





Abstract
Nucleic acid-based therapies are being rapidly developed for prevention and management of cardiovascular diseases (CVD). Remarkable advancements have been achieved in the delivery, safety, and effectiveness of these therapeutics in the past decade. These therapies can also modulate therapeutic targets that cannot be sufficiently addressed using traditional drugs or antibodies. Among the nucleic acid-targeted therapeutics under development for CVD prevention are RNA-targeted approaches, including antisense oligonucleotides (ASO), small interfering RNAs (siRNA), and novel genome editing techniques.
Genetic studies have identified potential therapeutic targets that are suggested to play a causative role in development and progression of CVD. RNA- and DNA-targeted therapeutics can be particularly well delivered to the liver, where atherogenic lipoproteins and angiotensinogen (AGT) are produced. Current targets in lipid metabolism include proprotein convertase subtilisin/kexin type 9 (PCSK9), apolipoprotein A (ApoA), apolipoprotein C3 (ApoC3), angiopoietin-like 3 (ANGPTL3). Several large-scale clinical development programs for nucleic acid-targeted therapies in cardiovascular prevention are under way, which may also be attractive from a therapy adherence point of view, given the long action of these therapeutics. In addition to genome editing, the concept of gene transfer is presently under assessment in preclinical and clinical investigations as a potential approach for addressing low-density lipoprotein receptor deficiency. Furthermore, ongoing research is exploring the use of RNA-targeted therapies to treat arterial hypertension by reducing hepatic angiotensinogen (AGT) production.
This review summarizes the rapid translation of siRNA and ASO therapeutics as well as gene editing into clinical studies to treat dyslipidemia and arterial hypertension for CVD prevention. It also outlines potential innovative therapeutic options that are likely relevant to the future of cardiovascular medicine.
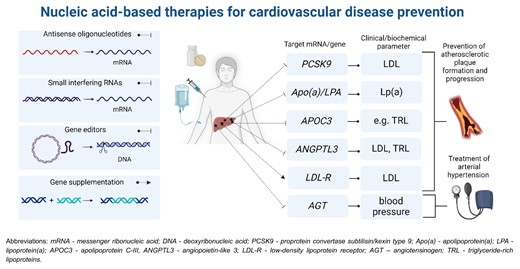
Nucleic acid-based therapies in cardiovascular prevention. Blunt arrows indicate inhibition of the target mRNA/gene (antisense oligonucleotides, small interfering RNAs, and gene editors) and a sharp arrow indicates gene supplementation.
1. Introduction
Large-scale genetic and epidemiological studies have unveiled novel targets that are thought to play pivotal roles in the development of atherosclerotic cardiovascular disease (ASCVD). Some of these targets, such as lipoprotein(a) (Lp(a)), prove challenging to tackle effectively using small-molecule drugs. In contrast, RNA- and DNA-targeted therapies offer the potential to target virtually any gene of interest. Recent advances in nucleic acid-based therapies have substantially enhanced their effectiveness, delivery methods, and safety profiles. Use of such drugs could reshape the prevailing approach to treating lipid disorders, particularly since long-term use of lipid-lowering drugs is often required to achieve prognostic benefits.
The concept of nucleic acid-targeting traces back to 1978 when the antisense approach was proposed.1 Another common RNA-targeting strategy—RNA interference (RNAi), which involves the regulation of gene expression through the silencing of specific genes using small RNA molecules—was described in 1990s. This discovery led to Andrew Z. Fire and Craig C. Mello being awarded the Nobel Prize in Physiology or Medicine in 2006.2 It facilitated extensive research efforts aimed at developing, refining, and implementing nucleic acid-targeted therapies. The first RNA-targeted drug to receive regulatory approval was the antisense oligonucleotide (ASO) fomivirsen used to treat cytomegalovirus infection in patients with acquired immunodeficiency syndrome (AIDS). In 2014, the clinical development of the first RNA-targeted drug for treating hypercholesterolemia, PCSK9 mRNA targeting siRNA inclisiran, was initiated. Inclisiran received approval for clinical use from the EMA in 2020 and from the Food and Drug Administration (FDA) in 2021. An ASO targeting APOC3 mRNA, volanesorsen, was approved by the EMA in 2019. There are ongoing preclinical and clinical studies assessing further nucleic acid-based therapeutics targeting Apo(a), ANGPTL3, PCSK9, and APOC3 for potential therapeutic applications. In addition to lipid-lowering drugs, RNA-targeting drugs for treating arterial hypertension are currently undergoing clinical development (Table 1).
Summary of nucleic acid-targeted drugs in the treatment of dyslipidemia and arterial hypertension
| Target | Drug name | Mechanism of action | Application | Company | Stage of development |
|---|---|---|---|---|---|
| Dyslipidemia | |||||
| PCSK9 | Inclisiran | siRNA | 284 mg s.c. Q6M (except for the second injection after 3 months) | Novartis Pharmaceuticals | Approved for clinical use (EMA and FDA) |
| CiVi008 | ASO | To be defined | CiVi Biopharma | Preclinical | |
| AZD6615 | ASO | – | AstraZeneca | Clinical (Phase 1, terminated) | |
| AZD8233 | ASO | Range 15–90 mg s.c. on days 1, 8, 29, and 57 | AstraZeneca | Clinical (Phase 2) | |
| AZD0780 | ASO | To be defined | AstraZeneca | Clinical (Phase 1) | |
| Verve-101 | Gene editing | Range 0.1–0.6 mg/kg | Verve Therapeutics | Clinical (Phase 1) | |
| CTX330 | Gene editing | To be defined | CRISPR Therapeutics | Preclinical | |
| Apo(a) | Pelacarsen | ASO | 80 mg s.c. Q1M | Novartis Pharmaceuticals | Clinical (Phase 3) |
| Olpasiran | siRNA | 225 mg s.c. Q12W | Amgen | Clinical (Phase 3) | |
| Lepodisiran (LY3819469) | siRNA | Range 4–608 mg s.c. (interval to be defined) | Eli Lilly and Company | Clinical (Phase 2) | |
| Zerlasiran (SLN360) | siRNA | To be defined | Silence Therapeutics plc | Clinical (Phase 2) | |
| CTX320 | Gene editing | To be defined | CRISPR Therapeutics | Preclinical | |
| ANGPTL3 | Vupanorsen | ASO | Range 80–329 mg s.c. Q1M | Pfizer/Ionis Pharmaceuticals | Discontinued |
| ARO-ANG3 | siRNA | Range 100–300 mg s.c. Q12W | Arrowhead Pharmaceuticals | Clinical (Phase 2) | |
| Verve-201 | Gene editing | To be defined | Verve Therapeutics | Preclinical | |
| CTX310 | Gene editing | To be defined | CRISPR Therapeutics | Preclinical | |
| APOC3 | Volanesorsen | ASO | 285 mg s.c. Q1W, after 3 months Q2W | Ionis Pharmaceuticals | Approved for clinical use (EMA) |
| Olezarsen | ASO | 50 mg, 80 mg s.c. Q4W | Ionis Pharmaceuticals | Clinical (Phase 3) | |
| Plozasiran (ARO-APOC3) | siRNA | 50 mg s.c. Q12W | Arrowhead Pharmaceuticals | Clinical (Phase 3) | |
| APOB | Mipomersen | ASO | 200 mg Q1W | Genzyme/Isis | Approved for clinical use (FDA) |
| LDL-R | Not denoted | gene therapy | 2.5E12–7.5E12 GC (genome copies)/kg | REGENXBIO Inc. | Clinical (Phase 1/2, terminated early) |
| Arterial hypertension | |||||
| AGT | Zileberisan | siRNA | Range 150–600 mg s.c. Q6M or 300 mg Q3M | Alnylam Pharmaceuticals | Clinical (Phase 2) |
| IONIS AGT-LRx | ASO | 80 mg s.c. Q1W | Ionis Pharmaceuticals | Clinical (Phase 2) | |
| Target | Drug name | Mechanism of action | Application | Company | Stage of development |
|---|---|---|---|---|---|
| Dyslipidemia | |||||
| PCSK9 | Inclisiran | siRNA | 284 mg s.c. Q6M (except for the second injection after 3 months) | Novartis Pharmaceuticals | Approved for clinical use (EMA and FDA) |
| CiVi008 | ASO | To be defined | CiVi Biopharma | Preclinical | |
| AZD6615 | ASO | – | AstraZeneca | Clinical (Phase 1, terminated) | |
| AZD8233 | ASO | Range 15–90 mg s.c. on days 1, 8, 29, and 57 | AstraZeneca | Clinical (Phase 2) | |
| AZD0780 | ASO | To be defined | AstraZeneca | Clinical (Phase 1) | |
| Verve-101 | Gene editing | Range 0.1–0.6 mg/kg | Verve Therapeutics | Clinical (Phase 1) | |
| CTX330 | Gene editing | To be defined | CRISPR Therapeutics | Preclinical | |
| Apo(a) | Pelacarsen | ASO | 80 mg s.c. Q1M | Novartis Pharmaceuticals | Clinical (Phase 3) |
| Olpasiran | siRNA | 225 mg s.c. Q12W | Amgen | Clinical (Phase 3) | |
| Lepodisiran (LY3819469) | siRNA | Range 4–608 mg s.c. (interval to be defined) | Eli Lilly and Company | Clinical (Phase 2) | |
| Zerlasiran (SLN360) | siRNA | To be defined | Silence Therapeutics plc | Clinical (Phase 2) | |
| CTX320 | Gene editing | To be defined | CRISPR Therapeutics | Preclinical | |
| ANGPTL3 | Vupanorsen | ASO | Range 80–329 mg s.c. Q1M | Pfizer/Ionis Pharmaceuticals | Discontinued |
| ARO-ANG3 | siRNA | Range 100–300 mg s.c. Q12W | Arrowhead Pharmaceuticals | Clinical (Phase 2) | |
| Verve-201 | Gene editing | To be defined | Verve Therapeutics | Preclinical | |
| CTX310 | Gene editing | To be defined | CRISPR Therapeutics | Preclinical | |
| APOC3 | Volanesorsen | ASO | 285 mg s.c. Q1W, after 3 months Q2W | Ionis Pharmaceuticals | Approved for clinical use (EMA) |
| Olezarsen | ASO | 50 mg, 80 mg s.c. Q4W | Ionis Pharmaceuticals | Clinical (Phase 3) | |
| Plozasiran (ARO-APOC3) | siRNA | 50 mg s.c. Q12W | Arrowhead Pharmaceuticals | Clinical (Phase 3) | |
| APOB | Mipomersen | ASO | 200 mg Q1W | Genzyme/Isis | Approved for clinical use (FDA) |
| LDL-R | Not denoted | gene therapy | 2.5E12–7.5E12 GC (genome copies)/kg | REGENXBIO Inc. | Clinical (Phase 1/2, terminated early) |
| Arterial hypertension | |||||
| AGT | Zileberisan | siRNA | Range 150–600 mg s.c. Q6M or 300 mg Q3M | Alnylam Pharmaceuticals | Clinical (Phase 2) |
| IONIS AGT-LRx | ASO | 80 mg s.c. Q1W | Ionis Pharmaceuticals | Clinical (Phase 2) | |
Summary of nucleic acid-targeted drugs in the treatment of dyslipidemia and arterial hypertension
| Target | Drug name | Mechanism of action | Application | Company | Stage of development |
|---|---|---|---|---|---|
| Dyslipidemia | |||||
| PCSK9 | Inclisiran | siRNA | 284 mg s.c. Q6M (except for the second injection after 3 months) | Novartis Pharmaceuticals | Approved for clinical use (EMA and FDA) |
| CiVi008 | ASO | To be defined | CiVi Biopharma | Preclinical | |
| AZD6615 | ASO | – | AstraZeneca | Clinical (Phase 1, terminated) | |
| AZD8233 | ASO | Range 15–90 mg s.c. on days 1, 8, 29, and 57 | AstraZeneca | Clinical (Phase 2) | |
| AZD0780 | ASO | To be defined | AstraZeneca | Clinical (Phase 1) | |
| Verve-101 | Gene editing | Range 0.1–0.6 mg/kg | Verve Therapeutics | Clinical (Phase 1) | |
| CTX330 | Gene editing | To be defined | CRISPR Therapeutics | Preclinical | |
| Apo(a) | Pelacarsen | ASO | 80 mg s.c. Q1M | Novartis Pharmaceuticals | Clinical (Phase 3) |
| Olpasiran | siRNA | 225 mg s.c. Q12W | Amgen | Clinical (Phase 3) | |
| Lepodisiran (LY3819469) | siRNA | Range 4–608 mg s.c. (interval to be defined) | Eli Lilly and Company | Clinical (Phase 2) | |
| Zerlasiran (SLN360) | siRNA | To be defined | Silence Therapeutics plc | Clinical (Phase 2) | |
