myotubes induced by palmitate (Figure 2, G and H). Taken together, these data demonstrate that CLOCK and BMAL1 can regulate muscle insulin sensitivity.
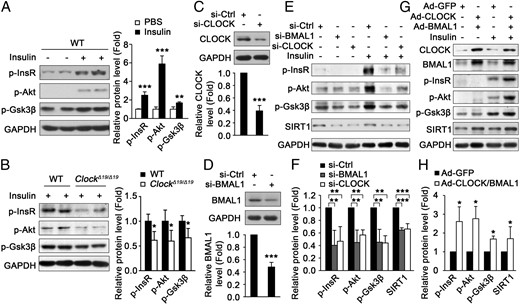
CLOCK and BMAL1 regulate insulin sensitivity in mouse skeletal muscle and cultured myotubes. A and B, The insulin-induced phosphorylation of InsR, Akt, and Gsk3β in skeletal muscle of wild-type (WT) mice at ZT7 (A), which was decreased in the muscle of ClockΔ19/Δ19 mice (B) (n = 4 for each group). C and D, The CLOCK (C) and BMAL1 (D) protein levels were determined by Western blot after transfection of siRNA for 96 hours. E, Knockdown of CLOCK or BMAL1 induced insulin resistance in C2C12 myotubes as measured by Western blot for insulin-induced phosphorylation of InsR, Akt and Gsk3β. F, Quantification of the protein levels in panel E. G, Ectopic expression of CLOCK and BMAL1 by adenovirus attenuated insulin resistance in C2C12 myotubes induced by palmitate. H, Quantification of the protein levels in panel G stimulated with insulin. *, P < .05; **, P < .01; ***, P < .001.
CLOCK and BMAL1 regulate muscle SIRT1 expression
To reveal the underlying mechanism for how CLOCK and BMAL1 regulate muscle insulin sensitivity, SIRT1, an important regulator of insulin sensitivity and involved in CLOCK/BMAL1-regulated hepatic insulin sensitivity (15), was studied. As shown in Figure 3A, we found that SIRT1 protein levels in skeletal muscle were increased at ZT13 compared with those at ZT1, which is similar with the circadian change of PER2, a well-known transcription target of CLOCK/BMAL1. Consistently, SIRT1 activity in skeletal muscle as indicated by the acetylation levels of RelA was also increased at ZT13 compared with those at ZT1 (Figure 3A). Moreover, SIRT1, PER2, and acetylated RelA protein levels in the skeletal muscle of ClockΔ19/Δ19 mice were very similar at ZT1 and ZT13 (Figure 3B), suggesting that SIRT1 is regulated by CLOCK with a similar mechanism as PER2. To further confirm this conclusion, we measured the SIRT1 protein level and activity in skeletal muscle of wild-type and ClockΔ19/Δ19 mice. As shown in Figure 3C, SIRT1 protein level and activity in ClockΔ19/Δ19 mice were markedly down-regulated compared with those in wild-type mice, suggesting the CLOCK-dependent expression of SIRT1. Consistently, knockdown of CLOCK or BMAL1 by siRNA in C2C12 myotubes decreased the SIRT1 protein level and activity as indicated by the acetylation levels of p53 (Figure 3D). In addition, the ectopic expression of CLOCK and BMAL1 by adenovirus in C2C12 myotubes increased the SIRT1 protein level and activity (Figure 3E). Taken together, these results show that CLOCK and BMAL1 regulate muscle SIRT1 protein level and activity.
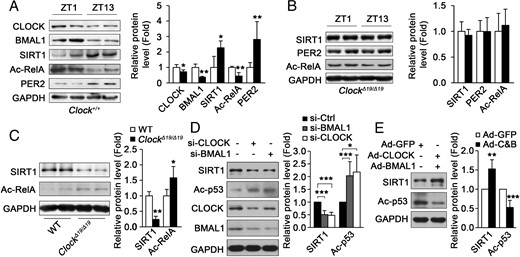
CLOCK and BMAL1 regulate SIRT1 protein levels in mouse skeletal muscle and cultured myotubes. A, The protein levels of CLOCK, BMAL1, SIRT1, Ac-RelA, and PER2 in the muscle of C57BL/6 mice at ZT1 and ZT13 fasted for 12 hours. Ac-RelA was monitored to indicate the activity of SIRT1 (n = 4 for each group). B, SIRT1, Ac-RelA, and PER2 protein levels in the muscle of ClockΔ19/Δ19 mice at ZT1 and ZT13 fasted for 12 hours (n = 4 for each group). C, SIRT1 and Ac-RelA protein levels in the muscle of ClockΔ19/Δ19 mice at ZT7 (n = 4 for each group). D, Knockdown of CLOCK or BMAL1 in C2C12 myotube down-regulated SIRT1 protein level and activity as monitored by acetylated p53. E, Ectopic expression of CLOCK and BMAL1 by adenovirus (Ad-C&B) in C2C12 myotubes up-regulated SIRT1 protein level and activity. *, P < .05; **, P < .01; ***, P < .001.
