





Abstract
Aldosterone’s role in the kidney and its pathophysiologic actions in hypertension are well known. However, its role or that of its receptor [minieralocorticoid receptor (MR)] in other cardiovascular (CV) disease are less well described. To identify their potential roles in six CV conditions (heart failure, myocardial infarction, atrial fibrillation, stroke, atherosclerosis, and thrombosis), we assessed these associations in the following four areas: (i) mechanistic studies in rodents and humans; (ii) pre-clinical studies of MR antagonists; (iii) clinical trials of MR antagonists; and (iv) genetics. The data were acquired from an online search of the National Library of Medicine using the PubMed search engine from January 2011 through June 2021. There were 3702 publications identified with 200 publications meeting our inclusion and exclusion criteria. Data strongly supported an association between heart failure and dysregulated aldosterone/MR. This association is not surprising given aldosterone/MR’s prominent role in regulating sodium/volume homeostasis. Atrial fibrillation and myocardial infarction are also associated with dysregulated aldosterone/MR, but less strongly. For the most part, the data were insufficient to determine whether there was a relationship between atherosclerosis, stroke, or thrombosis and aldosterone/MR dysregulation. This review clearly documented an expanding role for aldosterone/MR’s dysregulation in CV diseases beyond hypertension. How expansive it might be is limited by the currently available data. It is anticipated that with an increased focus on aldosterone/MR’s potential roles in these diseases, additional clinical and pre-clinical data will clarify these relationships, thereby, opening approaches to use modulators of aldosterone/MR’s action to more precisely treat these CV conditions.
1. Introduction
When discovered in 1953 by Simpson et al.,1 aldosterone was described as a hormone that help regulated salt, volume, and potassium homeostasis primarily by modulating the kidney’s handling of sodium and potassium absorption and excretion. The structural mediator of this action—the mineralocorticoid receptor (MR)—was not identified until more than one-third of a century later.2 Yet, evidence for the presence of extra-renal effects of aldosterone, specifically in the cardiovascular (CV) system, was evident nearly a decade before its discovery. Selye3 reported the formation of perivascular granulomas in various tissues in rats fed with deoxycorticosterone acetate (DOCA), an intermediate in the biosynthetic pathway of aldosterone with mineralocorticoid activity. Later, they reported the development of myocardial necrosis and fibrosis after DOCA administration to animals.4 However, it was not until approximately 30 years later when Brilla and Weber5 in Karl Weber’s group essentially repeated Selye’s experiment using aldosterone rather than DOCA that the growth of data supporting the extra-renal role of aldosterone/MR in the CV system began to expand.
At about the same time as Weber’s group was performing their experiments, an expansion of our understanding of how activation of steroid receptors induces cellular changes was occurring. During the latter third of the 20th century, the prevailing dogma was that steroid receptor activation resulted in a direct genomic modification. In contrast, peptide receptor activation resulted in a more rapid change in cellular function with the potential of a secondary, delayed indirect genomic effect. However, an increasing body of recent data has documented that activation of steroid receptors leads to both a genomic and a rapid non-genomic effect similar to what had been seen with peptide receptors. While some data suggest that the mediator of a steroid’s non-genomic effect is via a second receptor, in most cases, the data support the hypothesis that the non-genomic effect is mediated by the same receptor that triggers the direct genomic effect. Thus, aldosterone mediates its effects via MR-mediated genomic and rapid non-genomic pathways.6 The reason for the two pathways is not clear and may differ depending on the steroid. For oestrogen receptor α and the MR, the non-genomic pathway appears to damping the potential adverse effects of activating the genomic pathway.7,8
Herein, we will review the physiologic and pathologic roles of aldosterone and MR in the CV system with data obtained from four assessment areas: (i) mechanistic studies in rodents and humans; (ii) pre-clinical studies of MR antagonists (MRAs); (iii) clinical trials of MRAs; and (iv) genetic predisposing factors. We will not review their roles in essential hypertension or primary aldosteronism because of several recent extensive reviews on these topics.9,10 Thus, we will focus on six other CV conditions: heart failure (HF); myocardial infarction (MI); atrial fibrillation (AF); atherosclerosis; stroke; and thrombosis.
1.1 Search strategies and determining the strength of evidence
We acquired the data used for this review from an online search of the National Library of Medicine using the PubMed search engine from January 2011 through June 2021 (see Supplementary material online, FigureS1). We provide our detailed search strategy and methods of providing the strength of evidence in the supplementary material online, TableS1.
2. Aldosterone, MR, and the regulation of aldosterone secretion
2.1 Aldosterone biosynthesis
Aldosterone is mainly synthesized in zona glomerulosa cells of the adrenal cortex. Cholesterol is the substrate for aldosterone synthesis (Figure 1A). The initial step is the transfer of cholesterol, using Steroidogenic Acute Regulatory (StAR) protein, from the mitochondrial outer membrane to the intermembrane where it is acted on by cholesterol side-chain cleavage enzyme (CYP11A1) to form pregnenolone before converting to progesterone and 11-deoxycorticosterone, respectively. The last two steps in aldosterone biosynthesis are mediated by aldosterone synthase (CYP11B2).11

Aldosterone biosynthesis. (A) Cholesterol is the substrate for aldosterone biosynthesis. The last two steps in aldosterone biosynthesis are mediated by CYP11B2. Enzymes localized in the mitochondria are in orange boxes. (B) Regulation of aldosterone via the RAAS. Renin converts angiotensinogen to angiotensin I. ACE converts angiotensin I to angiotensin II. Angiotensin II directly stimulates aldosterone production in the adrenal gland and suppresses renin release. Aldosterone then exerts its role in sodium reabsorption in the kidney, in which the increase in volume acts as a negative feedback to the renin secretion. Potassium stimulates aldosterone secretion, which increases renal potassium loss. Finally, aldosterone binds to the MR and acts as a negative feedback to aldosterone secretion through a short negative feedback loop. (C) Cortisol is converted to the inactive cortisone via 11β-HSD2, whereas the cortisone is converted to active cortisol via 11β-HSD1. CYP11A1, cytochrome P450 side-chain cleavage; CYP11B2, aldosterone synthase; ACE, angiotensin-converting enzyme; MR, mineralocorticoid receptor; 11β-HSD1, 11 β-hydroxysteroid dehydrogenase type 1; 11β-HSD2, 11 β-hydroxysteroid dehydrogenase type 2.
2.2 Regulation of aldosterone secretion
Three primary factors modify aldosterone secretion—angiotensin II, potassium, and adrenocorticotropin hormone (ACTH). The most extensively studied of these have been angiotensin II and its generating mechanism: the renin–angiotensin system (Figure 1B). While renin can be synthesized in many cells, its primary source for this review is the juxtaglomerular apparatus of the kidney. Its release is regulated by both stimulatory (e.g. volume depletion, β-adrenergic stimulation, and prostaglandins) and inhibitory (e.g. angiotensin II, atrial natriuretic peptide, dopamine, potassium, and α-adrenergic stimulation) factors. Renin converts angiotensinogen, primarily synthesized in the liver, but also in other cell, to angiotensin I. Angiotensin-converting enzyme (ACE) converts angiotensin I to the biologically active angiotensin II. Angiotensin II directly stimulates aldosterone production by the zona glomerulosa cell and also can directly suppress renin release. As noted above, aldosterone binds to the MR in the renal cortical collecting duct and the connecting tubule. Aldosterone then exerts its primary role in sodium reabsorption in the kidney and colon,12 in which the volume increase acts as a long negative feedback loop to modulate renin secretion. In addition, activation of the MR on the zona glomerulosa cell acts as a negative feedback to aldosterone secretion through an ultra-short feedback loop.13
Potassium ions directly stimulate aldosterone secretion, independent of any effect they may have on the circulating renin–angiotensin–aldosterone system (RAAS). The glomerulosa cell is susceptible to potassium. An acute increase in serum potassium of 0.1 mEq/L can produce as much as a 25% increase in serum aldosterone levels and vice versa.14 Thus, potassium is involved in a short feedback loop with aldosterone. The relationship between the RAAS and potassium is complex. However, aldosterone’s primary role in mineral homeostasis likely is to control the renal loss of potassium with varying levels of sodium intake.15
ACTH is a potent acute aldosterone secretagogue. However, an ACTH infusion has only a short-lived effect in humans and experimental animals unless a pulsatile infusion is given.16 With continuous ACTH stimulation, there is a paradoxical decrease in aldosterone secretion. Other secretagogues are endothelin, serotonin, parathyroid hormone, very-low-density lipoprotein, and leptin.17–20
2.3 Mineralocorticoid receptor
MR is expressed in a wide range of tissues and cell types, both epithelial and non-epithelial cells. Its ligands include mineralocorticoids (e.g. aldosterone), glucocorticoids, and progesterone. MR has a high affinity for both aldosterone and cortisol (corticosterone in rodents). In normal circumstances, aldosterone binds and activates MR although at a 100- to 1000-fold lower circulating level than cortisol in large part due to the presence of 11-β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) in many epithelial tissues. 11β-HSD2 converts cortisol to the inactive cortisone (Figure 1C). In some non-epithelial cells, such as cardiomyocytes and inflammatory cells, there is a lack of 11β-HSD2. Thus, in cardiomyocytes, glucocorticoids can occupy and activate the MR rather than aldosterone because of its higher circulating levels.