| CTX320 | Gene editing | To be defined | CRISPR Therapeutics | Preclinical | |
| ANGPTL3 | Vupanorsen | ASO | Range 80–329 mg s.c. Q1M | Pfizer/Ionis Pharmaceuticals | Discontinued |
| ARO-ANG3 | siRNA | Range 100–300 mg s.c. Q12W | Arrowhead Pharmaceuticals | Clinical (Phase 2) | |
| Verve-201 | Gene editing | To be defined | Verve Therapeutics | Preclinical | |
| CTX310 | Gene editing | To be defined | CRISPR Therapeutics | Preclinical | |
| APOC3 | Volanesorsen | ASO | 285 mg s.c. Q1W, after 3 months Q2W | Ionis Pharmaceuticals | Approved for clinical use (EMA) |
| Olezarsen | ASO | 50 mg, 80 mg s.c. Q4W | Ionis Pharmaceuticals | Clinical (Phase 3) | |
| Plozasiran (ARO-APOC3) | siRNA | 50 mg s.c. Q12W | Arrowhead Pharmaceuticals | Clinical (Phase 3) | |
| APOB | Mipomersen | ASO | 200 mg Q1W | Genzyme/Isis | Approved for clinical use (FDA) |
| LDL-R | Not denoted | gene therapy | 2.5E12–7.5E12 GC (genome copies)/kg | REGENXBIO Inc. | Clinical (Phase 1/2, terminated early) |
| Arterial hypertension | |||||
| AGT | Zileberisan | siRNA | Range 150–600 mg s.c. Q6M or 300 mg Q3M | Alnylam Pharmaceuticals | Clinical (Phase 2) |
| IONIS AGT-LRx | ASO | 80 mg s.c. Q1W | Ionis Pharmaceuticals | Clinical (Phase 2) | |
| Target | Drug name | Mechanism of action | Application | Company | Stage of development |
|---|---|---|---|---|---|
| Dyslipidemia | |||||
| PCSK9 | Inclisiran | siRNA | 284 mg s.c. Q6M (except for the second injection after 3 months) | Novartis Pharmaceuticals | Approved for clinical use (EMA and FDA) |
| CiVi008 | ASO | To be defined | CiVi Biopharma | Preclinical | |
| AZD6615 | ASO | – | AstraZeneca | Clinical (Phase 1, terminated) | |
| AZD8233 | ASO | Range 15–90 mg s.c. on days 1, 8, 29, and 57 | AstraZeneca | Clinical (Phase 2) | |
| AZD0780 | ASO | To be defined | AstraZeneca | Clinical (Phase 1) | |
| Verve-101 | Gene editing | Range 0.1–0.6 mg/kg | Verve Therapeutics | Clinical (Phase 1) | |
| CTX330 | Gene editing | To be defined | CRISPR Therapeutics | Preclinical | |
| Apo(a) | Pelacarsen | ASO | 80 mg s.c. Q1M | Novartis Pharmaceuticals | Clinical (Phase 3) |
| Olpasiran | siRNA | 225 mg s.c. Q12W | Amgen | Clinical (Phase 3) | |
| Lepodisiran (LY3819469) | siRNA | Range 4–608 mg s.c. (interval to be defined) | Eli Lilly and Company | Clinical (Phase 2) | |
| Zerlasiran (SLN360) | siRNA | To be defined | Silence Therapeutics plc | Clinical (Phase 2) | |
| CTX320 | Gene editing | To be defined | CRISPR Therapeutics | Preclinical | |
| ANGPTL3 | Vupanorsen | ASO | Range 80–329 mg s.c. Q1M | Pfizer/Ionis Pharmaceuticals | Discontinued |
| ARO-ANG3 | siRNA | Range 100–300 mg s.c. Q12W | Arrowhead Pharmaceuticals | Clinical (Phase 2) | |
| Verve-201 | Gene editing | To be defined | Verve Therapeutics | Preclinical | |
| CTX310 | Gene editing | To be defined | CRISPR Therapeutics | Preclinical | |
| APOC3 | Volanesorsen | ASO | 285 mg s.c. Q1W, after 3 months Q2W | Ionis Pharmaceuticals | Approved for clinical use (EMA) |
| Olezarsen | ASO | 50 mg, 80 mg s.c. Q4W | Ionis Pharmaceuticals | Clinical (Phase 3) | |
| Plozasiran (ARO-APOC3) | siRNA | 50 mg s.c. Q12W | Arrowhead Pharmaceuticals | Clinical (Phase 3) | |
| APOB | Mipomersen | ASO | 200 mg Q1W | Genzyme/Isis | Approved for clinical use (FDA) |
| LDL-R | Not denoted | gene therapy | 2.5E12–7.5E12 GC (genome copies)/kg | REGENXBIO Inc. | Clinical (Phase 1/2, terminated early) |
| Arterial hypertension | |||||
| AGT | Zileberisan | siRNA | Range 150–600 mg s.c. Q6M or 300 mg Q3M | Alnylam Pharmaceuticals | Clinical (Phase 2) |
| IONIS AGT-LRx | ASO | 80 mg s.c. Q1W | Ionis Pharmaceuticals | Clinical (Phase 2) | |
Another significant breakthrough in the field occurred in 2012, when CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9) was first proposed as a potential therapeutic approach to target disease-causing genes. Although still in early stages of clinical development, CRISPR-based therapies hold tremendous promise. A first genome editing drug to reduce low-density lipoprotein cholesterol (LDL-C) in patients with atherosclerotic cardiovascular disease (ASCVD) and heterozygous familial hypercholesterolemia (HeFH)—a base editor (BE) targeting PCSK9—entered a clinical trial in 2022.
The purpose of this review is to cover basic principles of RNA- and DNA-targeted therapeutic strategies as well as update the reader on rapidly developing therapeutics in the field of cardiovascular prevention with focus on dyslipidemia and arterial hypertension.
2. Overview of nucleic acid-targeted therapies
2.1 General principles
Nucleic acid-targeted therapeutics represent a broad class of agents that inhibit gene expression or function, aiming to suppress disease-associated proteins selectively. The core principle behind the therapeutic use of nucleic acids, aside from gene supplementation therapy, involves suppressing DNA or RNA expression. This action may effectively stop the synthesis of disease-associated proteins without impacting the production of other proteins.
2.2 Delivery methods
The delivery of nucleic acid-targeting drugs poses a significant challenge, requiring sophisticated strategies to ensure that these therapeutic agents reach their intended target cells within the body. These drugs must navigate through the complex biological environment to exert their action at the cellular level. Effective delivery systems are crucial for overcoming barriers such as enzymatic degradation in the bloodstream, nonspecific uptake by cells, and the need to cross cell membranes to access intracellular targets. Lipid nanoparticles (LNPs), viral vectors, and conjugation with targeting ligands, such as GalNAc (N-Acetylgalactosamin), are among the innovative approaches developed to enhance the stability, specificity, and intracellular delivery of nucleic acid-based therapies. These delivery technologies not only improve the pharmacokinetics and biodistribution of nucleic acid drugs but also minimize off-target effects. Here, we will briefly discuss delivery systems for RNA and DNA therapeutics, whereas this topic was reviewed extensively elsewhere.3–5
2.2.1 Nanoparticles
The most common forms of nanoparticles for drug delivery are LNPs. LNPs are 50–100 nm particles, consisting of ionizable lipids, which facilitate endosomal escape and nucleic acid delivery into the cytoplasm; structural lipids, like phospholipids, which stabilize the LNP structure; cholesterol, which supports membrane fluidity and stability; and polyethylene glycol (PEG)-lipids, which extend circulation time by reducing immune detection.6 LNP-mRNA formulations protect mRNA from degradation, enabling its delivery into the targeted cells. Upon entering cells via endocytosis, LNPs facilitate the release of mRNA into the cytoplasm.6 An example of RNA-targeting drug delivered via LNPs is patisiran (marketed as Onpattro) to treat hereditary ATTR.7,8 To note, two well-known COVID-19 vaccines, Pfizer-Biontech and Moderna utilize mRNA-LNP drug delivery system.9
2.2.2 Conjugation-based delivery systems
One notable and successful example of ligand conjugate is GalNAc. GalNAc binds to the asialoglycoprotein receptor (ASGPR) that is abundantly expressed on the sinusoidal surface of hepatocytes (∼500 000 receptors per cell), while the extrahepatic expression is minimal. Upon binding, hepatocytes quickly internalize sialyl-GalNAc molecules through endocytosis.10–12 The acidic conditions within the endosome prompt the release of GalNAc-conjugates from ASGPR, allowing the receptor to return to the liver cell surface. This process involves GalNAc binding to ASGPR, initiating endocytosis within approximately 15 min, during which a small fraction of the siRNA or ASO cargo is delivered into the hepatocyte cytoplasm, triggering the desired RNA drug effects. The ASGPR mechanism facilitates the entry of siRNAs/ASOs into liver cells at a rate about 100 times higher than that achieved by any other receptor–ligand interaction. However, a significant hurdle remains: less than 1% of siRNA/ASO successfully escapes the endosome to reach the hepatocyte cytoplasm. Despite this low endosomal escape rate, the abundant expression and rapid recycling of ASGPR on hepatocyte surfaces enable effective targeting.4,12 This unique capacity of ASGPR, unmatched by any other ligand–receptor system, highlights the challenge of enhancing endosomal escape—a critical focus for developing next-generation RNA-based therapeutics.12
2.3 RNA-targeted approaches
2.3.1 ASOs
The ASOs were first described in 1978, when they were proposed as a potential therapeutic approach for inhibiting the Rous sarcoma virus.1 The first FDA approved ASO drug was fomivirsen (in 1998), to treat cytomegalovirus retinitis in patients with AIDS. It was, however, withdrawn in 2001 due to the significant decline in the occurrence of opportunistic infections in patients with AIDS.13
The ASOs are short, single-stranded nucleic acid polymers complementary to the specific RNA through base-pairing. Depending on the mechanism of action, ASOs can be divided into two subcategories: RNase-H competent (Figure 1A) and steric block (‘occupancy-only’). In the first case, the RNA–DNA heteroduplex complex is formed when ASOs bind to mRNA with further recruitment of the endogenous enzyme RNase-H, which catalyzes mRNA degradation (Figure 1B). Steric block ASOs do not bind to RNase-H and therefore do not result in direct degradation of mRNA. But they can modulate the gene expression by interfering with the interaction between mRNA and cellular machinery, such as splicing factors.4,14
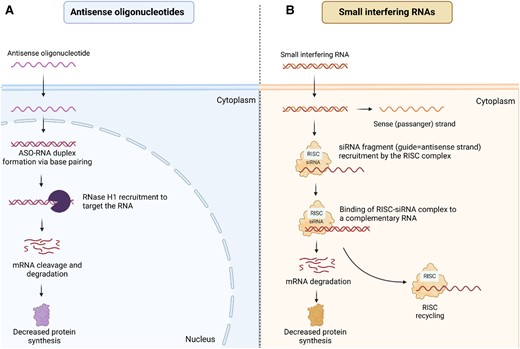
Mechanism of action of ASOs and siRNAs. (A) the RNA–DNA heteroduplex complex is formed when ASOs bind to mRNA with further recruitment of the endogenous enzyme RNase-H, which catalyzes mRNA degradation. (B) siRNA molecules are introduced into the cytoplasm, upon entering the cytoplasm, siRNA molecules bind to argonaute (AGO) proteins, which are part of the RNA-induced silencing complex (RISC). Within the RISC complex, the passenger strand of siRNA is separated from the guide strand, which remains bound to the AGO protein as the mature RISC complex. The guide strand within the RISC complex uses its sequence to recognize complementary mRNA molecules, usually within the cytoplasm of the same cell. Once the target mRNA is recognized, the AGO protein within the RISC complex cleaves the mRNA molecule, leading to its degradation and subsequent gene silencing. This cleavage activity is carried out by the RNAse H1 domain present in AGO proteins. ASO, antisense oligonucleotide; siRNA, small interfering RNA; RISC, RNA-induced silencing complex.