Two E-box elements in the Sirt1 promoter are required for its CLOCK- and BMAL1-dependent transcription in muscle cells
To figure out how CLOCK and BMAL1 regulate SIRT1 protein level, we measured Sirt1 mRNA levels in the skeletal muscle of wild-type mice and ClockΔ19/Δ19 mice. As shown in Figure 4A, Sirt1 mRNA levels in ClockΔ19/Δ19 mice were significantly down-regulated compared with those in wild-type mice. Moreover, knockdown of CLOCK or BMAL1 by siRNA in C2C12 myotubes markedly decreased Sirt1 mRNA level (Figure 4B). Consistently, ectopic expression of CLOCK and BMAL1 by adenovirus in C2C12 myotubes increased Sirt1 mRNA level (Figure 4C). These data demonstrate that CLOCK and BMAL1 regulate Sirt1 mRNA level in muscle cells.
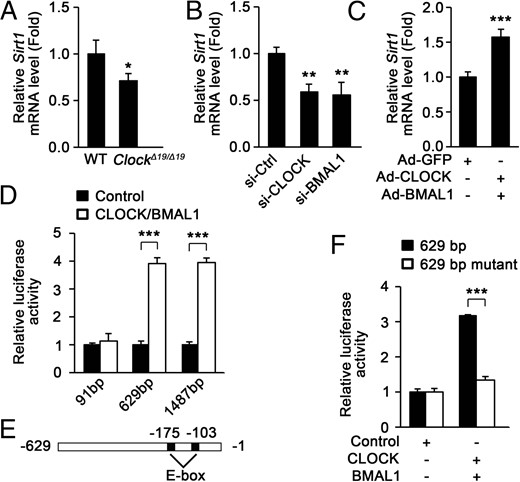
CLOCK and BMAL1 bind to Sirt1 promoter and increase its transcription. A, Sirt1 mRNA level was decreased in the muscle of ClockΔ19/Δ19 mice (n = 4). B, Knockdown of CLOCK or BMAL1 in C2C12 myotubes down-regulated Sirt1 mRNA level. Ctrl, control. C, Ectopic expression of CLOCK and BMAL1 by adenovirus in C2C12 myotubes up-regulated Sirt1 mRNA level. D, The effect of CLOCK and BMAL1 on luciferase expression under the control of the indicated Sirt1 promoter fragments in C2C12 myoblasts. E, Location of the predicted two E-boxes in Sirt1 promoter. F, Mutation of the two E-boxes attenuated the activation of the 629 bp Sirt1 promoter by CLOCK and BMAL1 in C2C12 myoblasts. *, P < .05; **, P < .01; ***, P < .001.
To investigate whether the change of Sirt1 mRNA level in muscle cells is regulated at the transcriptional level, luciferase reporters with the 91-, 629-, or 1487-bp Sirt1 promoter were used to analyze their transcriptional activity. As expected, ectopic expression of CLOCK and BMAL1 markedly increased the transcriptional activity of the 629- and 1487-bp Sirt1 promoter but did not affect the activity of the 91-bp Sirt1 promoter (Figure 4D). CLOCK and BMAL1 have been shown to bind to E-box elements and activate transcription (44). As shown in Figure 4E, there are two E-box elements, 103 or 175 bp upstream of the initiation codon of Sirt1. Mutation of the two E-boxes in the 629-bp Sirt1 promoter disrupted the effect of CLOCK and BMAL1 on its transcriptional activity (Figure 4F).
These data suggest that CLOCK and BMAL1 bind to the E-boxes in Sirt1 promoter and increase its transcription in muscle cells.