2.4 Aldosterone actions
Aldosterone acts through caveolae-mediated intracellular events and exerts its action via classic genomic and rapid non-genomic pathways (Figure 2). In the classic genomic pathway, aldosterone binds to the MR at the cell membrane and is translocated to the nucleus via the action of heat-shock proteins, where it binds to specific regions of the DNA resulting in gene transcription.
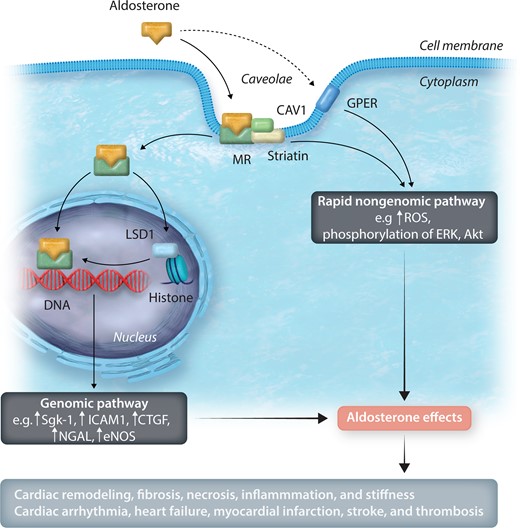
Aldosterone actions via genomic and rapid non-genomic pathways. Aldosterone acts through caveolae-mediated intracellular events and exerts its actions via genomic and rapid non-genomic pathways. MR is complexed with CAV1 and striatin in a caveolae. In the genomic pathway, aldosterone bound to the MR is translocated to the nucleus, where it binds to specific regions of the DNA and leads to gene transcription. LSD1, along with other epigenetic factors, can modulate this effect. There are two main players in the non-genomic pathway. The interaction between the MR, CAV1, and striatin or the activation via the GPER pathway activates the rapid non-genomic pathway, usually via phosphorylation of tyrosine kinase receptors and ERK. MR, mineralocorticoid receptor; CAV1, caveolin-1; LSD1, lysine-specific demethylase 1; Sgk-1, serum and glucocorticoid regulated kinase 1; ICAM1, intracellular adhesion molecule 1; CTGF, connective tissue growth factor; NGAL, neutrophil gelatinase-associated lipocalin; eNOS, endothelial nitric oxide synthase; ROS, reactive oxygen species; ERK, extracellular signal-regulated protein kinase; Akt, protein kinase B; GPER, G protein-coupled estrogen receptor.
A recently described epigenetic modulator of this process is lysine-specific demethylase 1 (LSD1), a flavin-containing protein. It can demethylate two different histone sites. Depending on the site demethylated, LSD1 can serve as a co-promoter or a co-repressor of gene transcription.21 An activated steroid receptor can modulate the histone where the LSD1 binds.22 Thus, there is a cross-talk between these two molecules in modifying aldosterone’s genomic effect. Interestingly, the level of salt intake modulates both the level of aldosterone secretion and the level of LSD1.23
Aldosterone also exerts its action via a rapid non-genomic pathway in which the response [increased reactive oxygen species (ROS), extracellular signal-regulated protein kinase 1 and 2 (ERK1/2), Akt, regulation of sodium and pH]7,24–28 can be seen in seconds or minutes, similar to what is seen with cell-surface receptors, e.g. angiotensin II, insulin, and catecholamines. There are two main players in the non-genomic pathway. First, caveolin-1 (CAV1) and striatin in caveolae are complexed with the MR on the cell surface. When aldosterone interacts with this complex, a rapid non-genomic pathway is activated29–31 with phosphorylation of tyrosine kinase receptors and ERK1/2.27 Striatin is critical in modulating aldosterone’s non-genomic effect as the pathway is not activated in its absence.27 However, in the absence of striatin, aldosterone/MR’s genomic pathway is still activated. An in-depth review of the roles of CAV1 and striatin in mediating aldosterone’s non-genomic effects is provided in a series of publications and an extensive review by Baudrand et al.6,23,28–30,32–34 Second, it has been proposed that aldosterone mediates its non-genomic effects via activating a second receptor—the G-protein-coupled oestrogen receptor (GPER).24,25,35,36 Aldosterone stimulation via GPER also increased ERK1/2 phosphorylation.24,25 It is uncertain whether CAV1 is involved in GPER’s mechanism of action. Activation of the GPER pathway does not modify the MR/dependent genomic pathway.36 Thus, both potential non-genomic pathway mechanisms are independent of MR’s direct genomic effect.
3. Aldosterone, MR, and the CV system
Extensive mechanistic studies in animals and humans have supported the roles of aldosterone and MR in CV diseases. Three general results have come from these studies. First, a liberal sodium intake enhances aldosterone/MR’s adverse effects. Second, often hypertension does not appear to be a critical mediator of the damage, as many studies have demonstrated no significant blood pressure (BP) changes associated with dramatic changes in CV damage.37–43 Third, deleterious effects of aldosterone occur even when its circulating levels are not high as MR can be increased and/or activated by other mineralocorticoids or ligand-independent mechanisms.33,44 Here, we review roles of aldosterone or MR activation in the following five CV-associated cell types: cardiomyocytes, vascular smooth muscle cells (VSMCs), endothelial cells (ECs), inflammatory cells, and fibroblasts.
3.1 Cardiomyocytes
Cardiomyocyte MR activation promotes cardiac fibrosis, hypertrophy, remodelling, and arrhythmia (Figure 3). Several important MR target genes in cardiac fibrosis and remodelling include plasminogen activator inhibitor 1 (PAI-1), connective tissue growth factor (CTGF), and neutrophil gelatinase-associated lipocalin (NGAL)—an important regulator of matrix metalloproteinase (MMP9).45–47 There was a marked increase in tissue fibrosis in the deoxycorticosterone acetate/dietary salt (DOCA/salt mice model).48,49 DOCA/salt increased T-cell chemoattractant CC chemokine receptor 5 (CCR5) and NADPH oxidase (NOX) mRNA levels.48 Cardiomyocyte MR-null mice had less tissue fibrosis and a higher cardiac MMP2/MMP9 activity, which is likely to promote collagen degradation. There was no significant difference in profibrotic gene expression (CTGF, collagen type III, and fibronectin) between cardiomyocyte MR-null and wild-type (WT) mice. Cardiomyocyte MR-null mice had an upregulation in the CTGF/transforming growth factor α (TGF α)-inhibitory protein Decorin.48
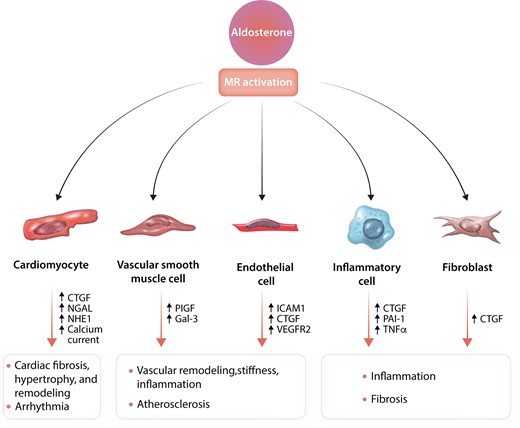
Aldosterone and effects of MR activation in various cells involved in cardiovascular diseases. Detailed discussion of this figure is given in Section 3. MR, mineralocorticoid; CTGF, connective tissue growth factor; NGAL, neutrophil gelatinase-associated lipocalin; NHE1, Na+–H+ exchange isoform 1; ICAM1, intracellular adhesion molecule 1; VEGFR2, vascular endothelial growth factor type 2 receptor; PIGF, placental growth factor; Gal-3, galectin 3; PAI-1, plasminogen activator inhibitor 1; TNFα, tumour necrosis factor α.