Nuclease degradation in vivo was the major problem in the early stages of ASO development. Phosphorothiorate linkages in the ASO backbone were the first modification introduced to overcome this limitation (first generation ASOs).14 Sugar backbone moieties were further modified to reduce nonspecific off-target effects. As a result, second-generation ASOs have higher potency and stability. Third generation ASOs have even higher stability in vivo as they are practically resistant to degradation by enzymes. Overall, during the past decades, different chemical modifications were introduced to increase the potency, stability, and target affinity.14,15 Further, conjugation to GalNAc enabled significant improvement in tissue-directed delivery. Asialoglyprotein receptor, which is abundantly expressed on the surface of hepatocytes (∼500 000 receptors per cell), possesses strong binding affinity to GalNAc-terminated oligosaccharides, which enables GalNAc-conjugated therapeutics to achieve their pharmacodynamic effect at lower doses and with infrequent administrations.16
2.3.2 siRNAs
RNA interference (RNAi) is a natural biological process that inhibits gene expression by inducing mRNA degradation. The mechanism was first described in 1998 by Fire and Mello, who received the Nobel prize in 2006 for their discovery.17,18 RNA interference is a natural defensive mechanism against endogenous parasitic and exogenous pathogenic nucleic acids. Different therapeutics, such as siRNAs and micro-RNAs (miRNAs) can act in an analogous manner and silence the expression of target genes.
siRNAs contain 20–24 nucleotides and induce RNAi via targeted mRNA degradation (Figure 1B). After entering the cytoplasm, siRNA is incorporated into argonaute protein (AGO) as wells other proteins [Dicer, transactivation response element RNA-binding protein, Argonaute 2 (AGO 2)], forming a complex, which precedes RNA-induced silencing complex (RISC). In the next step, the passenger strand of siRNA is detached from the complex, while the guide strand and AGO protein form RISC–siRNA complex. RISC uses siRNA to recognize complementary mRNA, which then cleaved by cellular exonucleases.19
Like antisense drugs, siRNAs have some unfavorable properties, such as low stability and possible off-target effects. Modifications in phosphate backbone and sugar moiety as well as base modifications are some examples of structural changes introduced to overcome these limitations.20 Furthermore, conjugation with GalNAc increases the selective targeting and facilitates siRNA uptake by hepatocytes.21,22
2.4 CRISPR-based genome editing approaches
2.4.1 CRISPR/Cas9
Although RNA-targeted therapeutics have strongly evolved in the past years to increase both efficacy and safety, the concept of long-lasting effect remains very attractive. Gene editing tools allow to ‘cut’ the genes of interest in irreversible manner, allowing long-term effect without need for repeated drug administrations.23 Unquestionably, the discovery of CRISPRs catalyzed the progress in the field of genome editing. The first description of short palindromic repeat DNA sequences goes back to 1987,24 although the name was attributed later, in 2002. CRISPRs were initially thought to be a novel DNA repair mechanism in thermophilic archaea and bacteria. The similar sequences were then found in bacteriophages, viruses, and plasmids.25 In 2012, Jennifer Doudna and Emmanuelle Charpentier developed a groundbreaking technology that allows precise delivery of CRISPR/Cas system to specific sites in the human genome.26 In recognition of this significant achievement, they were awarded Nobel Prize in Chemistry in 2020.27
Recently, the FDA approved the first CRISPR/Cas9-based drug, Casgevy (exagamglogene autotemcel) for the treatment of sickle-cell disease (SCD) in patients aged ≥ 12 years.28–30 The drug also received a positive opinion from the EMEA (European Medicines Evaluation Agency) in December 2023 for the treatment of SCD and transfusion-dependent β-thalassemia.29,30 Casgevy is a cell-based therapy utilizing autologous CD34+ hematopoietic stem cells (HSCs) edited via CRISPR/Cas9 to target the BCL11A gene, which normally suppresses γ-globin and fetal hemoglobin (HbF) production in erythroid cells. The process involves collecting the patient's HSCs to isolate CD34+ cells for ex vivo gene editing with the CRISPR/Cas9 complex through electroporation. This modification reduces BCL11A expression, enhancing HbF production, which is associated with better outcomes in SCD due to elevated HbF levels.
A notable example of an advanced CRISPR/Cas9-based therapy in the field of cardiovascular therapy is NTLA-2001, which targets hepatic transthyretin production. This CRISPR/Cas9-based drug has been in a phase 3 trial for patients with transthyretin-mediated amyloidosis (ATTR) cardiomyopathy since December 2023, aiming to enroll more than 700 participants.31
Most CRISPR/Cas systems consist of several elements: (i) CRISPR arrays—repetitive DNA sequences, (ii) Cas proteins (most well-known Cas9), (iii) guide RNA (gRNA), (iv) protospacer adjacent motifs (PAMs), and (v) repair template. The process of editing the genome with CRISPR/Cas involves three primary stages: recognition, cleavage, and repair.32 The gRNA is responsible for the navigation of Cas nuclease and recognition of the target sequence in the gene of interest. Cas nuclease cleaves target DNA by inducing DNA double-strand break located downstream of the cut site, varying in length (2–5 base pairs) across bacterial species. Based on the structure and functions of Cas protein, CRISPR–Cas systems are divided into Class I and Class II.33,34 Class I systems include multi-subunit Cas-protein complexes, and Class II consists of a single Cas protein. The first used and the most common Cas protein is extracted from Streptococcus pyogenes (SpCas9). The PAM sequence, a short DNA sequence located downstream of the cut site, enables the Cas protein to specifically recognize a host's PAM sequence.32 After recognizing the target PAM, Cas9 induces local DNA melting, forming RNA–DNA hybrid. Subsequently, the DNA can be cleaved. Introduced double-strand breaks are repaired the cellular machinery: non-homologous end-joining (NHEJ) or homology directed repair (HDR), Figure 2A. HDR enables the incorporation of sequence from the exogenous DNA template at the DNA double stranded break site. HDR allows precise editing but has a low editing efficiency due to restriction to the G2 and S phases of the cell cycle.35 On the other hand, NHEJ is generally considered to be more efficient compared to HDR but can result in indels (insertions of deletions) at unintended genomic locations, leading to off-target mutations. HDR can be used for introducing specific mutations in the presence of an exogenous DNA template. However, due to lower efficiency, NHEJ remains the dominant repair pathway. To enhance HDR's precision and efficacy, strategies like inhibiting the NHEJ are undertaken.36,37
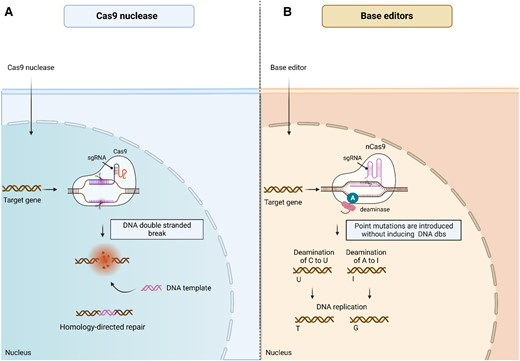
Gene editing by CRISPR-based drugs. (A) Conventional CRISPR–Cas9 system contains sgRNA, which is complementary to the target DNA sequence and a Cas9 enzyme. The CRISPR/Cas9 complex binds to the target DNA sequence and cuts it at a specific site, creating a DBS. DNA repair pathways include non-homologous end joining (not shown) or homology-directed repair. NHEJ can introduce small mutations, while HDR allows for precise DNA sequence changes using DNA template delivered by the CRISPR/Cas9 system. The resulting repaired DNA leads to desired gene edits. (B) Base editing is a form of gene editing that allows for precise changes to individual nucleotide bases in the DNA molecule. It involves fusing the Cas9 nickase with a cytidine deaminase enzyme (for C-to-T edits) or an adenine deaminase enzyme (for A-to-G edits) to create a base editor complex. The complex is guided to the target DNA sequence by a sgRNA. Once bound to the DNA, the base editor modifies the target base without creating a DSB.