CLOCK and BMAL1 regulate muscle insulin sensitivity through SIRT1
Given those findings above, we presumed that CLOCK and BMAL1 regulate insulin sensitivity in muscle cells in a SIRT1-dependent way. As shown in Figure 5, A–C, CLOCK and BMAL1 failed to attenuate insulin resistance induced by palmitate in C2C12 myotubes with knockdown of SIRT1. Furthermore, ectopic expression of SIRT1 by adenovirus ameliorated insulin resistance induced by the knockdown of BMAL1 or CLOCK in C2C12 myotubes (Figure 5, D–F). These data show that CLOCK and BMAL1 regulate muscle insulin sensitivity via SIRT1.
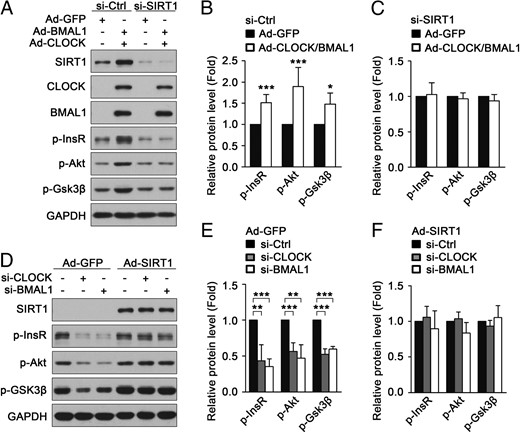
CLOCK and BMAL1 regulate insulin sensitivity through SIRT1 in cultured myotubes. A, CLOCK and BMAL1 failed to attenuate insulin resistance induced by palmitate in C2C12 myotubes with knockdown of SIRT1. Ctrl, control. B and C, Quantification of p-InsR, p-Akt, and p-Gsk3β protein levels in panel A. D, Ectopic expression of SIRT1 ameliorated insulin resistance induced by knockdown of BMAL1 or CLOCK in C2C12 myotubes. E and F, Quantification of p-InsR, p-Akt, and p-Gsk3β protein levels in panel D. *, P < .05; **, P < .01; ***, P < .001.
Resveratrol increases SIRT1 activity and attenuates muscle insulin resistance induced by circadian misalignment
To test the effect of circadian misalignment on SIRT1 and insulin sensitivity, we used light deprivation as a model of circadian misalignment. After being maintained in constant darkness for 2 weeks, the circadian expression pattern of BMAL1 protein in mouse skeletal muscle was impaired (Supplemental Figure 3, A and B), and both BMAL1 and SIRT1 protein levels in the muscle were markedly decreased at ZT/CT7, whereas CLOCK protein levels were almost the same (Figure 6A). Resveratrol has been reported to induce the activation of SIRT1 and ameliorate insulin resistance associated with decreased SIRT1 (24, 39). To study whether resveratrol can also improve insulin resistance associated with decreased SIRT1 caused by constant darkness, mice were fed with resveratrol at a dose of 25 mg/kg · d under a normal condition for 14 days. Then the mice were fed continuously with resveratrol and kept in constant darkness for 22 days. Subsequently, SIRT1 activity was assessed by detecting the acetylation levels of p53 and RelA, two known targets of SIRT1. As shown in Figure 6B, the acetylation levels of p53 and RelA were markedly increased in the muscle of mice under constant darkness at ZT/CT7, indicating the decreased SIRT1 activity. As expected, feeding with resveratrol relieved the negative effect of constant darkness on SIRT1 activity in muscle at ZT/CT7 (Figure 6B). Moreover, constant darkness-induced impaired insulin signaling in muscle at ZT/CT7, including the decreased insulin-induced phosphorylation of InsR, Akt, and Gsk3β, was also reversed by feeding with resveratrol (Figure 6C). When the mice were fed continuously with resveratrol and still maintained under normal condition for 22 days, resveratrol had slight but not significant effect on insulin signaling in skeletal muscle at ZT7 (Supplemental Figure 4). Similarly, resveratrol also significantly increased SIRT1 protein level and activity in mouse skeletal muscle and attenuated the muscle insulin resistance induced by constant darkness at ZT/CT1 and ZT/CT13 (Figure 6, D–G). Taken together, these data demonstrate that resveratrol can improve muscle insulin sensitivity under constant darkness.