Apart from the aldosterone-MR effect via the classic genomic pathway, aldosterone increases the sodium-hydrogen ion exchanger isoform 1 (NHE-1) stimulation, leading to cardiac hypertrophy.26,50 However, cardiac hypertrophy may not be mediated solely by cardiomyocyte MR. Evidence in the HF model rodent supports this as there was a lack of attenuation in cardiac hypertrophy in cardiomyocyte MR-null mice.51 An increase in aldosterone also stimulates ventricular myocyte apoptosis both in vivo and in vitro.52
Cardiomyocyte MR activation can lead to ventricular arrhythmia and sudden cardiac death. In vitro studies showed an increase in T-type calcium channel expression and spontaneous beating frequency in rat ventricular myocytes stimulated with aldosterone.53 An increase in aldosterone levels correlated with the amplitude of L-type calcium current and impacted action potential duration.54 Aldosterone increased ryanodine activity in which an enhanced diastolic leak of calcium via ryanodine receptor generates delayed afterdepolarizations.55 In addition, aldosterone increased store-operated calcium entry (SOCE) activities in neonatal rat ventricular myocytes due to an increase in Orai1, transient receptor potential canonical (TRPC)1, TRPC4, TRPC5, and stromal interaction molecule1 (STIM1).56 These data were supported by the study in adult rat ventricular myocytes.57 Aldosterone increased SOCE, TRPC1, TRPC5, and STIM1 mRNA expression and protein level. Aldosterone increased calcium entry in the ventricular myocyte, which was prevented by co-treatment with RU-28318 (a specific MRA).57 Furthermore, aldosterone regulates potassium channels. For example, (i) aldosterone induced the opening of mitochondrial ATP-dependent potassium channels via EGFR transactivation and increased mitochondrial ROS production in rat ventricular myocyte.58 (ii) Aldosterone induced the upregulation of Kv1.5 (potassium channel) expression in neonatal rat atrial myocytes, in which spironolactone reversed it.59 Some of these activities may be related to activation of the MR/non-genomic pathway.58
There is also an interaction between cardiomyocytes and ECs via a paracrine mechanism.60 For example, in cardiomyocyte-specific overexpression of human MR mice, there was a decrease in NO-mediated relaxing response to acetylcholine in coronary arteries. Furthermore, cardiac levels of ROS, NOX, and expression of NOX subunit gp91phox increased.60
Finally, there is an important synergy between aldosterone and cortisol (corticosterone) in cardiomyocytes. As noted above, cortisol and aldosterone bind equally well to the MR. Furthermore, cortisol’s concentration in plasma is an order of magnitude higher than aldosterone. What protects the MR to allow specific binding with aldosterone is 11β-HSD2 that inactivates cortisol. However, the cardiomyocyte lacks 11β-HSD2.61 Thus, under normal conditions, MR is primarily occupied by cortisol. This binding appears to protect the cardiomyocyte from activation by aldosterone that is harmful, as transgenic mice overexpressing 11β-HSD2 have cardiac hypertrophy, fibrosis, and HF.62 In addition, balanced cardiomyocyte glucocorticoid receptor (GR) and MR signalling are important.63 For example, (i) mice with cardiomyocyte-specific overexpression human MR had ventricular arrhythmia and sudden death.64 (ii) Mice with cardiomyocyte-specific overexpression of human GR had bradycardia and atrio-ventricular block.65 In addition, cardiomyocyte MR-null mice had normal heart morphology and function. They did not display a significant increase in the expression of pathological markers, including b-myosin heavy chain (Myh7), skeletal muscle a-actin (Acta1), brain natriuretic peptide (Nppb), and smooth muscle a-actin (Acta2).63 In contrast, the cardiomyocyte GR-null mice had significantly increased Acta1 and Nppb expression, developed cardiac hypertrophy, LV systolic dysfunction, and pre-mature HF death. Interestingly, in mice with cardiomyocyte-specific deletion of both GR and MR, all four pathological markers increased. However, these mice had normal cardiac morphology, systolic function, and did not die pre-maturely from HF, unlike the cardiomyocyte GR-null mice.
3.2 Vascular smooth muscle cells
VSMC MR plays an essential role in vascular remodelling, stiffness, and atherosclerosis (Figure 3).37–41,66,67 Aldosterone stimulates various gene expressions following endothelial damage and in segments of vessel prone to atherosclerosis under non-laminar flow conditions.41 Aldosterone regulates several important vascular MR target genes, including placental growth factor (PlGF), a vascular endothelial growth factor (VEGF) family member.37,38 PlGF binds to VEGF type 1 receptor expressed on endothelial and inflammatory cells to promote angiogenesis.37 PlGF promotes VSMC proliferation and monocyte chemotaxis, which are vital to form atherosclerotic plaques. VSMC MR-null mice had a reduced vascular fibrotic response to aldosterone treatment and a wire-induced carotid injury.67 In VSMC MR-null mice, the induction of α5-integrin expression by aldosterone and salt intake was prevented by altering the fibronectin/α5 subunit integrin ratio.66
Galectin 3 (Gal-3) also is involved in vascular remodelling. In vitro and in vivo studies showed that aldosterone increase Gal-3 expression. The upregulation of Gal-3 specifically increased collagen type I production and inhibition of Gal-3 blocks the effect of aldosterone-induced inflammation, collagen type I deposition, vascular fibrosis, and remodelling.68
3.3 Endothelial cells
Activation of MR in Ecs impairs vascular reactivity, promotes inflammation, and contributes to cardiac dysfunction. Aldosterone induces the expression of intracellular adhesion molecule 1 (ICAM1), CTGF, and other proinflammatory genes (Figure 3).69,70 ICAM1 expression promotes leukocyte adhesion and is necessary for the formation of early atherosclerotic plaque.69 After DOCA/salt treatment (which activates the MR), there is increased macrophage recruitment and proinflammatory genes in WT but not in EC MR-null mice.71 Furthermore, in mice with ApoE–/–/ICAM1–/–, aldosterone does not induce an increase in plaque size, lipid content, and macrophage infiltration.69 In humans, aldosterone regulates ICAM1 expression in human Ecs and human umbilical vein Ecs (HUVECs).71,72 However, following pressure-overload in mice, LV ICAM1 expression and CD4+ T-cell and CD11b+ monocyte infiltration increased and were not affected by EC MR deletion. Interestingly, EC MR-null mice were protected from systolic dysfunction independent of increased cardiac adhesion molecule expression and leukocyte infiltration induced by LV pressure overload.73 Activation of MR in EC leads to vascular stiffness via activation of the endothelium sodium channel (EnNaC), promoting sodium entry into the EC. Increased sodium entry also results in more inflammation, NOX2, reduced endothelium NO synthase and NO production, and a subsequent increase in vascular stiffness.74 Furthermore, aldosterone decreases coronary flow and reduces coronary vasodilation to adenosine and acetylcholine.75In vitro study showed that aldosterone increased stiffness in HUVECs culture.76
Activation of MR in EC impairs angiogenesis via downregulation of VEGFR2. In response to hypoxia-induced factor 1α, cardiomyocytes produce VEGF, which binds to VEGFR2 and reduces angiogenesis. In contrast, in EC MR-null mice, the number of capillaries per myocyte area was higher than in WT mice following DOCA/salt treatment because angiogenesis was not reduced. These data were supported by an in vitro study in HUVECs, demonstrating an upregulation of VEGFR2 after MR knockdown.70
3.4 Inflammatory cells
Immune systems have an essential role in mediating inflammatory response and fibrosis (Figure 3). There is no 11β-HSD2 expression in inflammatory cells.77,78 Thus, it would be anticipated that cortisol or corticosterone would preferentially bind to the MR over aldosterone as well as binding to the GR. In vitro studies in rodents demonstrated that corticosterone stimulates proinflammatory response in macrophages that could be secondary to MR or GR activation. However, blocking the GR but not the MR suggests that corticosterone’s proinflammatory effect is via activating the GR and not MR.77
Following L-NG-nitro-arginine-methyl-ester (L-NAME)/salt or DOCA/salt, macrophage MR-null mice were protected from increasing interstitial collagen deposition. There was an increase in CTGF, PAI-1, inducible NOS, and collagen type III expression in both macrophage MR-null and WT mice.79,80 Recruitment of macrophages in the heart was also increased in both genotypes.79,80 However, inducible NOS positive macrophage infiltration and tumour necrosis factor α (TNFα) expression in WT was substantially greater than in MR-null mice, demonstrating that the loss of MR reduces macrophages with classically-activated or proinflammatory phenotypes (M1).79 These data were supported by another study from Usher et al.77In vitro, aldosterone increased TNFα expression and other M1 markers. MRA decreased proinflammatory gene expression and increased markers of alternatively activated states or M2 phenotypes, including Arg1 and Fizz1, which play an important role in wound healing and tissue repair. In vivo, the L-NAME/salt-treated myeloid MR-null mice were protected against cardiac hypertrophy, fibrosis, and vascular fibrosis. As would be anticipated, collagen type III, TGFβ, TNFα, and PAI-1 expression were also reduced in myeloid MR-null mice vs. WT mice. Similar to the in vitro response to MRA, macrophages isolated from myeloid MR-null mice showed a reduction in most M1 markers and increased in many M2 markers.