2.4.2 Base editors
Base editors (BEs) were invented in 2016 by the research group of David Liu at the Harvard University.38 BEs have emerged as a gene editing methodology that does not require the induction of DNA double-strand breaks (DSBs), unlike the conventional CRISPR/Cas9 system. BEs consist of a fusion protein that combines a deaminase enzyme with an impaired Cas nuclease generated from Streptococcus pyogenes Cas9 (SpCas9). These proteins are joined together using the SpCas9 gene. The impaired Cas nuclease can either be a catalytically dead Cas9 (dCas9) carrying mutations that render it unable to cleave DNA double strands, or a Cas9 nickase (nCas9) with the D10A mutation that nicks the non-edited DNA strand. The fusion protein is recruited to the target DNA in a way that is programmed by gRNA, but without induction of DSBs. The deaminase enzyme, either cytosine deaminase (in cytosine BEs) or adenine deaminase (in adenine BEs), is grafted onto the dysfunctional Cas9 gene. The ability of the cytosine BEs to convert cytidine (C) to thymidine (T) and the ability of the adenine BEs to convert adenosine (A) to guanosine (G) enables the exact editing of nucleotides in the DNA sequence (Figure 2B). Because they do not require the induction of DSBs, BEs are more convenient and refined for accurate gene editing. They also provide a method that is template-free, which means that they do not require a donor template for HDR. This makes BEs particularly useful for editing non-dividing cells or difficult-to-transfect cells that may have low HDR efficiency. Studies have shown that BEs cause less off-target effects compared to conventional Cas9 nucleases.39 However, BEs also have certain limitations. First, BEs are not able to install certain transversion mutations, which could be required for specific gene editing applications. Secondly, an off-target editing induced by deaminases that is similar to Cas9 off-target editing has been reported.40 To overcome the limitations of BEs and Cas9 nuclease systems, other approaches, such as prime editing, have been developed.41,42
2.5 Gene supplementation
The initial meaning of the term ‘gene therapy’ refers to the administration of nucleic acid sequences to tissues and organs, with the aim of restoring the function of genes that are relevant to the pathophysiology of certain diseases and are either downregulated or absent. In recent years, the term has been expanded with gene editing technologies to target und ‘cut’ specific genes, such as CRISPR-based therapies. Therefore, it is important to distinguish between these two terms, as the first refers to the supplementation of gene copies without altering the intrinsic genome, while the second term involves modifications in the inherit DNA. The term ‘gene supplementation’ or ‘gene transfer therapy’ is used to describe the process of gene copy supplementation/addition.
To date, alipogene tiparvovec (Glybera®) is the only gene transfer drug in the cardiovascular field, which received approval from the EMEA. The drug was used to treat lipoprotein lipase (LPL) deficiency, but was subsequently withdrawn from the market due to very high costs.43 Most of the gene therapies that have gained approval from the regulatory agencies are utilized for the treatment of hematologic and neuromuscular disorders.44
Due to its potential in addressing gene deficiencies, the gene transfer is investigated as a viable strategy within the field of FH treatment. Just recently, a phase 1/2a trial (NCT02651675) evaluated recombinant adeno-associated virus (rAAV)-based liver-directed human LDL-R gene therapy in patients with homozygous familial hypercholesterolemia (HoFH).45 The study was terminated early as the drug development was discontinued due to financial reasons.
3. Nucleic acid-targeted drugs in treatment of lipid disorders
Elevated LDL-C levels are a causal risk factor for CVD.46 According to the EAS/ESC guidelines, it is recommended to lower LDL-C levels to specific targets, which vary based on an individual’s risk category.47 Despite the effectiveness of existing lipid-lowering medications like statins, ezetimibe, bempedoic acid, and PCSK9 antibodies in reducing LDL-C levels, there is still a significant challenge in achieving and maintaining these target levels.48,49 Nucleic acid-targeted drugs have the potential to improve patient adherence and support the long-term achievement of LDL-C goals. Furthermore, some atherogenic lipoproteins, such as Lp(a), cannot be addressed using traditional small molecule drugs or antibodies. Presently, there are no approved drugs for reducing Lp(a) concentrations. In this context, innovative siRNAs and ASOs designed to target LPA mRNA present a unique opportunity to tackle hyperlipoproteinemia(a).
Four major targets in nucleic acid-targeted treatment of dyslipidemias are PCSK9, Apo(a), APOC3, and ANGPTL3. These proteins are produced in the liver and play pivotal roles in the regulation of lipid metabolism. Rationale for each target identification and corresponding therapeutics is discussed in respective sections.
3.1 PCSK9
PCSK9 is a protease that is produced in the liver and plays a role in the regulation of plasma LDL-C levels. When secreted into the circulation, PCSK9 binds to the epidermal growth factor-like A (EGF-A) domain of the LDL-R on the hepatocyte surface. Normally LDL-R attaches LDL particle and ensures its transport into the endosome, where LDL is degraded by the lysosomes. At the same time, LDL-R is recycled into the surface of hepatocyte and can bind more LDL particles. PCSK9 prevents this process by binding to LDL-R, as PCSK9–LDL-R undergoes internalization and degradation upon formation (Figure 3).
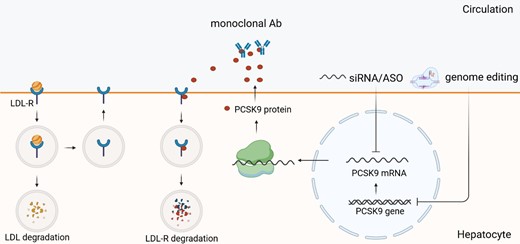
PCSK9 synthesis, secretion, and inhibition by nucleic acid-targeted therapies. PCSK9 is synthesized in the liver. After secretion, it binds to LDL-R, thus leading to LDL-R internalization and degradation (‘coupled degardation’), and preventing it from recycling. Therefore, inhibiting PCSK9 results in LDL-C reduction. LDL-R, low-density lipoprotein receptor; PCSK9, proprotein convertase subtilisin/kexin type 9.
Loss-of-function mutations have been reported in apparently healthy individuals with LDL levels ∼14–16 mg/dL. Furthermore, the analysis of PCSK9 sequence variants showed that individuals with PCSK9 non-sense mutations had significantly lower risk of coronary heart disease50 as well as calcific aortic disease.51 This has led to the development of various approaches targeting PCSK9 as a potential therapeutic strategy for reducing LDL-C concentrations and minimizing cardiovascular risk.22,52
Due to the flat and featureless nature of the PCSK9: EGF-A interaction surface, the development of small molecules targeting PCSK9 protein has been a challenge.53 Monoclonal antibodies—evolocumab and alirocumab—were the first PCSK9 inhibitors that were developed. They are administered through subcutaneous injections, usually given twice a month. They have shown a substantial reduction in LDL-C and major cardiovascular outcomes in patients with ASCVD in large randomized trials.54,55 Besides monoclonal antibodies, there are ongoing developments of other drug classes aimed at reducing the interaction between PCSK9 and LDL-R by targeting the PCSK9 protein. These include, but are not limited to, cyclic peptides and adnectins.53,56
The predominant expression of PCSK9 in hepatocytes makes it a highly appealing candidate for nucleic acid-targeted interventions. Inclisiran, the first siRNA to target PCSK9, has been approved in 2020. Other siRNAs and ASOs too are being currently developed. Genome editing technologies, such BEs, have been employed to directly modify the PCSK9 gene.
3.1.1 PCSK9 mRNA targeting
3.1.1.1 siRNA inclisiran
Inclisiran, a GalNAc-modified siRNA that inhibits PCSK9 production, has been approved by the EMEA in 202057 and by the FDA in 2021.58 It is indicated in familial hypercholesterolemia or as an add-on to statins in patients who do not achieve optimal LDL-C reduction on maximally tolerated statin therapy. Results from the two phase 3 trials, ORION-10 and ORION-11, demonstrated a placebo-adjusted LDL-C reduction by ∼50% from baseline to day 510 with biyearly subcutaneous injections.59 Just recently, a long-term open label extension study of phase 2 trial, ORION-3, confirmed sustained LDL-C reduction over 4 years without any significant safety concerns.60 Further, ORION-9 study demonstrated LDL-C lowering efficacy of inclisiran in patients with heterozygous familial hypercholesterolemia, showing 39% reduction in LDL-C.61
While inclisiran effectively reduced LDL-C in previous trials, its effect on cardiovascular and total outcomes is currently being explored in large long-term trial, ORION-4 (NCT03705234), which will include 15 000 participants to receive inclisiran vs. placebo.62 In the meanwhile, a prespecified exploratory patient-level analysis of ORION-9, ORION-10, and ORION-11 trials showed a significant reduction in major cardiovascular outcomes [OR 0.74 (95% CI 0.58–0.94)] over 18 months in 3655 patients.63 Further, VICTORION program should assess inclisiran efficacy and safety in different populations and clinical settings. The program includes VICTORION-2 PREVENT trial (NCT05030428), which investigates whether inclisiran (used as add-on to statins) will prevent major cardiovascular events (MACE) in patients with ASCVD.62 Further trials in the VICTORION program include (but are not limited to): VICTORION-INITIATE (patients with ASCVD, receiving inclisiran as ‘first step’ strategy),64 VICTORION-INCEPTION (patients with acute coronary syndrome),65 and VICTORION-PLAQUE (a study using computed tomography angiography to assess the effect of inclisiran in addition to maximally tolerated statin therapy on atherosclerotic plaque progression).66
3.1.1.2 ASO CIVI 007
CIVI 007 was a third generation ASO. A phase 2a trial to assess the safety, tolerability, pharmacodynamics and pharmacokinetics of the drug was completed in the late 2020 (NCT04164888).67 However, no results or changes were reported on clinicaltrials.gov since 2020. According to the CiVi Biopharma website, an agent called CiVi008 is currently undergoing preclinical assessment.68
3.1.1.3 ASO ION 449
ION449 is a PCSK9-targeting ASO, which is formulated in two ways: AZD8233 for subcutaneous administration, and AZD6615 for oral administration. A phase 2 dose-finding trial, ETESIAN (NCT04641299), demonstrated a 79% reduction in LDL-C levels and 44.7% reduction in Lp(a) levels with the maximum dose of AZD8233.69 The first clinical trial to investigate the oral formulation, AZD6615, was terminated due to the changes in the cohort design (NCT04055168).70 As of the submission date of this publication, there are no further trials registered on clinicaltrials.gov that aim to investigate AZD6615.
3.1.1.4 ASO AZD0780
Another orally administered PCSK9-targeting ASO, AZD0780, is currently in a phase 1 trial (NCT05384262).71 Oral ASOs could provide an alternative option for individuals who prefer taking medications orally instead of through subcutaneous injections. This also could help avoid side effects commonly associated with injection-site reactions.