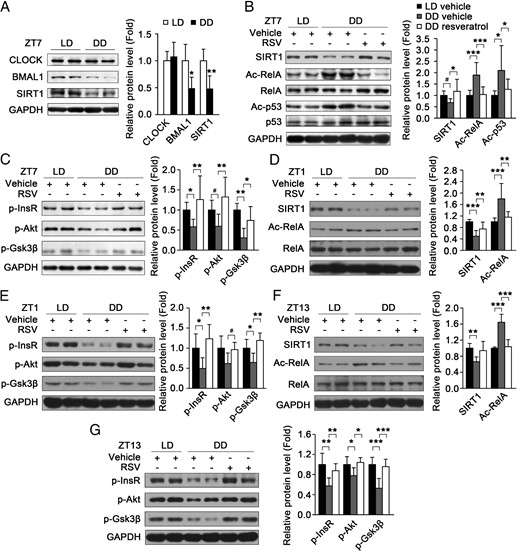
Resveratrol increases SIRT1 activity and attenuates the muscle insulin resistance induced by circadian misalignment. A, The protein levels of CLOCK, BMAL1, and SIRT1 in the muscle of mice under light/dark (LD) and constant dark (DD) conditions at ZT/CT7 (n = 5 for each group). B, Resveratrol (RSV) increased SIRT1 protein level and activity in the muscle of mice under constant dark (DD) condition at CT7. SIRT1 activity was indicated by the acetylation levels of p53 and RelA with Western blot (n = 8 for each group). C, The insulin-induced phosphorylation of InsR, Akt, and Gsk3β in the muscle of mice treated with vehicle or resveratrol under LD or DD condition at ZT/CT (n = 8–9 for each group). D–G, Resveratrol increased SIRT1 protein level and activity (D and F) and attenuated the muscle insulin resistance induced by constant darkness (E and G) at ZT/CT1 and ZT/CT13 (n = 5–8 for each group). *, P < .05, **, P < .01, ***, P < .001 by ANOVA; #, P > .05 by ANOVA, P < .05 by Student's t test.
Discussion
In this study, we show that the down-regulation of CLOCK or BMAL1 protein level is associated with muscle insulin resistance. Our observation is consistent with the previous report that muscle-specific Bmal1 ablation causes impaired insulin-dependent glucose uptake in skeletal muscle (23). Moreover, our observation is also consistent with the fact that circadian misalignment is associated with insulin resistance in humans (12), and hepatic expression of CLOCK or BMAL1 is down-regulated under insulin-resistant conditions (15). Furthermore, we show that the knockdown of CLOCK or BMAL1 by siRNAs induces muscle insulin resistance in C2C12 myotubes, and constant darkness down-regulates BMAL1 and induces insulin resistance in muscle. Similarly, it has been reported that insulin sensitivity in skeletal muscle represented by glucose uptake also displays circadian oscillations (45, 46), and muscle-specific deletion of Bmal1 impairs insulin sensitivity in skeletal muscle (23). These results are also in accordance with the observations that circadian misalignment induces insulin resistance in humans (10, 11), and circadian misalignment by shortening the light/dark cycle or exposed to constant light leads to insulin resistance or the loss of circadian rhythms of insulin sensitivity in mice (13, 14). Moreover, we demonstrate that ectopic expression of CLOCK and BMAL1 in C2C12 myotubes improves insulin resistance induced by palmitate. In accordance with this observation, tail vein injection of adenoviruses expressing CLOCK and BMAL1 improves insulin tolerance in db/db mice (15), and mice fed with a high-fat diet in a time-restricted manner with equal caloric intake exhibit an increase in the peak to trough ratio of BMAL1 and CLOCK mRNA levels, improved glucose tolerance, and a decreased fasting insulin level (47). Constant darkness has been considered as a free-running circadian state (48, 49). In addition, it has been reported that time-restricted feeding can amplify the amplitude of circadian clock and prevent metabolic diseases in mice fed a high-fat diet (47), and we observed that constant darkness decreased the muscle BMAL1 protein level at ZT/CT7. So it is possible that constant darkness might also reduce the amplitude of circadian clock in addition to inducing the free running of circadian clock and thus finally lead to insulin resistance in mouse skeletal muscle. All these findings show that CLOCK and BMAL1 are involved in the regulation of muscle insulin sensitivity and suggest that the maintenance of the normal circadian expression of CLOCK and BMAL1 would be helpful for preventing metabolic disorders in muscle.