It is likely that the aldosterone/MR-mediated inflammatory effect is mediated by monocyte chemoattractant protein 1. The chemokine ligand 2 (CCL2) gene encodes this protein that is required for tissue macrophage recruitment. Thus, DOCA-induced macrophage recruitment is substantially reduced in CCL2-null mice. Furthermore, compared with WT mice, CCL2-null mice had less cardiac fibrosis. Finally, there was a substantial suppression of cardiac inflammatory markers and a greater collagen breakdown by MMP9 without significantly affecting cardiac hypertrophy in CCL2-null mice treated with DOC.49
3.5 Fibroblasts
Fibroblasts play an essential role in forming tissue fibrosis (Figure 3), which is later followed by remodelling. In rat cardiac fibroblasts, aldosterone increases hydroxyproline content and CTGF expression; spironolactone or finerenone reverses these effects. 11β-HSD2 is not expressed in cardiac fibroblasts. Therefore, cortisol can bind to the MR. However, cortisol did not increase CTGF expression levels in cultured cardiac fibroblasts.81 In addition, aldosterone induces fibroblast proliferation in which spironolactone or eplerenone (MR blockade) can reverse this effect. However, mifepristone (a GR antagonist), also can reduce fibroblast proliferation, suggesting that GR activation had an antiproliferative effect.82 However, these positive in vitro data have not led to potential effects in vivo. In the pressure-overload HF model, MR knockout fibroblast did not change the phenotype.51 A search for other in vivo models was not found.
4. Aldosterone, MR, and pathologic roles in CV diseases
This section reviews the relationship between aldosterone/MR and the six focused CV conditions. Each review is divided into four subsections: pre-clinical studies; clinical studies; genetic association studies; and strength of evidence between aldosterone, MR, and each CV disease.
4.1 Heart failure
In pre-clinical studies, mineralocorticoid receptor antagonists (MRAs) demonstrated CV protection beyond diuretic effects. From 1999, several large randomized controlled trials (RCTs) have shown clinical benefits of MRAs in reducing mortality in HF patients.83–85
4.1.1 Pre-clinical studies
In rodents with pressure-overload cardiac hypertrophy and HF (commonly induced by transverse aortic constriction), there were increased cardiac hypertrophy, dilation, fibrosis, inflammation, gap junction remodelling, arrhythmia, and decreased cardiac function and coronary flow reserve.86,87 MR activation in various cells plays an important role in the pathogenesis of cardiac hypertrophy and dysfunction. For example, Rac1, a member of the Rho family of GTPases, is an intracellular modulator that can activate the MR. In mice with transverse aortic constriction, Rac1 is activated resulting in increased nuclear accumulation of MR and increased expression of MR target genes, e.g. NOX4 gene induction and ROS overproduction.44,88 In contrast, Rac1 deletion in cardiomyocytes suppresses MR signalling, attenuates cardiac hypertrophy and dysfunction.44 Furthermore, the type of damage Rac1 activation of MR produces varies depending in which cardiac cell type the MR is activated. Thus, compared with WT mice, (i) cardiomyocyte MR-null mice had attenuated cardiac dilation and improved cardiac function but still had cardiac hypertrophy, fibrosis, and inflammation.51 (ii) VSMC MR-null mice had improved cardiac hypertrophy, cardiac function, dilation, fibrosis, inflammation, and increased coronary flow reserve.86 (iii) T-cell MR-null animals had similar CV protective benefits as VSMC MR-null mice, except coronary flow reserve was not improved.86,89 (iv) EC MR-null mice had improved cardiac function.73 (v) In myeloid MR-null mice, cardiac hypertrophy and fibrosis were reduced.90 However, fibroblast MR knockout did not show CV benefits when subjected to transverse aortic constriction.51
In rodents with HF, MRAs reduce cardiac hypertrophy, dysfunction, and damage.26,43–45,82,91–94 For example, eplerenone reduced myocardial fibrosis, apoptosis, and mortality.91 Spironolactone inhibited cardiac fibroblast proliferation,81,93 blunted pathological gap junction remodelling, and reduced ventricular pre-mature beats.87 However, it did not diminish cardiac hypertrophy. Several studies in rodents with HF have compared the efficacy of two MRAs: finerenone and eplerenone.92,95 Compared with eplerenone, finerenone treatment led to a significant reduction of left ventricular (LV) wall thickness and mass.92 Furthermore, finerenone showed better antifibrotic activity than eplerenone in a mice model of cardiac fibrosis.95 In addition to the phenotypic effects of MRAs, MRAs also had a positive effect on molecular consequences of HF. For example, they prevented an increase in CTGF and NOX4 expression.44,45,96
4.1.2 Clinical studies
4.1.2.1 Mechanistic studies and RCTs/meta-analyses
In patients with HF with reduced ejection fraction, RCTs and meta-analyses documented consistent improvement when MRAs were given. In 1999, the Randomized Aldactone Evaluation Study (RALES) was the first large RCT to demonstrate CV benefit in chronic HF patients with LV ejection fraction (LVEF) ≤35%. There was a significant reduction in mortality in the spironolactone group (Table 1).83 In 2003, the Eplerenone Post Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) was the second large RCT to demonstrate a significant reduction in deaths and other primary endpoints in patients with acute MI and systolic dysfunction.84 Almost 10 years later, in 2011, eplerenone showed a reduction in death from CV causes or hospitalization for HF in the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF).85 A meta-analysis from these three RCTs further showed a lower risk of sudden cardiac death in the MRA-treated than placebo-treated patients after a mean follow-up of 18 months (hazard ratio of 0.77, 95% confidence interval 0.66–0.89).97 In support of these results, spironolactone also improves biomarkers of collagen metabolism, inflammation, and thrombosis in patients at risk for HF.98
RCTs of MRAs and CV diseases
| Studiesa | Participants | n | Treatment (in addition to standard therapy) | Primary outcome | Main findings |
|---|---|---|---|---|---|
| RALES (1999)83 | Patients with chronic HF (NYHA FC IV within 6 months) and were in NYHA FC III or IV at the time of enrolment and LVEF ≤35% | 1663 | Spironolactone or placebo | Death from any cause |
|
| Category: HF |
| ||||
| EPHESUS (2003)84 | Patients with acute MI and LVEF ≤40% | 6632 | Eplerenone or placebo | Time to death from any cause and time to death from CV causes or first hospitalization for a CV event, including HF, acute MI, stroke, or ventricular arrhythmia |
|
| Category: HF |
| ||||
| EMPHASIS-HF (2011)85 | Patients with chronic HF (NYHA FC class II) and LVEF ≤30% (or LVEF >30–35% with wide QRS complex) | 2737 | Eplerenone or placebo | Composite of death from CV causes or a first hospitalization for HF |
|
| Category: HF, AF | |||||
| TOPCAT (2014)99 | Patients with symptomatic HF and LVEF ≥45% | 3445 | Spironolactone or placebo | Composite of death from CV causes, aborted cardiac arrest, or hospitalization for HF |
|
| Category: HF |
| ||||
| REMINDER (2014)123 | Patients with acute MI without clinical HF | 1012 | Eplerenone (within 24 h of STEMI) or placebo | Composite endpoint for time to the first occurrence of CV mortality, re-hospitalization, or extended initial hospital stay due to diagnosis of HF or sustained VT or VF, and LVEF ≤40% at 1 month or later post-randomization or high BNP |
|
| Category: MI |
| ||||
| ALBATROSS (2016)124 | Patients with ischaemic symptoms within 72 h with ≥1 of the following indicators of MI (ST-segment elevation or Q waves in ≥2 contagious leads, new/undated left bundle branch block, elevated troponin levels, or Thrombolysis In Myocardial Infarction score ≥3) | 1603 | Potassium canrenoate followed by spironolactone or standard treatment alone | Composite of death, resuscitated cardiac arrest, significant ventricular arrhythmia, class IA indication for an implantable defibrillator, or new or worsening HF during 6-month follow-up |
|
| Category: MI |
|
| Studiesa | Participants | n | Treatment (in addition to standard therapy) | Primary outcome | Main findings |
|---|---|---|---|---|---|
| RALES (1999)83 | Patients with chronic HF (NYHA FC IV within 6 months) and were in NYHA FC III or IV at the time of enrolment and LVEF ≤35% | 1663 | Spironolactone or placebo | Death from any cause |
|
| Category: HF |
| ||||
| EPHESUS (2003)84 | Patients with acute MI and LVEF ≤40% | 6632 | Eplerenone or placebo | Time to death from any cause and time to death from CV causes or first hospitalization for a CV event, including HF, acute MI, stroke, or ventricular arrhythmia |
|
| Category: HF |
| ||||
| EMPHASIS-HF (2011)85 | Patients with chronic HF (NYHA FC class II) and LVEF ≤30% (or LVEF >30–35% with wide QRS complex) | 2737 | Eplerenone or placebo | Composite of death from CV causes or a first hospitalization for HF |
|
| Category: HF, AF | |||||
| TOPCAT (2014)99 | Patients with symptomatic HF and LVEF ≥45% | 3445 | Spironolactone or placebo | Composite of death from CV causes, aborted cardiac arrest, or hospitalization for HF |
|
| Category: HF |
| ||||
| REMINDER (2014)123 | Patients with acute MI without clinical HF | 1012 | Eplerenone (within 24 h of STEMI) or placebo | Composite endpoint for time to the first occurrence of CV mortality, re-hospitalization, or extended initial hospital stay due to diagnosis of HF or sustained VT or VF, and LVEF ≤40% at 1 month or later post-randomization or high BNP |
|
| Category: MI |
| ||||
| ALBATROSS (2016)124 | Patients with ischaemic symptoms within 72 h with ≥1 of the following indicators of MI (ST-segment elevation or Q waves in ≥2 contagious leads, new/undated left bundle branch block, elevated troponin levels, or Thrombolysis In Myocardial Infarction score ≥3) | 1603 | Potassium canrenoate followed by spironolactone or standard treatment alone | Composite of death, resuscitated cardiac arrest, significant ventricular arrhythmia, class IA indication for an implantable defibrillator, or new or worsening HF during 6-month follow-up |
|
| Category: MI |
|
NYHA FC, New York Heart Association functional classification; LVEF, left ventricular ejection fraction; HF, heart failure; MI, myocardial infarction; STEMI, ST-elevation myocardial infarction; CV, cardiovascular; MRA, mineralocorticoid receptor antagonist; RR, relative risk; HR, hazard ratio; CI, confidence interval; VT, ventricular tachycardia; VF, ventricular fibrillation.