3.1.2 PCSK9 gene editing
3.1.2.1 CRISPR/Cas9
Considering that loss-of-function mutations in PCSK9 have been observed in healthy individuals, the gene has emerged as a promising target for DNA-targeted therapies. In initial in vivo studies, CRISPR/Cas9 and gRNA were delivered using adenovirus vectors to target the PCSK9 gene, resulting in a substantial 90% decrease in plasma protein levels. However, adenovirus vectors contain viral coding sequences that may trigger an immune response, posing potential immunogenicity concerns. Subsequent studies have utilized adenovirus-associated vectors, which exhibit lower protein expression levels and the potential for sustained gene expression. Adeno-associated viruses (AAVs) are believed to be non-pathogenic in humans and do not typically induce significant immune responses, making them an attractive alternative for gene delivery in comparison to adenovirus vectors.72
Prolonged expression of Cas9 protein has been associated with higher rate of off-target cleavage, cytotoxicity, and immunogenicity. To tackle this issue in context of PCSK9 gene targeting, Li et al. have engineered an all-in-one self-cleavage AAV-CRISPR/Cas9 system, in which Cas9 cuts both genomic locus and the AAV vector, leading to self-limitation of Cas9 expression.73 The system resulted in stable and sufficient reductions of PCSK9 and cholesterol levels. However, 40% of the modified Cas9 (SaCas9) protein was still expressed after 24 weeks, indicating that complete removal of Cas9 could not be achieved. Nevertheless, according to the analysis of off-target effects via next-generation sequencing, there was a 20-fold reduction in off-target activity at 24 weeks after AAV administration. Currently, several PCSK9-gene targeting CRISPR/Cas9 drugs are in the preclinical stage of development, such as CTX330 by CRISPR Therapeutics. Another candidate PBGENE-PCSK9 from Precision Biosciences/Iecure was discontinued at preclinical stage.74,75
3.1.2.2 BE Verve-101
First base editing system targeted against PCSK9 gene was described in 2019 by Musunuru et al.76 The deoxyadenosine deaminase domain was combined with Streptococcus pyogenes nickase Cas9, creating a system that chemically modifies adenosine nucleosides on one DNA strand, resulting in highly efficient A-T to G-C transition mutations at the target site. This approach achieved approximately 60% editing efficiency and a 60% reduction in both PCSK9 protein and LDL-C levels, which remained sustainable over a long-term period. The BE demonstrated an 83% PCSK9 reduction and 69% LDL-C reduction in non-human primates. The effects were durable, lasting up to 476 days after dosing.77 These data supported the initiation of the first-ever human phase 1b trial, HEART-1, to investigate the drug, later named Verve-101, in New Zealand in 2022 (NCT05398029).78 The study aims to recruit 44 participants with familial hypercholesterolemia, and the first participant received the injection in July 2022. Recently, at the AHA Scientific Sessions, interim results were presented. Nine participants diagnosed with HeFH—comprising 7 males and 2 females—received a single intravenous infusion of VERVE-101 at various doses. LDL-C was reduced by 39 and 48% in the two participants receiving 0.45 mg/kg of the drug and by 55% in a sole participant who received the maximal dose—0.6 mg/kg.79 During the trial, four participants experienced reactions related to the infusion process. Additionally, three individuals reported upper respiratory infections, including COVID-19. One participant, who received a 0.6 mg/kg dosage, showed a temporary rise in liver enzymes, suggesting potential liver inflammation or damage. Two participants experienced cardiovascular events: one died from cardiac arrest five weeks after receiving VERVE-101 (0.3 mg/kg), and the other had myocardial infarction one day after the drug administration (0.45 mg/kg). Both cases were interpreted in the context of underlying severe ASCVD. An independent safety board reviewed the cases and recommended that trial enrollment be continued without changes to the drug protocol.79,80 Nonetheless, in the recent update, VERVE Therapeutics announced that the trial was temporarily paused due to a serious adverse in a patient who received 0.45 mg/kg dosage. The participant suffered a Grade 3 transient elevation in serum alanine aminotransferase and a serious Grade 3 event of drug-induced thrombocytopenia within the first 4 days following treatment.81
3.2 Apo (a)/Lp(a)
Lipoprotein(a) [Lp(a)] distinguishes itself from LDL particles due to the presence of Apo(a) covalently bound to ApoB (Figure 4). The Apo(a) protein is encoded by the LPA gene, which is highly similar to plasminogen, and contains a series of kringle domains.82,83 The plasma concentrations of Lp(a) are predominantly determined by LPA gene expression, with over 90% heritability in populations of European and African descent.84 Since its initial description in 1963,85 numerous epidemiological, genome-wide association, and mendelian randomization studies have provided compelling evidence supporting the causal role of Lp(a) in the development of atherosclerotic disease and calcific aortic disease.86–90 An analysis of Lp(a) concentrations and the incidence of ASCVD in the UK Biobank revealed a linear relationship between the two, both in the context of primary and secondary prevention.91,92
As per data from global epidemiological studies, more than 25% of patients with vascular disease have Lp(a) levels surpassing the threshold for increased cardiovascular risk (50 mg/dL or 125 nmol/L).93 At present, there are no approved drugs specifically targeting high Lp(a) levels. According to the recommendations of the European Atherosclerosis Society panel, individuals with elevated Lp(a) should receive optimal treatment to mitigate the risk of ASCVD.92 This involves addressing other contributing factors, such as LDL-C, blood pressure, glucose levels, and lifestyle. In cases where patients have very high Lp(a) levels and progressive ASCVD despite optimal correction of other risk factors, lipoprotein apheresis may be recommended as a treatment option.
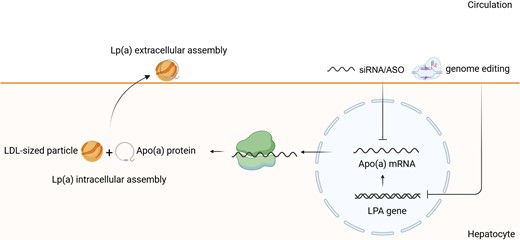
Lp(a) assembly, secretion, and inhibition by nucleic acid-targeted therapies. Lp(a) is an LDL-like particle bound to APOA protein encoded by LPA gene. After transcription of LPA gene, Apo(a) mRNA is translated into Apo(a) protein. The first step of Lp(a) assembly occurs within the cell, where Apo(a) protein is coupled with LDL-sized particle. Upon secretion into the surface, Lp(a) extracellular assembly takes place. Inhibition of Apo(a) mRNA or LPA gene results in Lp(a) reduction. Lp(a), lipoprotein(a); Apo(a), apolipoprotein(a); LPA, lipoprotein gene.
Antibodies against PCSK9 and siRNA targeting PCSK9 have been shown to moderately reduce Lp(a) levels by approximately 25% from baseline.94,95 However, it is important to note that these drugs are currently only indicated for the treatment of hypercholesterolemia and are not specific for the treatment of elevated Lp(a) levels. In the absence of drugs that substantially reduce Lp(a) levels, nucleic acid-based therapies represent a promising strategy to reduce the contribution of elevated Lp(a) to onset and development of ASCVD and calcific aortic disease.
3.2.1 Apo(a) mRNA targeting
3.2.1.1 ASO pelacarsen (previously denoted IONIS-Apo(a)Rx or Apo(a)-LRX)
Pelacarsen is a second-generation GalNAc-conjugated ASO that targets and binds to Apo(a) mRNA in liver cells.96 A dose ranging trial involving 286 patients with in patients with established ASCVD and Lp(a) levels ≥60 mg/dL, demonstrated a dose-dependent Lp(a) reduction up to 80% (NCT03070782).97,98 Eighty one percent of patients attained Lp(a) levels ≤50 mg/dL or ≤125 nmol/L with 60 mg pelacarsen injected every 4 weeks, and 98% achieved same target levels with 20 mg pelacarsen administered every week. Oxidized phospholipids on ApoB and Apo(a) were also reduced by up to 88 and 70%, respectively. LDL-C was moderately reduced, up to 25%. Injection site reactions were most common adverse effect (27 vs. 6% in placebo group), while myalgia or arthralgia as well as post-injection malaise were the most common causes for therapy discontinuation.
To assess the effect of pelacarsen on major cardiovascular outcomes, a phase 3 trial, HORIZON, was launched in 2019 (NCT04566445).99 More than 8000 participants with established CVD and elevated Lp(a), defined as ≥70 mg/dL, were enrolled in the study between 2019 and 2022. The study is expected to be completed in 2025.99
3.2.1.2 siRNA olpasiran
Olpasiran (formerly known as AMG 890) is currently undergoing phase 3 clinical trials for the treatment of elevated Lp(a) levels. A phase 2 trial, the OCEAN(a)-DOSE (NCT04270760), including 281 adults with established ASCVD and elevated Lp(a) levels (>150 nmol/L), demonstrated placebo-adjusted Lp(a) reduction of 101% with the highest dosage (225 mg every 12 weeks). The drug was well-tolerated, injection site reactions being the most common side effect. The OCEAN(a) cardiovascular outcome study was initiated in 2022 and is expected to be completed in 2026.100,101 Recently, at the European Society of Cardiology Congress 2023, further data from the phase two trial were presented, showing a lasting effect on Lp(a) reduction in the off-treatment extension period, nearly a year after the last dose.102
3.2.1.3 siRNA Lepodisiran (LY3819469)
Another siRNA lepodisiran (LY3819469), targeting Lp(a), is currently undergoing a phase 2 clinical trial (NCT05565742) to evaluate its effect in participants with elevated Lp(a) levels. The phase 1 trial in healthy volunteers (NCT04914546) demonstarted a favourable safety profile, and dose-dependent, long-duration Lp(a) reduction of 94% at day 337103.
3.2.1.4 siRNA muvalaplin (known as LY3473329)
Muvalaplin is an oral Lp(a) lowering drug, which is currently being tested in the phase 2b KRAKEN trial (NCT05563246). A phase 1 randomized trial (NCT04472676) was recently published, showing the absence of tolerability concerns and Lp(a) reduction by 65% following daily administration for 14 days.104
3.2.1.5 siRNA Zerlasiran (SLN360)
SLN360 is a GalNAc-conjugated siRNA that reduced LPA mRNA expression by >90% and serum Lp(a) levels by 65–90% in preclinical studies.105Phase 1 trial was completed in November 2022106 and phase 2 trial is underway.107
3.2.2 LPA gene editing
First successful in vivo gene editing to silence LPA was reported by Doerfler et al. in 2022.108 They utilized a gRNA that was designed to specifically target the first kringle domain in the LPA gene; and used AAV as a delivery system to introduce the gRNA and the Cas9 protein into the liver of transgenic mice. As a result, very efficient Apo(a) protein removal from the circulation was achieved.108 Just recently, Verve Therapeutics and Eli Lilly and Company announced a partnership to develop an LPA gene editing drug.109
3.3 ANGPTL3
ANGPTL3 is a small (45 kDa) protein produced by the liver. In vivo studies have demonstrated that ANGPTL3 impairs the clearance of triglyceride (TG)-rich lipoproteins by inhibiting LPL, Figure 5. Targeted deletions of ANGPTL3 result in increased activity of LPL and reduced plasma TG levels.110 ANGPTL3 inhibits LPL through two potential mechanisms: by unfolding LPL and destabilizing it, or by causing the release of LPL from glycosylphosphatidylinositol-anchored HDL-binding protein 1 and the capillary endothelium in an inactive form.110,111 In addition, ANGPTL3 has been shown to inhibit endothelial lipase (EL), which leads to increased levels of HDL-C.112 Previous studies exploring the mechanisms how ANGPTL3 reduces LDL-C were not conclusive. The impact of ANGPTL3 on hepatic very low-density lipoprotein (VLDL) production and lipoprotein clearance was examined in earlier studies, but there were conflicting results between mice studies and genetic studies in humans. In mice, ANGPTL3 was associated with lower VLDL–TG secretion but had no impact on VLDL–APOB production, while human ANGPTL3 loss-of-function carriers had reduced VLDL–APOB production rates. According to Adam et al., ANGPTL3 inhibition results in the derepression of EL, which then drives the remodeling and clearance of VLDL in cases where LDL-R is absent. This depletion of the LDL precursor pool then limits the production of LDL particles and ultimately reduces plasma LDL-C levels.113
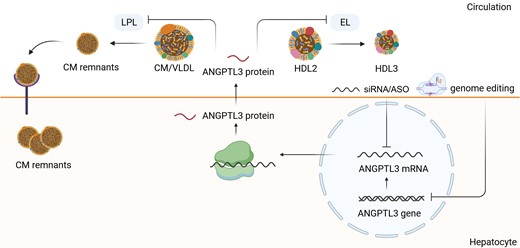
ANGPTL3 synthesis, secretion, and inhibition by nucleic acid-targeted therapies. ANGPTL3 is synthesized in the hepatocyte and impairs the clearance of TG-rich lipoproteins by inhibiting LPL. LPL catalyzes the hydrolysis of the triacylglycerol component of circulating chylomicrons and VLDL, thereby providing non-esterified fatty acids and 2-monoacylglycerol for tissue utilization. The inhibition of ANGPTL3 mRNA/ANGPTL3 gene results in the increased clearance of CMs and VLDL and promotes the CM uptake by hepatocytes. This leads to the decrease of TG-rich lipoproteins in the circulation. LPL, lipoprotein lipase; CM, chylomicrone; VLDL, very low-density lipoprotein; IDL, intermediate-density lipoprotein, LDL, low-density lipoprotein, TG, triglycerides.