SIRT1, a master metabolic regulator, has been shown to be regulated by various transcriptional factors (50). In this study, we demonstrate that the expression of SIRT1 is regulated by two core circadian transcription factors, CLOCK and BMAL1, and two E-box elements in Sirt1 promoter are required for its CLOCK- and BMAL1-dependent transcription in muscle cells. It has been reported that SIRT1 binds to CLOCK, BMAL1, and PER2 and deacetylates BMAL1 and PER2 to regulate the circadian clock (35, 36). Thus, CLOCK/BMAL1-SIRT1 might form a feedback loop to link circadian clock and metabolism in muscle. Studies have shown that overexpression of SIRT1 retards muscle differentiation through deacetylating and repressing MyoD, a master regulator of myogenesis (51). CLOCK/BMAL1 has been reported to bind to the core enhancer of MyoD and up-regulate its transcription and expression and to be necessary for the maintenance of skeletal muscle phenotype and function (52). In combination with our findings, CLOCK/BMAL1 and SIRT1 might coordinate to regulate MyoD and skeletal muscle differentiation and function. In skeletal muscle, SIRT1 has been reported to regulate the acetylation and transcription of PGC-1α (53, 54), which can stimulate the expression of CLOCK and BMAL1 in C2C12 myotubes (55). Thus, SIRT1, PGC-1α, and CLOCK/BMAL1 might form a positive feedback loop to orchestrate metabolism, mitochondrial biogenesis, and circadian clock in muscle.
SIRT1 has been reported to be an important regulator of insulin sensitivity in key insulin target tissues (24–26) and is required for the normal circadian change of hepatic insulin sensitivity (15). Here we demonstrate that SIRT1 mediates CLOCK/BMAL1-regulated muscle insulin sensitivity in cultured muscle cells. Similarly, overexpression of SIRT1 in C2C12 myotubes prevents palmitate-induced insulin resistance (24), and overexpression of SIRT1 in rat skeletal muscle by electroporation attenuates muscle insulin resistance induced by a high-fat diet (56). All these findings show that SIRT1 can improve muscle insulin sensitivity under different insulin-resistant conditions. Resveratrol can activate SIRT1 and protect mice against diet-induced obesity and insulin resistance (24, 31, 32). Moreover, resveratrol can alleviate insulin resistance in C2C12 myotubes via SIRT1 (24). Similarly, we show that resveratrol supplementation attenuates muscle insulin resistance in mice induced by constant darkness. All these findings show that CLOCK/BMAL1-dependent SIRT1 transcription is involved in the regulation of muscle insulin sensitivity and suggest that the activation of SIRT1 by small molecules, such as resveratrol, might be beneficial to attenuate muscle insulin resistance.
Taken together, this study shows that the down-regulation of CLOCK/BMAL1 is correlated with muscle insulin resistance, and CLOCK/BMAL1 regulates muscle insulin sensitivity via SIRT1 by enhancing its transcription. Moreover, resveratrol supplementation improves muscle insulin sensitivity in mice under a constant dark condition. Our findings provide insights for the coordination of the circadian clock and metabolism in muscle and a potential strategy for combating muscle insulin resistance correlated with circadian misalignment.
Appendix
Acknowledgments
We thank Kazuhiro Yagita (Department of Anatomy and Brain Science, Kobe University School of Medicine, Kobe, Japan) for providing the plasmids expressing CLOCK and BMAL1.
This work was supported by Grants 31470768, 81321062, 31200595, 31200591, and 81472181 from the National Natural Science Foundation of China; National Basic Research Program of China Grant 2014CB542300; the Knowledge Innovation Program of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences Grant 2014KIP106; and the Chinese Academy of Sciences/State Administration of Foreign Experts Affairs International Partnership Program for Creative Research Teams.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- Ad
adenovirus
- BMAL
brain and muscle aryl hydrocarbon receptor nuclear translocator-like protein
- CLOCK
circadian locomotor output cycles kaput
- CT
circadian time
- DD
constant darkness
- dT
deoxythymidine
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- Gsk3β
glycogen synthase kinase 3β
- InsR
insulin receptor
- LD
12-hour light, 12-hour dark cycle
- MTT
dimethylthiazol-diphenyltetra zolium-bromide
- p
phosphorylated
- PER2
period 2
- siRNA
small interfering RNA
- SIRT1
sirtuin 1
- WT
wild type
- ZT
zeitgeber time.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}