We included only studies with >1000 participants (not directly focused on diabetes mellitus or chronic kidney disease), and the primary outcome was related to our focused cardiovascular conditions.
RCTs of MRAs and CV diseases
| Studiesa | Participants | n | Treatment (in addition to standard therapy) | Primary outcome | Main findings |
|---|---|---|---|---|---|
| RALES (1999)83 | Patients with chronic HF (NYHA FC IV within 6 months) and were in NYHA FC III or IV at the time of enrolment and LVEF ≤35% | 1663 | Spironolactone or placebo | Death from any cause |
|
| Category: HF |
| ||||
| EPHESUS (2003)84 | Patients with acute MI and LVEF ≤40% | 6632 | Eplerenone or placebo | Time to death from any cause and time to death from CV causes or first hospitalization for a CV event, including HF, acute MI, stroke, or ventricular arrhythmia |
|
| Category: HF |
| ||||
| EMPHASIS-HF (2011)85 | Patients with chronic HF (NYHA FC class II) and LVEF ≤30% (or LVEF >30–35% with wide QRS complex) | 2737 | Eplerenone or placebo | Composite of death from CV causes or a first hospitalization for HF |
|
| Category: HF, AF | |||||
| TOPCAT (2014)99 | Patients with symptomatic HF and LVEF ≥45% | 3445 | Spironolactone or placebo | Composite of death from CV causes, aborted cardiac arrest, or hospitalization for HF |
|
| Category: HF |
| ||||
| REMINDER (2014)123 | Patients with acute MI without clinical HF | 1012 | Eplerenone (within 24 h of STEMI) or placebo | Composite endpoint for time to the first occurrence of CV mortality, re-hospitalization, or extended initial hospital stay due to diagnosis of HF or sustained VT or VF, and LVEF ≤40% at 1 month or later post-randomization or high BNP |
|
| Category: MI |
| ||||
| ALBATROSS (2016)124 | Patients with ischaemic symptoms within 72 h with ≥1 of the following indicators of MI (ST-segment elevation or Q waves in ≥2 contagious leads, new/undated left bundle branch block, elevated troponin levels, or Thrombolysis In Myocardial Infarction score ≥3) | 1603 | Potassium canrenoate followed by spironolactone or standard treatment alone | Composite of death, resuscitated cardiac arrest, significant ventricular arrhythmia, class IA indication for an implantable defibrillator, or new or worsening HF during 6-month follow-up |
|
| Category: MI |
|
| Studiesa | Participants | n | Treatment (in addition to standard therapy) | Primary outcome | Main findings |
|---|---|---|---|---|---|
| RALES (1999)83 | Patients with chronic HF (NYHA FC IV within 6 months) and were in NYHA FC III or IV at the time of enrolment and LVEF ≤35% | 1663 | Spironolactone or placebo | Death from any cause |
|
| Category: HF |
| ||||
| EPHESUS (2003)84 | Patients with acute MI and LVEF ≤40% | 6632 | Eplerenone or placebo | Time to death from any cause and time to death from CV causes or first hospitalization for a CV event, including HF, acute MI, stroke, or ventricular arrhythmia |
|
| Category: HF |
| ||||
| EMPHASIS-HF (2011)85 | Patients with chronic HF (NYHA FC class II) and LVEF ≤30% (or LVEF >30–35% with wide QRS complex) | 2737 | Eplerenone or placebo | Composite of death from CV causes or a first hospitalization for HF |
|
| Category: HF, AF | |||||
| TOPCAT (2014)99 | Patients with symptomatic HF and LVEF ≥45% | 3445 | Spironolactone or placebo | Composite of death from CV causes, aborted cardiac arrest, or hospitalization for HF |
|
| Category: HF |
| ||||
| REMINDER (2014)123 | Patients with acute MI without clinical HF | 1012 | Eplerenone (within 24 h of STEMI) or placebo | Composite endpoint for time to the first occurrence of CV mortality, re-hospitalization, or extended initial hospital stay due to diagnosis of HF or sustained VT or VF, and LVEF ≤40% at 1 month or later post-randomization or high BNP |
|
| Category: MI |
| ||||
| ALBATROSS (2016)124 | Patients with ischaemic symptoms within 72 h with ≥1 of the following indicators of MI (ST-segment elevation or Q waves in ≥2 contagious leads, new/undated left bundle branch block, elevated troponin levels, or Thrombolysis In Myocardial Infarction score ≥3) | 1603 | Potassium canrenoate followed by spironolactone or standard treatment alone | Composite of death, resuscitated cardiac arrest, significant ventricular arrhythmia, class IA indication for an implantable defibrillator, or new or worsening HF during 6-month follow-up |
|
| Category: MI |
|
NYHA FC, New York Heart Association functional classification; LVEF, left ventricular ejection fraction; HF, heart failure; MI, myocardial infarction; STEMI, ST-elevation myocardial infarction; CV, cardiovascular; MRA, mineralocorticoid receptor antagonist; RR, relative risk; HR, hazard ratio; CI, confidence interval; VT, ventricular tachycardia; VF, ventricular fibrillation.
We included only studies with >1000 participants (not directly focused on diabetes mellitus or chronic kidney disease), and the primary outcome was related to our focused cardiovascular conditions.
In contrast, spironolactone failed to show mortality benefits in patients with HF with preserved ejection fraction(HfpEF).99 In the Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) trial, spironolactone did not significantly reduce the primary endpoint (composite of death from CV causes, aborted cardiac arrest, or hospitalization for HF). However, in the post hoc analysis of TOPCAT, there was a significant reduction of the primary outcome in the spironolactone group in patients from the Americas (USA, Canada, Brazil, and Argentina), but not in patients randomized from Russia and Georgia.100 These data suggested that regional and racial heterogeneity should be taken to consideration. Furthermore, canrenone (an active metabolite of spironolactone) concentrations were undetectable in a significantly higher percentage of participants in the spironolactone group from Russia than from the USA and Canada.101 These findings suggested that the trial results obtained in Russia may not reflect the true therapeutic response to spironolactone. In addition, post hoc analysis of the TOPCAT-Americas trial suggested a potential reduction in all-cause mortality associated with spironolactone in women.102 However, a recent meta-analysis from RALES, EMPHASIS-HF, and TOPCAT-Americas trials showed a consistent and beneficial MRA effect, including the primary outcome of CV death or HF hospitalization and other secondary outcomes, regardless of sex.103
A recent meta-analysis also demonstrated that patients with HfpEF and HF with mid-range ejection fraction did not have an improvement in mortality although the spironolactone arm did reduce hospitalization, improving functional class and 6 min walk test.104 There are two ongoing large clinical trials in patients with mid-ranged or preserved EF that met our criteria, including the SPIRIT-HF (NCT04727073) and the FINEARTS-HF (NCT04435626) that may provide data to resolve the patient population with HF who will benefit from MRA treatment.