In 1991, during a screening for cardiovascular disease, nine families living in a small Italian town were identified to be having low LDL-C and apoB plasma concentrations.114 All of them had ANGPTL3 mutation, but only homozygous probands had complete ANGPTL3 deficiency and displayed hypolipidemic phenotype. They were apparently healthy individuals without any increasing risk for tumors or liver disease. Further studies confirmed that individuals with ANGPTL3 loss-of-function mutations have lower plasma TG and LDL-C.115–118 In 2017, a case–control study consisting of 58 000 individuals showed that carriers of loss-of-function mutations had reduced TG, LDL-C, and HDL-C concentration, which translated into 41% reduction in rate of ASCVD. Other studies demonstrated lower plaque burden in coronary arteries, measured by computer tomography, associated with 34% reduction in odds for coronary heart disease.116 In contrast to mutations in APOB and MTTP (microsomal triglyceride transfer) genes that can cause hepatic steatosis due to hypobetalipoproteinemia,119,120 ANGPTL3 deficiency does not lead to hepatic steatosis.121 These findings suggest that ANGPTL3 deficiency has a protective role, and its complete deficiency is benign.
Evinacumab, a monoclonal antibody targeting ANGPTL3, has shown promising results in clinical trials. Evinacumab entered clinical trials in patients with HoFH, as it was shown to lower LDL-C levels in mice lacking LDL-R. Remarkably, LDL-C reduction of 47% was achieved in 65 patients with HoFH.122 Furthermore, in 15 subjects with null-null LDL-R mutations, LDL-C was lowered by 43%. Evinacumab is currently the only lipid-lowering agent that does not require the presence of LDL-R, and it has been approved by the FDA for the treatment of HoFH.
3.3.1 ANGPTL3 mRNA targeting
3.3.1.1 ASO vupanorsen (IONIS-ANGPTL3-LRX)
Second-generation GalNAc-conjugated ASO, Vupanorsen, targets hepatic ANGPTL3 mRNA causing degradation of ANGPTL3 protein. In phase 1 and phase 2 trials vupanorsen resulted in ∼50% reduction in TGs and up to 28% reduction in non-HDL cholesterol.123,124 However, further clinical development of vupanorsen was discontinued due to insufficient magnitude of non-HDL and TG reduction and due to safety concerns (liver enzyme elevation and increase in hepatic fat fraction up to 76%).125
3.3.1.2 siRNA ARO-ANG3
In a phase 1 trial (NCT03747224), ARO-ANG3 reduced ANGPTL3 (mean −45 to −78%) and TG (median −34 to 54%) levels 85 days after dose.126 The cohort consisted of 40 participants with various dyslipidemias, 22.5% of whom had hepatic steatosis. The drug was well-tolerated without any abnormal laboratory changes or meaningful decrease in platelet count. The end-of-study results from ARCHES-2, a phase 2b double-blind placebo-controlled trial (NCT04832971) evaluating the efficacy and safety of ARO-ANG3 in patients with mixed dyslipidemia, were reported at the annual AHA Congress 2023.127 At week 24, ARO-ANG3 demonstrated a significant reduction of up to 56% in TG levels and up to 20% in LDL-C levels. Importantly, unlike vupanorsen, ARO-ANG3 did not lead to increased liver fat; it rather showed a reduction in the liver fat fraction (liver fat decreased in the placebo group too). Adverse effects were reported as ‘consistent with those expected in this patient population and with associated underlying comorbidities’.127 Another phase 2 trial (Gateway, NCT05217667) has recruited patients with genetic or clinical diagnosis of HoFH and is expected to be completed in 2025.128
3.3.1.3 siRNA LY3561774
LY3561774 is a siRNA designed to target ANGPTL3, which is currently undergoing a phase 2 clinical trial (PROLONG-ANG3; NCT05256654), including 175 participants with mixed dyslipidemia and on a stable dose of statin.129 The study is expected to be completed in 2024.
3.3.2 ANGPTL3 gene editing
ANGPTL3 is considered an appealing target for gene editing due to the benign nature of loss-of-function mutations, like PCSK9, making it an attractive candidate for gene editing interventions. Using an adenovirus encoding a BE and gRNA targeting ANGPTL3, Chadwick et al. demonstrated a 35% editing rate, a 49% reduction in ANGPTL3 protein, a 31% reduction in TG, and a 19% reduction in cholesterol in wild-type mice, within one week.130 An LNP system delivering CRISPR/SpCas9 mRNA and ANGPTL3-targeting gRNA led to a 38% editing efficiency, a 65% reduction in circulating ANGPTL3 protein, a 56% reduction in LDL-C, and a 29% reduction in TGs.131
Adenine BE inactivates the ANGPTL3 gene, Verve-201 was nominated as a development candidate after screening and selection of 200 BEs and successful in vitro and in vivo experiments. Verve-201 targets a unique site (23 DNA base pair PAM sequence) not found elsewhere in human genome, which should minimize potential off-target editing.132 According to the 2 years data from preclinical studies on wild-type monkeys and non-human primates, Verve-201 reduces circulating ANGPTL3 by an average of ∼90%, with >60% liver editing.132,133 Importantly, no change in liver biomarkers was observed.
CRISPR therapeutics is currently in the process of developing a CRISPR/Cas9 product called CTX310, which targets ANGPTL3. In preclinical studies, CTX310 has shown promising results by effectively reducing ANGPTL3 protein levels by more than 85%.75,134 The drug is expected to progress into clinical trials in 2023.135
3.4 APOC3
Apolipoprotein C3, a glycoprotein predominantly synthesized in the liver, plays a significant role in regulating plasma levels of LDL-C and triglyceride-rich lipoprotein (TRK) cholesterol.136 APOC3 is linked to atherosclerosis in several respects, including, but not limited to inhibition of LPL activity (which leads to increase in plasma TG levels), and delayed clearance of cholesterol-rich remnants (Figure 6).
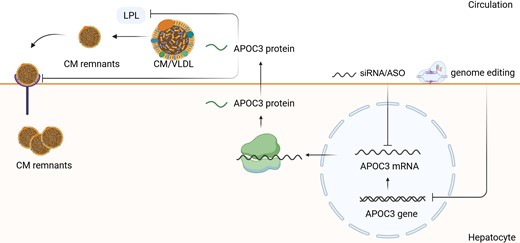
APOC3 synthesis, secretion, and inhibition by nucleic acid-targeted therapies. APOC3 impairs the clearance of TG-rich lipoproteins by inhibiting LPL, which stimulates the hydrolysis nascent CMs and VLDL. It also inhibits the CM remnant uptake into the hepatocytes. The inhibition of APOC3 mRNA/APOC3 gene results in the increased clearance of CMs and VLDL and promotes the CM uptake by hepatocytes. This leads to the decrease of TG-rich lipoproteins in the circulation. LPL, lipoprotein lipase; CM, chylomicrone; VLDL, very low-density lipoprotein; IDL, intermediate-density lipoprotein, LDL, low-density lipoprotein, TG, triglycerides.
Human studies with complete or partial loss-of-function variants in APOC3 have demonstrated increased clearance of VLDL triglyceride and APOB100, indicating increased LPL-mediated lipolysis of VLDL triglycerides.137,138 There is also evidence for APOC3 direct involvement in atherosclerosis: APOC3 increases the affinity of LDL for the proteoglycans in the arterial wall and alters the lipid composition of LDL-C, which introduces conformational changes in apoB100.139,140
In the Exome Sequencing Project, which involved 3734 participants, mutations in the APOC3 gene were found to be associated with a significant decrease of 39% in TG levels and 16% in LDL-C levels.141 Carriers of APOC3 loss-of-function mutations were found to have a 40% lower risk of coronary heart disease compared to noncarriers. There was no presence of homozygosity or compound heterozygosity. Lower APOC3 plasma levels were associated with a reduced incidence of coronary heart disease in cox regression analysis, but this association was not significant when adjusted for additional cardiovascular risk factors. However, in the Verona Heart Study, a significant association between APOC3 levels and cardiovascular death was observed, even when adjusted for other risk factors.142
Pharmacological inhibition of APOC3 has shown efficacy in reducing plasma TG levels. APOC3 inhibition has particularly been proven beneficial in patients diagnosed with familial chylomicronemia syndrome (FCS), a condition characterized by markedly elevated TG levels and recurrent episodes of pancreatitis.143
3.4.1 APOC3 mRNA targeting
3.4.1.1 ASO volanesorsen
Volanesorsen is a second-generation ASO (without GalNAc conjunction), targeting APOC3 mRNA and reducing TG levels in plasma. Volanesorsen was approved in the EU in 2019 for FCS, severe hypertriglyceridemia, and familial partial lipodystrophy.144 Familial chylomicronemia syndrome is rare recessive genetic disorder, associated with accumulation of chylomicrons, large TG-rich lipoprotein particles.143 Underlying mechanism is impaired clearance of chylomicrons due to LPL deficiency caused by mutations in LPL—(lipoprotein lipase), APOA5 (apolipoprotein A5), APOC2 (apolipoprotein C2), LMF1 (Lipase maturation factor 1), and other genes.145 Patients with familial chylomicronemia exhibit high TG levels (in case of monogenic cause TG levels are severely increased) and often episodes of acute pancreatitis.
In patients with multifactorial hyperchylomicronemia, a phase 2 study, COMPASS, demonstrated a significant TG reduction of 70%.146 A phase 2 APPROACH trial showed a 53% placebo-adjusted decrease in TG levels and 80% reduction in APOC3 in patients with FCS after weekly injections of 300 mg volanesorsen over 52 weeks.147 Most severe complication of hypertriglyceridemia—acute pancreatitis—was observed in placebo group, but not in volanesorsen arm. TG reduction of similar extent was observed in phase BROADEN trial (phase 2/III), where it was administered to patients with partial lipodystrophy.148
Injection-site reaction is the most common side effect of volanesorsen. Decrease in platelet count was observed in 33% of patients in volanesorsen arm compared to 3% in placebo arm in APPROACH trial. Severe thrombocytopenia (0 to <25 000/µL) occurred in 6% of patients vs. none in placebo group. It is hypothesized that the natural history of FCS is associated with thrombocytopenia, which then may be exacerbated by volanesorsen.149,150 Therefore, thrombocyte count should be monitored during the treatment and the frequency of injections should be corrected depending on the severity of thrombocytopenia and according to the manufacturer’s instructions. Due to safety concerns, the FDA denied approval of volanesorsen in 2018.151
A recent meta-analysis of three randomized controlled trials investigated volanesorsen in patients with hypertriglyceridemia and analyzed the pooled incidence of acute pancreatitism.152 Among a total of 207 patients, with 121 receiving volanesorsen and 86 receiving placebo, pancreatitis occurred in 4 patients in the volanesorsen group and 18 patients in the placebo group. The pooled odds ratio was 0.18 (95% CI: 0.04–0.82), indicating a significantly lower occurrence of pancreatitis in the volanesorsen group compared to the placebo group.
3.4.1.2 ASO olezarsen
Olezarsen (formerly known as AKCEA-APOC3-LRx) is an ASO similar to volanesorsen in mechanism of action. The only difference between these drugs is presence of GalNAc moiety in olezarsen, which, due to high affinity to hepatocytes, facilitates lower dosing and less frequent injections. In phase 2 dose-ranging study, olezarsen injections over 6–12 months resulted in TG reduction by 23–60%. APOC3, VLDL, non-HDL, apoB were also significantly reduced. The safety profile was more favorable than that of volanesorsen, with only mild decrease in platelet count (140 000 to 100 000/µL) occurring in ∼10% of cases.149 A phase 2 trial (NCT05355402), to evaluate reduction of TG levels at 6 and 12 months in patients with hypertriglyceridemia and cardiovascular risk, was launched in May 2022 and is expected to be completed in 2023.153
In a recently published phase 2b randomized, controlled trial (Bridge–TIMI 73a, NCT05355402) involving patients with moderate to severe hypertriglyceridemia and elevated cardiovascular risk, olezarsen led to significant reductions in TG levels compared to placebo—49.3% for the 50 mg dose and 53.1% for the 80 mg dose.154 Additionally, the drug notably decreased levels of apolipoprotein B and non-HDL cholesterol. The safety profile was consistent across all groups, showing no significant risks of hepatic, renal, or platelet abnormalities.
In a phase 3 trial (BALANCE, NCT04568434) enrolling patients with FCS, 71% of whom had a history of acute pancreatitis episodes in the last 10 years, olezarsen was tested at doses of 50 and 80 mg administered subcutaneously every 4 weeks for 53 weeks.155 Eighty milligram of the drug reduced fasting TG levels by 43.5%, while the reduction with the 50 mg dose was not statistically significant. Additionally, the rate of acute pancreatitis episodes by 53 weeks was substantially lower in the olezarsen-treated groups, with 11 episodes in the placebo group vs. one in each treatment group.
Furthermore, ongoing phase 3 trials are evaluating olezarsen in patients with severe HTG, defined as TG ≥ 500 mg/dL (CORE CS5, NCT05079919; CORE2 CS6 NCT05552326); moderate HTG (150–499 mg/dL) with or at risk for ASCVD (CS8, NCT05355402; ESSENCE CS9, NCT05610280).
3.4.1.3 siRNA plozasiran (known as ARO-APOC3)
Plozasiran, or as previously denoted, ARO-APOC3, is an siRNA which inhibits APOC3 production in the liver. In the recently published phase 2b SHASTA-2 trial, participants with severe hypertriglyceridemia who received two doses of plozasiran or placebo (dosed on Day 1 and Week 12) produced a 77% reduction in APOC3 and 57% reduction in TG levels at Week 24, which persisted at Week 48.156 Over 90% of the treated patients reached TG levels below 500 mg/dL, a threshold for the risk of acute pancreatitis. The treatment was well-tolerated.
Interim results of a phase 2b trial, MUIR (NCT04998201), were presented at the AHA Meeting 2023, showing mean reduction of TG by 52–64% at Week 24, when dosed on Day 1 and Week 24 in patients with mixed dyslipidemia.157 The ongoing phase 3 trial PALISADE (NCT05089084) is further investigating the effect of plozasiran on TG reduction in patients with FCS.158
3.4.2 APOC3 gene editing
APOC3 knockout mice exhibit marked reductions in VLDL and TG, along with reduced atherosclerotic plaque formation.159 Genetic studies analyzing loss-of-function mutations in APOC3 have provided evidence for protective role against atherosclerosis.141,160 However, there is still a gap in evidence when it comes to specific mechanisms by which APOC3 loss-of-function might be atheroprotective. One example of APOC3 gene editing is ARCUS-APOC3, a meganuclease, which is currently in preclinical development. In transgenic mice, ARCUS-APOC3 led to a significant reduction of 74% in APOC3 protein levels, resulting in a corresponding 62% reduction in TG levels.161
3.5 ANGPTL3 vs. APOC3 inhibition
While the range of options for reducing LDL-C is extensive, there remains an unaddressed gap in managing hypertriglyceridemia and mixed dyslipidemia. Inhibition of ANGPTL3 and APOC3 may represent promising strategies to bridge this gap.
ANGPTL3 inhibition leads to decreases in both TG levels and LDL-C, whereas APOC3 inhibition results in significant reductions in TG levels but does not substantially lower LDL-C levels. This is due to the targeted action of APOC3 inhibitors on APOC3, which inhibits LPL and hepatic lipase—key enzymes in TG metabolism. Thus, APOC3 inhibition is particularly effective in treating severe (monogenic) hypertriglyceridemia. Conversely, ANGPTL3 inhibition may be better suited for patients with mixed dyslipidemia due to its broader lipid-modulating effects.
These two approaches have been thoroughly reviewed and compared elsewhere.147,162
3.6 LDL-R gene therapy
Mutations in the LDL-R gene account for approximately 85% of cases of familial hypercholesterolemia (FH), including both heterozygous and homozygous forms.163 Unfortunately, effective treatment options for HoFH are limited, with the monoclonal antibody evinacumab currently being the only drug that can decrease LDL-C in the complete absence of LDL-R.122 Therefore, correcting the underlying genetic defect using gene editing technologies represents a promising approach for achieving sufficient and long-term LDL-C reduction in patients with LDL-R deficiency.
The main limitation in using LDL-R as a target for gene editing therapy is that over 2000 pathogenic and likely pathogenic variants in LDL-R gene exist, which makes it difficult to identify and correct all potential mutations through gene editing. This variability also makes it challenging to predict the efficacy and safety of gene therapy in individual patients, as the effects of different LDL-R mutations may vary widely. Therefore, while gene editing for LDL-R shows promise, alternative approaches may need to be explored for effective and personalized treatment of patients with LDL-R deficiency.
LDL-R gene supplementation, as a potential approach in the treatment of FH, has been studied since the 1990s.164,165 Several delivery methods were tested to transfer the LDL-R gene into the hepatocytes, including, but not limited to AAV vectors, adenovirus vectors, RNA-based vehicles, and lentiviral vectors. Among these approaches, AAV-mediated gene transfer was shown to be a viable approach to long-term transgene expression in preclinical models of HoFH.166 A phase 1/2a trial (NCT02651675) evaluated gene supplementation using recombinant AAV (rAAV)-based liver-directed human LDL-R gene therapy (denoted as RGX-501) in nine patients with HoFH. However, clinical development of RGX-501 was discontinued resulting in the early termination of the study.45
3.6.1 Historical perspective: apolipoprotein B100
Apolipoprotein B100 (ApoB100) is a backbone component of LDL, VLDL, and Lp(a). ApoB concentrations have a high sensitivity and specificity in predicting cardiovascular events.167 A mendelian randomization study implied that the benefit of TG and LDL-C lowering may be proportional to the absolute change in ApoB100.46 The only ApoB-targeting nucleic acid-based drug investigated in human clinical trial is mipomersen. A meta-analysis of 13 clinical trials and 729 subjects treated with mipomersen (including phase 2 and phase 3 trials), showed significant reduction in ApoB and other atherogenic lipoproteins. However, mipomersen treatment was associated with hepatic liver enzyme elevation and hepatic steatosis.168 Mipomersen was approved by the FDA in 2013 for treatment of HoFH.169 The EMEA rejected mipomersen’s approval due to concerns about liver enzyme elevation, increase in liver fat, flu-like symptoms, and cardiovascular events.170 All taken together indicates that despite the role in the development of CVD, the safety issues associated with inhibiting ApoB100 hinder its viability as a target, especially in the light of alternative targets. Therefore, the focus on ApoB100 targeting has gradually diminished, as attention has shifted toward other potential therapeutic avenues.
4. Nucleic acid-based drugs in the treatment of arterial hypertension
Systemic arterial hypertension is a modifiable risk factor for all-cause mortality and morbidity and is associated with increased risk for CVD. Globally, the age-standardized prevalence of hypertension is 24% in men and 20% in women.162 This condition contributes to more than 8 million deaths worldwide, due to stroke, coronary heart disease, other vascular diseases, and renal disorders.171
Despite the significant amount of unmet clinical needs in the treatment of hypertension, the development of novel agents or non-pharmacologic strategies in this area was somewhat stagnant in the last two decades. Achieving satisfactory control of high blood pressure levels has proven to be a challenging task and highly dependent on therapy adherence to daily complex therapeutic schemes which include lifestyle changes and multiple drugs. The use of nucleic acid-targeted approaches may offer a solution, as these drugs could provide control over hypertension with infrequent injections.
4.1 Angiotensinogen targeting
The renin–angiotensin–aldosterone system (RAAS) plays a crucial role in the development of hypertension and the consequent target organ damage, Figure 7. Briefly, renin, a protease produced by juxtaglomerular cells in the terminal afferent arteriole of the kidneys, cleaves the substrate angiotensinogen (AGT) to angiotensin I (Ang-I), which is then converted to angiotensinogen II (Ang-II) by angiotensin-converting enzyme (ACE). The produced biological effector Ang-II interacts with the AT1-subtype receptor which activates intracellular pathways contributing to increase smooth muscle cell vascular tone and to promote cellular growth thus producing hypertension and hypertension-induced end-organ damage. Currently available antihypertensive drugs acting on RAAS-system, such as ACE-inhibitors or AT1-blockers, can cause a compensatory rise in renin due to reduction of blood pressure (BP) and loss of negative feedback on AGT. This leads to the restoration of Ang-II levels and Ang-II competition at the AT1-receptor level (phenomenon known as RAAS escape). Therefore, targeting the RAAS upstream at the level of the step limiting stage of the cascade by interfering with AGT nucleic acid is an alternative approach to decrease BP even in the presence of a compensatory rise in renin.20 One specific concern regarding the AGT RNA inhibition is represented by its long-acting RAAS-inhibiting and BP-lowering effect and the potential need for rapid and adequate restoration of BP in hypovolemic/hypotensive setting, or in case of acute kidney injury.20 This issue was addressed by Uijl et al. who first treated hypertensive rates with AGT-targeting siRNA, and then with fludcortisone or high-salt diet.172 This resulted in the normalization of BP with return to baseline levels on Day 5 and Day 7. Alternative strategy is based on the REVERSIR platform which comprises an oligonucleotide complementary to the siRNA anti-sense strand, that can bind to the RISC-bound siRNA, which may in turn result in the reverse of siRNA-mediated gene silencing.173
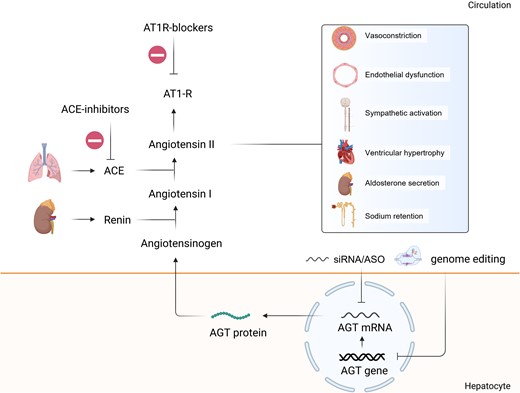
AGT and RAAS pathway and its inhibition by nucleic acid-targeted therapies. Renin acts upon its substrate angiotensinogen, which is produced by the liver, to produce angiotensin I. ACE plays a crucial role in the pathway by converting angiotensin I into angiotensin II. ACE is primarily located on the surface of endothelial cells in the lungs, where it carries out this conversion. Besides vasocontriction, angiotensin II has effects on several tissues and organs. It promotes endothelial dysfunction, sympathetic activation, ventricular hypertrophy; stimulates aldorsterone secretion and sodium retention, ultimately causing increase in blood pressure. New drugs can inhibit the secretion of AGT protein by targeting the AGT mRNA. AT1R, angiotensin II receptor 1; ACE, angiotensin converting enzyme.