Association studies
Observational studies have shown a positive association between aldosterone levels or log-aldosterone with increased mortality in populations with symptomatic HF, or with coronary artery disease (CAD), or without obvious CV diseases.105–108 For example, baseline aldosterone level was a significant predictor of increased mortality risk in symptomatic HF patients during a 2 year follow-up period.107,109 Furthermore, in the Jackson Heart Study, log-serum aldosterone level was positively associated with increased mortality and incident CV diseases including HF.105 Finally, a higher baseline aldosterone level was associated with HF incidence in MI patients during a 2 year follow-up period110 and concentric LV hypertrophy measured by echocardiography in patients at risk of developing diastolic dysfunction or HfpEF.111
4.1.3 Genetics
Polymorphic variants in various genes have been reported to be associated with aldosterone dysregulation.112 However, no genetic association study in HF met our criteria (see Supplementary material online).
4.1.4 Strength of evidence
Level A.
4.2 Myocardial infarction
4.2.1 Pre-clinical studies
Following coronary artery ligation, a common MI model in rodents, MR mRNA expression and protein level in the LV increased, suggesting an enhanced expression of the MR in the heart during the post-infarct state.113 Furthermore, there were increased aldosterone and corticosterone levels (in rodents) after MI.114,115 As mentioned above, both steroids can bind to the MR on the myocyte but only corticosterone can bind to the GR. In addition, aldosterone and cortisol administration can increase the infarct area in cardiomyocytes.116 This effect is probably mediated through activation of the MR since spironolactone but not mifepristone can reverse it.116 MR plays an important role in mediating infarct size and vessel formation following MI. First, cardiomyocyte MR-null mice had increased capillary density, reduced accumulation of extracellular matrix proteins in the surviving myocardium, reduced cardiac hypertrophy, and attenuated pulmonary oedema.86 These cardiomyocyte MR-null mice also had reduced infarct expansion and enhanced neo-vascular formation at 7 days after MI. In addition, they had: (i) reduced mitochondrial oxygen production; (ii) blunted upregulation of NOX2 and NOX4; and (iii) suppressed NF-kB.114 Moreover, VSMC MR-null mice showed improved LV dysfunction, endothelial function, and reduced oxidative stress after MI.117 Finally, myeloid MR-null mice displayed improved cardiac function and remodelling associated with enhanced infarct neovascularization and scar maturation. In addition, single administration of MRAs to macrophages at the onset of MI improved the healing response and protected against cardiac remodelling and functional deterioration.118
In rats with coronary artery ligation, ventricular arrhythmia occurred because of the increase in hyperpolarization-activated and cyclic nucleotide-gated (HCN)2 and HCN4 levels. Spironolactone reduced the occurrence of arrhythmia by preventing an increase in HCN4.119
In rats with MI, there was an impaired diastolic function and increased collagen content in the LV interstitium and the aorta.120 MRAs reduced the infarct area and abnormal LV remodelling.115,116,121 In addition to an improved LV compliance and elastance, finerenone treatment reduced interstitial fibrosis in mice with MI.117 Early eplerenone treatment enhanced neovascularization in the healing myocardium,115 improved echocardiographic and hemodynamic evidence of diastolic dysfunction, myocardial interstitial collagen, and fibrosis.120,122
4.2.2 Clinical studies
4.2.2.1 RCTs/meta-analyses
The benefits of early MRAs in MI patients have been evaluated in several large RCTs (Table 1). For example, the Role of Eplerenone in Acute Myocardial Infarction–Double-Blind, Early Treatment Initiation, Randomized, placebo-controlled, multi-center study (REMINDER) documented that patients receiving eplerenone within 24 h after ST-elevated MI (STEMI) had a significant reduction in the brain natriuretic peptide (BNP)/N-terminal pro b-type natriuretic peptide (NT-proBNP) levels and the composite primary endpoint.123 The Early Aldosterone Blockade in Acute Myocardial Infarction (ALBATROSS) trial evaluated the effect of the MRA in patients with biochemical and clinical evidence of acute MI. These patients were treated with usual/standard care with or without MRA for 6 months. The composite endpoint was not significantly different between the two limbs. However, in those subjects with STEMI, MRA treatment significantly reduced the odds of death.124 Finally, a meta-analysis of 11 258 participants supported the benefits of MRA after MI as there was a reduction in overall and CV mortality in the MRA group compared to placebo or standard therapy alone.125
As mentioned above, pre-clinical studies in rodents showed the benefits of MRAs in reducing the infarct size following MI. However, the same effect was not observed in an RCT [Mineralocorticoid Receptor Antagonist Pre-treatment and Early Post-treatment to Minimize Reperfusion Injury after ST-elevation Myocardial Infarction (MINIMIZE STEMI)]. In patients treated acutely before reperfusion with an MRA for 3 months, LV remodelling was improved but a reduction in MI size was not observed.126 However, this was a small study which could have reduced the ability to observe the change in the size of the MI.
Association studies
Observational studies have shown a positive association between aldosterone levels and CAD. For example, increased baseline aldosterone levels were associated with: (i) acute ischaemic events, including MI, in CAD patients with preserved LV function;108 (ii) future vascular events in stable CAD patients during a median follow-up time of 4.7 years;127 (iii) future adverse events (reinfarction, HF, ventricular fibrillation, and cardiogenic shock) in MI patients during a 2 year follow-up.110 Yet, in patients undergoing carotid endarterectomy, during a median follow-up of 3 years, baseline aldosterone levels were not associated with the occurrence of future vascular events (composite of MI, stroke, vascular death, coronary revascularization or peripheral intervention, or leg amputation).128
4.2.3 Genetics
Polymorphic variants in various genes have been reported to be associated with aldosterone dysregulation. Polymorphic variants in two of these genes have been shown to be associated with MI—CYP11B2 and CAV1.
The human CYP11B2 gene is located at chromosome 8q22129 and although there were numerous common CYP11B2 polymorphisms, the rs1799998 C/T single nucleotide polymorphism (SNP), referred to as ‘–344,’ is the most reported SNP.130 Although the data are conflicting regarding which allele (C or T) is related to adverse CV outcomes,131–133in vitro studies suggested the possibility of both alleles relating to negative consequences in humans.134,135 The –344CC genotype was associated with CAD in Taiwanese women.136 A meta-analysis from Liu et al.137 demonstrated the significant association of the –344C/T polymorphism and CAD risk in the subgroup of Asians, although it did not show the same effect in the overall study population. There were several studies in Caucasians which were done before 2011. One study on middle-aged men supported the association of the –344C/T polymorphism with CAD, while two studies did not.
A CAV1 rs3807989 variant was associated with a significant risk of CAD and MI in three independent cohorts and in a meta-analysis from China.138 We could not find a report of the polymorphic variants of CAV1 in other ethnicities that fit our criteria (see Supplementary material online).
4.2.4 Strength of evidence
Level B.
4.3 Atrial fibrillation
4.3.1 Pre-clinical studies
Aldosterone promotes AF in rats. Aldosterone administration for 1–2 months was associated with left atrial fibrosis, increased atrial fibroblast, interstitial collagen, and atrial myocyte hypertrophy, longer P-wave duration, doubled the AF time and shortened the atrial action potential.139,140 Blocking the MR with spironolactone prevented the increase in atrial collagen content and the reduction in atrial action potential but did not affect the AF time.139In vitro study using the mouse HL-1 atrial myocytes showed that rapid depolarization increased MR expression through a calcium-dependent mechanism. Aldosterone increased the expression of α-1G and -1H subunits of the T-type calcium channel, calcium current, and intracellular calcium load.141 In rat cardiac fibroblasts cultured with aldosterone, fibronectin was upregulated secondary to increased CTGF and lysyl oxidase (LOX) levels. In support that these effects are mediated by aldosterone, spironolactone reduced LOX levels in mice with cardiac overexpression of constitutively active Rac1.81 Finally, eplerenone reduced the frequency of AF in a model of atrial dilation and fibrosis without HF.