4.1.1 siRNA zilebesiran (known as ALN-AGT01)
Zilebesiran is first-in-class Gal-NAc-conjugated siRNA that is currently undergoing phase 2 clinical trial for the treatment of hypertension. In phase 1 study zilebesiran reduced systolic BP by 10 mmHg and diastolic BP by 5 mmHg by Week 8 after the injection of single-dose drug (200 mg or more). The BP reductions were sustained at 24 weeks. The medication was deemed safe, with rare injection site reactions. No severe side effects, such as hyperkaliemia, hypotension, or worsening of renal function were observed.174 The drug is currently evaluated in two phase 2 trials: KARDIA-1(NCT04936035) and KARDIA-2 (NCT05103332). In KARDIA-1, patients with mild-to-moderate hypertension and without any other antihypertensive medications received the drug or placebo, whereas KARDIA-2 includes patients with inadequate BP control with standard medications. Results of KARDIA-1, recently presented at the 2023 AHA Scientific Sessions, showed dose-dependent reductions of AGT up to 90% (range of four dosages 150–600 mg) vs. placebo.175 This was associated with reductions of systolic BP in ambulatory BP monitoring (nadir −14 mmHg) and of office systolic BP (nadir −12.1 mmHg) vs. placebo after 2–4 weeks pharmacologic wash-out.
4.1.2 ASO IONIS AGT-LRx
Another AGT inhibitor, an ASO IONIS AGT-LRx has undergone clinical evaluation in phase 1 and phase 2 trials. In a phase 2 monotherapy study, it demonstrated a mean absolute change in BP of 8 mmHg (not significant). Similarly, in an add-on phase 2 trial (patients on stable regimen of two to three antihypertensive drugs failing to achieve BP goals), BP was numerically smaller in AGT-LRx group compared to placebo (−12 mmHg vs. −5 mmHg, respectively) without reaching statistical significance. However, the sample size estimation for both studies was calculated based on plasma AGT reduction, and not on BP changes. In both cases, AGT levels were significantly reduced (−54% in monotherapy study and −67 in add-on study).176 Further phase 2 clinical trials that are currently underway with IONIS AGT-LRx include: (1) a phase-2 trial with office systolic BP as primary endpoint (ASTRAAS, NCT04714320),177 (2) a phase-2 trial in patients with chronic heart failure with reduced ejection fraction (ASTRAAS-HF, NCT04836182).178
5. Safety considerations
5.1 On-target side effects
Mechanism-related, or on-target toxicities happen when the interaction between the drug and its intended target leads to negative effects.179 This can happen if the target gene or pathway plays a critical role in maintaining cellular or physiological functions. Disrupting these functions may cause problems such as disturbances in cell cycle regulation, apoptosis, or DNA repair, as well as trigger immune responses that may result in autoimmunity or inflammation. One example of such an on-target effect is MRX34 (liposomal mimic of microRNA-34a (miR-34a)) for the treatment of advanced solid tumours. MRX34 is taken up by white blood cells, where miR-34a are expressed and have an important biological function, and leads to the alteration of chemokine profiles, potentially impacting tumor lysis negatively.180,181 Another example is chemotherapy-induced neuropathy in case of XIAP (X-linked inhibitor of apoptosis protein)-targeting ASO AEG35156 (caspase inhibitor) that results in the down-regulation of XIAP in neuronal cells, rather than in tumour cells.181 Systemic administration and higher dosing increase the risk of on-target toxicity.
5.2 Off-target effects
5.2.1 RNA therapeutics
Off-target effects in RNA therapeutics may arise from various mechanisms. One such effect occurs when the passenger strand, rather than the guide strand, is loaded into the RISC, potentially leading to the silencing of unintended genes.181,182,183Strategies to enhance guide-strand loading into RISC include modifications to the passenger strand, such as incorporating locked nucleic acids, 2′-F-O-Me-phosphorodithioate modifications, trimming, or biotinylation, and designing Dicer-substrate siRNAs with asymmetrical ends. Another potential off-target effect is interaction with proteins, exemplified by the first-generation ASO Oblimersen, targeting BCL2 (B-cell lymphoma 2) mRNA, which displayed limited efficacy in clinical trials due to unforeseen off-target effects, including gene regulation alterations and direct binding to proteins like the VDAC (voltage-dependent anion channel), leading to apoptosis.
Another potentially important off-target effect is immunogenicity. The innate immune system can recognize external RNA via diverse PAMP-receptors as part of a viral defense mechanism. Toll-like receptors 3, 7, and 8 play a central role in extracellular recognition, mediated by myeloid differentiation factor 88 (MyD88).181 This results in the activation of pro-inflammatory pathways, activating the NF-kB and production of cytokines in macrophages and dendritic cells. The single-stranded RNA is more prone to immune response; therefore, modern RNA therapeutics are double-stranded. Chemical modifications applied to newer generations allowed to limit immunogenicity.
5.2.2 CRISPR–Cas systems
Off-target effects in CRISPR/Cas9 system may occur when Cas9 targets and cuts genomic sites other than the intended target site, potentially causing genomic instability.184,185 These unintended sites are frequently related to the sgRNA. The sgRNA binds to DNA sequences that are similar, but not identical, to the intended target sequence. This is particularly true if the mismatches between the sgRNA and the DNA occur away from the ‘seed’ region, the 10–12 bp adjacent to the PAM, which is crucial for initial binding specificity.184 Off-target effects can be reduced by increasing the nucleases cleavage specificity or reducing the time frame of functional activity for their applications. For instance, self-cleavage systems have been developed, in which Cas9 cuts the genome locus and the delivery vector, thus resulting in self-limitation of Cas9 expression. This approach was successfully established to target PCSK9 gene in animal models.73 The shortening of Cas9 functional activity can also be achieved by using electroporation, which has a shorter half-life compared to other vector systems.186 Using Cas9 ortholog produced from Staphylococcus aureus, SaCas9, may also enhance the precise editing. SaCas9 is 300 amino acids shorter than SpCas9. The unique PAM recognition pattern facilitates higher specificity compared to traditional SpCas9.187 SaCas9 has more proneness towards the NHEJ editing and higher fidelity compared to SpCas9. Another possibility is to increase the on-target specificity, which could be achieved by modifications of Cas proteins with enhanced on-target specificity, such as eSpCas9, HF-Cas9, HypaCas9, and SniperCas9.164,188 Most of these proteins show reduced off-target levels while retaining on-target activity. Modifications in gRNA, e.g. dual-gRNAs, by facilitating large deletions, prove more effective for knockout screens, significantly improving the generation of loss-of-function mutations.189 Adjusting the Cas9–sgRNA complex concentration may boost specificity but lower on-target action.190 Employing a mutant nickase Cas9 variant with paired sgRNAs reduces off-target activity, maintaining on-target efficiency.191 Techniques like fCas9 hybrids, which combine inactive Cas9 with FokI nuclease, and new methods like using inhibitors for precise editing and innovative sgRNA delivery approaches, promise lower toxicity and broader CRISPR/Cas9 applications, focusing on reducing off-target effects.184,185
6. Comparison with monoclonal antibodies
Compared to monoclonal antibodies, nucleic acid-targeting therapeutics have both benefits and drawbacks. Advantages include more efficient production due to simpler development and manufacturing processes, along with greater stability at room temperature, which enhances storage and distribution capabilities.192,193 A significant benefit is their ability to target virtually any gene of interest. Moreover, extended dosing intervals, such as the 6 month period seen with siRNA inclisiran, may offer an interesting option for those challenged by the necessity of biweekly antibody injections.194 However, these therapies may also have potential limitations, including the risk of on- and off-target effects.
7. Summary and outlook
The field of nucleic acid-targeted therapeutics has great promise for the prevention and treatment of CVD. There is a growing interest in the possibility of targeting production of proteins which are causally associated with CVD development and progression. Advances in technologies such as base editing and other gene editing techniques are expected to further expand the possibilities of nucleic acid-targeted therapies. Currently, a variety of RNA- and DNA-targeted drugs for the treatment of dyslipidemias and arterial hypertension are undergoing development in both preclinical and clinical studies. Among them, several have already received approval and are used in clinical practice. Examples include siRNA inclisiran, which targets PCSK9 mRNA, and ASO volanesorsen, directed at APOC3 mRNA. A major advantage of these treatments is their infrequent administration, which has the potential to enhance treatment adherence and ensure a lasting effect.
It is crucial to continue monitoring and addressing potential concerns related to long-term safety profiles. Efforts have been made to reduce off-target effects and accelerate the development of these therapeutics through innovative technologies, facilitating a rapid transition from bench to bedside. However, clinical experience with these medications, both in clinical trials and real-world settings, needs to be expanded.
Note Added in Proof: During the review and proofreading of the manuscript, the following updates were noted: 1) A new base editor targeting PCSK9, Verve-102, which uses a different LNP delivery system than VERVE-101, began a clinical trial in May 2024 (https://classic.clinicaltrials.gov/ct2/show/NCT06164730); 2) The results of the phase 2 trial for the siRNA targeting Apo(a), zerlasiran (SLN360), were published in April 2024 (Nissen SE, Wolski K, Watts GF, et al. Single Ascending and Multiple-Dose Trial of Zerlasiran, a Short Interfering RNA Targeting Lipoprotein(a): A Randomized Clinical Trial. JAMA. 2024;331(18):1534–1543. doi:10.1001/jama.2024.4504).
References
FDA News Release: FDA Approves First Gene Therapies to Treat Patients with Sickle Cell Disease. December 08, 2023. https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease (18 June 2024, date last accessed).
Food and Drug Administration (FDA) News & Events for Human Drugs. FDA approves add-on therapy to lower cholesterol among certain high-risk adults. https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-add-therapy-lower-cholesterol-among-certain-high-risk-adults. Published December 22, 2021. (1 February 2024, date last accessed).
Author notes
Conflict of interest: U.L. reports research grants to institution from Novartis and Amgen. E. S.-T. reports speaker fees, consulting fees and research grants from Daiichi Sankyo, Sanofi, Amgen, Novartis ans Sobi.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}