4.3.2 Clinical studies
4.3.2.1 Mechanistic studies
There was an increase in left atrial fibrosis in patients with AF.81,142 Data on MR expression are conflicting.81,141 However, there were higher mRNA expression levels of several aldosterone targeted genes in the left atrium of AF patients, e.g. LOX, NOX, and CTGF.88,142,143
4.3.2.2 RCTs/meta-analyses and association studies
There was no RCT in AF patients using MRAs reported. There were four association-type studies. (i) A post hoc study from EMPHASIS-HF (described in Section 4.1) showed a significant reduction in developing new-onset AF or atrial flutter in patients receiving eplerenone (hazard ratio of 0.58, 95% confidence interval 0.35–0.96) in which the effect was similar irrespective of atrial arrhythmia status at baseline.144 (ii) A recent meta-analysis supported the EMPHASIS-HF data as there was a significant overall reduction of AF risk in MRA-treated patients, including reducing new-onset AF and recurrent AF, but not post-operative AF.145 (iii) Baseline aldosterone level was associated with AF (odds ratio of 1.24, 95% confidence interval 1.01–1.53) in 1674 participants aged ≥45 years from a community-based cohort from Minnesota during a 12 year follow-up period.106 (iv) Furthermore, higher preoperative plasma aldosterone levels were associated with post-operative AF (odds ratio of 1.007, 95% confidence interval 1.003–1.012) in patients underwent coronary artery bypass graft.146 We could not find any specific data related to whether potassium levels influencing the AF incidence associated with MRA use or higher aldosterone levels. However, a currently in progress clinical trial (ALDOCURE, NCT03551548) may address this issue.147
4.3.3 Genetics
Polymorphic variants in various genes have been reported to be associated with aldosterone dysregulation. Polymorphic variants in the same two genes that have been reported to be associated with MI are shown to be associated with AF—CYP11B2 and CAV1.
Two studies showed conflicting results for the association of CYP11B2 polymorphic variant (–344C/T) and AF. One study in China showed that the –344 T allele was associated with AF.148 In contrast, another study showed that the –344C allele did not significantly affect AF in hypertensive Chinese populations, although the left atrial dimension of the C allele carriers was larger than the non-C allele carriers.149 However, the most recent meta-analysis of 5466 participants from Wang et al.150 supported the association of –344C/T polymorphism and AF in the overall population and the subgroup of East Asians. The majority of studies have been done in a Chinese population. No specific study that met our criteria has been performed in other ethnic groups.
Regarding the polymorphic variants in CAV1, several studies from Denmark and China demonstrated the significant association between the rs3807989 and AF.151–154
4.3.4 Strength of evidence
Level B–/C+.
4.4 Atherosclerosis
4.4.1 Pre-clinical studies
The roles of VSMCs and Ecs in atherosclerosis were discussed in detail in a previous section (see Section 3). In addition, NGAL expression increased in the atherosclerotic plaques of ApoE–/–/LDLR–/– mice with MI. Colocalization with MMP-9 in areas with high proteolytic activity suggests a role for NGAL/24p3 in modulating the MMP-9-mediated remodelling of plaques and infarcted hearts.47 In the ApoE–/– mice, the treatment with eplerenone demonstrated a reduction in the atherosclerotic area.39
4.4.2 Clinical studies
4.4.2.1 Mechanistic studies and RCTs/meta-analyses
No clinical trials relating an aldosterone effect to atherosclerosis met our criteria (see Supplementary material online). However, a recent meta-analysis showed an improvement in endothelial function measured by flow-mediated dilation with MRA treatment in patients with hypertension, obesity, or CAD.155 Finally, in vitro studies using the human aorta and HUVECs, spironolactone inhibited the expression of genes involved in promoting atherosclerosis, for example, ICAM1.41,69
4.4.2.2 Association studies
Observational studies have assessed the association between aldosterone levels and carotid plaque. For example, (i) aldosterone level was an independent predictor of carotid plaques in hypertensive patients.156 (ii) The highest quartile of aldosterone level was associated with carotid intima-media thickness and increased prevalence of carotid plaques assessed by ultrasonography.156 (iii) Baseline aldosterone level was an independent predictor of carotid plaque progression assessed by duplex ultrasonography in patients with symptomatic vascular disease during a 2 year-follow-up period.157 (iv) The highest aldosterone quartile during admission was associated with advanced intracranial atherosclerosis and arterial calcification assessed by computed tomography angiography in acute ischaemic stroke patients.158 Finally, one study found no significant relationship between aldosterone levels and carotid plaques as measured by ultrasonography. However, the aldosterone–renin ratio was related to carotid plaques, suggesting a link with aldosterone dysregulation.159
4.4.3 Genetics
Polymorphic variants in various genes have been reported to be associated with aldosterone dysregulation. One of the polymorphic variants has been reported to be associated with MI, AF, and atherosclerosis—CYP11B2. CYP11B2 polymorphic variant, –344C/T, was related to the severity of coronary atherosclerosis in Chinese subjects undergoing elective coronary angiography.160 No confirmatory published study that met our criteria were identified.
4.4.4 Strength of evidence
Level C.
4.5 Stroke
4.5.1 Pre-clinical studies
Under normal conditions, MR expression is found in the hippocampus and cerebral cortex and rarely in the striatum.161–163 After a stroke secondary to middle cerebral artery occlusion, MR expression is markedly increased in the striatum, particularly in astrocytes.161 Furthermore, the astrocytes are found to play an important role in brain remodelling after ischaemia.
Dorrance et al.164 reported a higher expression of epidermal growth factor and EGFR mRNA in cerebral vessels and aorta obtained from stroke-prone spontaneous hypertensive rats (SHRSP) than controls. Spironolactone treatment decreased infarct size and EGFR expression. These data support the deleterious effect of aldosterone via EGFR expression, which leads to increase cell proliferation, vascular remodelling, and a larger infarct.164 MR activation also has an important role in stroke as myeloid MR-null mice with transient middle cerebral artery occlusion had suppressed proinflammatory gene expression in the ischaemic hemisphere, resulting in a significantly reduced infarct size.165
In rodents, treatment with either eplerenone or spironolactone reduces ischaemic size, ischaemic volume, and neurological effects without affecting BP.161–165 In mice, spironolactone suppressed ROS production, reduced apoptosis in the infarct area, and stimulated VEGF expression from astrocytes.161 In spironolactone-treated SHRSP, the middle cerebral artery lumen and myogenic tone were increased, which may explain the protective role of MRA in ischaemic stroke.166 However, some findings suggested biological sex differences in response to MRA. For example, (i) unlike the male, spironolactone treatment did not affect infarct size in female SHRSP with stroke.167(ii) Eplerenone also significantly reduced infarct size in males but not in the female mice.162
4.5.2 Clinical studies
4.5.2.1 RCTs/meta-analyses and association studies
No clinical trials relating an aldosterone effect to stroke met our criteria (see Supplementary material online). However, in a meta-analysis by Elbers et al.,168 hypertensive HF patients treated with MRAs had similar odds ratio of fatal and non-fatal stroke compared with controls.
Observational studies have shown a positive association between aldosterone levels and stroke in CAD patients.108,127 For example, (i) an increase in log-aldosterone levels measured at baseline was associated with acute ischaemic events (acute MI or ischaemic stroke) during a median follow-up time of 15 months.108 (ii) Log-aldosterone levels measured at baseline was associated with significant vascular events, including stroke and death from ischaemic stroke, during a median follow-up of 4.7 years.127
4.5.3 Genetics
Polymorphic variants in various genes have been reported to be associated with aldosterone dysregulation. Two polymorphic variants in two of these genes have been reported to be associated with stroke—CYP11B2 and serum and glucocorticoid-regulated kinase 1 gene (SGK1).
Two studies from China explored the association of CYP11B2 –344C/T polymorphism and stroke with mixed results. One study confirmed a positive association.132 Another study did not.169 However, a third study (a meta-analysis) supports the association of the –344C/T polymorphism and ischaemic stroke in East and South Asians but not Caucasians.170
A study in two independent age- and sex-matched case–control groups in Sweden reported a significant association between the SGK1 rs1057293 polymorphism and ischaemic stroke.171
4.5.4 Strength of evidence
Level C.
4.6 Thrombosis
4.6.1 Pre-clinical studies
Aldosterone exerts prothrombotic actions in rodents.172–175 It is associated with platelet activation, fibrin production, and the release of procoagulant and antifibrinolytic factors. Acute aldosterone administration in rats increased the first stage of thrombus formation, the density of fibrin net, platelet aggregations, and reduced fibrinolysis. These effects were observed within 10 min and were partially suppressed by eplerenone.173 In addition, aldosterone increased the incidence of venous thrombosis and PAI-1, NOX, and ROS levels, but tissue plasminogen activator decreased.42,174 In mice, acute aldosterone administration enhanced platelet accumulation at the site of endothelial injury induced by laser.173 After 90 days of aldosterone treatment in mice, there were enhanced platelet activation and reduced fibrinolysis in which spironolactone blocked these effects.175
Spironolactone and eplerenone treatment decreased thrombus formation in both arteries and veins in aldosterone-treated rodents.42,174,175 Furthermore, eplerenone reduced bleeding time, PAI-1 expression, and ROS level in rats given aldosterone infusion.174
4.6.2 Clinical studies
4.6.2.1 RCTs/meta-analyses and association studies
No clinical trials relating an aldosterone effect to thrombosis met our criteria. However, there was one meta-analysis and one association study reported. The meta-analysis showed that in hypertensive HF patients treated with or without an MRA, there were no statistically significant differences in acute limb ischaemia, pulmonary embolism, deep vein thrombosis, or other arterial or venous thromboses.168 However, there are several limitations to this meta-analysis. First, the report of thrombosis in the study review was <1%, suggesting there may be significant under-reporting.176 Second, most patients were on ACE inhibitors which could reduce PAI-1 levels,177 thereby obscuring differences between control and treatment arms. In an association study, log-aldosterone levels were positively associated with log-fibrinogen, log d-dimer, and log PAI-1 levels in hypertensive patients.178
4.6.3 Genetics
Polymorphic variants in various genes have been reported to be associated with aldosterone dysregulation.112 However, no genetic association study in thrombosis met our criteria (see Supplementary material online).
4.6.4 Strength of evidence
Level C.
4.6.4.1 Mineralocorticoid receptor antagonists
In the previous section of this review, we have noted the response of patients with primary CV diseases to MRAs. In this section, we summarize the currently available MRAs and their characteristics regarding potency, specificity for the MR, binding mode, and pharmacokinetics. To date, MRAs have been developed using a renal collecting duct screening mechanism.179 Thus, the potential of hyperkalaemia with their use is a common risk based on the duration of action, the presence of active metabolites and the potency of the MRA, and the degree of renal insufficiency of the patient.180,181 There are two classes of MRAs—steroidal-based and non-steroidal-based. Because the steroidal-based MRAs have been in clinical use for a longer period of time, they have been used in individuals with the six CV conditions reviewed in this report.83–85,123,124 However, no studies using non-steroidal-based MRAs have been reported in these conditions to date.
Steroidal-based MRAs: There are three drugs in this class—spironolactone, eplerenone, and potassium canrenoate.180,182–185 Potassium canrenoate is seldom used currently.186 The other two are effective in patients with CV conditions, as discussed earlier (see Sections 4.1 and 4.2). In addition to the potential of hyperkalaemia as a class side effect, these agents also have the potential of interacting with other steroid receptors. This is particularly common with spironolactone interaction with the androgen and progesterone receptors,182 resulting in impotency and gynaecomastia in males and reproductive dysfunction in females. These side effects may limit their usefulness in treating chronic diseases, especially in patients with chronic kidney disease.
Non-steroidal-based MRAs: These are newer class and more selective for the MR than the steroidal class. The available agents vary in their potency and pharmacokinetics, similar to that reported for steroidal-based MRAs.95,187–196 However, they still have the potential side effect of hyperkalaemia. To date, there are five members of this class—finerenone, esaxerenone, apararenone, AZD9977, and KBP-5074. Only finerenone and esaxerenone have reported Phase III clinical trial results.197–199 For both agents, the study populations were diabetic with kidney disease. Esaxerenone also studied hypertensives.200 Main comparisons between steroidal-based and non-steroidal-based MRAs are shown in Table 2.
Comparison between steroidal and non-steroidal MRAs
| Steroidal MRAs | Non-steroidal MRAs | ||||
|---|---|---|---|---|---|
| Spironolactone | Eplerenone | Potassium canrenoate | Finerenone | Esaxerenone | |
| Potency and selectivity for MR188,193 | Potent, non-selective | Less potent, more selective than spironolactone | Potent, non-selective | Potent, selective | Potent, selective |
| Binding mode 188,193 | Partial agonist | Partial agonist | Partial agonist | Bulky antagonist and inverse agonist | Full antagonist with large side-chain rearrangements |
| Tissue distribution (kidney/heart)a189,196 | Kidney > heart | Kidney > heart | NA | Equal distribution | Equal distribution |
| Pharmacokineticsb181,183,184,185,190,194 | Prodrug with multiple active metabolites with long half-lives (>12 h) | No active metabolites; half-life 4–6 h | Prodrug with multiple active metabolitesc with long half-lives (>12 h) | No active metabolites and short half-life (2–3 h) | No active metabolites and long half-life (>18 h) |
| Clinical trials | RALES83, TOPCAT,99 ALBATROSS,124 SPIRIT-HF (NCT04727073, ongoing clinical trials) | EPHESUS,84 EMPHASIS-HF,85 REMINDER123 | Di Pasquale et al.186 | FIDELIO-DKD,197 FIGARO-DKD,198 FINEARTS-HF, (NCT04435626, ongoing clinical trials) | ESAX-DN,199 ESAX-HTN200 |
| Steroidal MRAs | Non-steroidal MRAs | ||||
|---|---|---|---|---|---|
| Spironolactone | Eplerenone | Potassium canrenoate | Finerenone | Esaxerenone | |
| Potency and selectivity for MR188,193 | Potent, non-selective | Less potent, more selective than spironolactone | Potent, non-selective | Potent, selective | Potent, selective |
| Binding mode 188,193 | Partial agonist | Partial agonist | Partial agonist | Bulky antagonist and inverse agonist | Full antagonist with large side-chain rearrangements |
| Tissue distribution (kidney/heart)a189,196 | Kidney > heart | Kidney > heart | NA | Equal distribution | Equal distribution |
| Pharmacokineticsb181,183,184,185,190,194 | Prodrug with multiple active metabolites with long half-lives (>12 h) | No active metabolites; half-life 4–6 h | Prodrug with multiple active metabolitesc with long half-lives (>12 h) | No active metabolites and short half-life (2–3 h) | No active metabolites and long half-life (>18 h) |
| Clinical trials | RALES83, TOPCAT,99 ALBATROSS,124 SPIRIT-HF (NCT04727073, ongoing clinical trials) | EPHESUS,84 EMPHASIS-HF,85 REMINDER123 | Di Pasquale et al.186 | FIDELIO-DKD,197 FIGARO-DKD,198 FINEARTS-HF, (NCT04435626, ongoing clinical trials) | ESAX-DN,199 ESAX-HTN200 |
MRAs, mineralocorticoid receptor antagonists; NA, not available.
Evaluated in rats.
Half-life in healthy volunteers.
Canrenone is a common metabolite of spironolactone and potassium canrenoate.
Comparison between steroidal and non-steroidal MRAs
| Steroidal MRAs | Non-steroidal MRAs | ||||
|---|---|---|---|---|---|
| Spironolactone | Eplerenone | Potassium canrenoate | Finerenone | Esaxerenone | |
| Potency and selectivity for MR188,193 | Potent, non-selective | Less potent, more selective than spironolactone | Potent, non-selective | Potent, selective | Potent, selective |
| Binding mode 188,193 | Partial agonist | Partial agonist | Partial agonist | Bulky antagonist and inverse agonist | Full antagonist with large side-chain rearrangements |
| Tissue distribution (kidney/heart)a189,196 | Kidney > heart | Kidney > heart | NA | Equal distribution | Equal distribution |
| Pharmacokineticsb181,183,184,185,190,194 | Prodrug with multiple active metabolites with long half-lives (>12 h) | No active metabolites; half-life 4–6 h | Prodrug with multiple active metabolitesc with long half-lives (>12 h) | No active metabolites and short half-life (2–3 h) | No active metabolites and long half-life (>18 h) |
| Clinical trials | RALES83, TOPCAT,99 ALBATROSS,124 SPIRIT-HF (NCT04727073, ongoing clinical trials) | EPHESUS,84 EMPHASIS-HF,85 REMINDER123 | Di Pasquale et al.186 | FIDELIO-DKD,197 FIGARO-DKD,198 FINEARTS-HF, (NCT04435626, ongoing clinical trials) | ESAX-DN,199 ESAX-HTN200 |
| Steroidal MRAs | Non-steroidal MRAs | ||||
|---|---|---|---|---|---|
| Spironolactone | Eplerenone | Potassium canrenoate | Finerenone | Esaxerenone | |
| Potency and selectivity for MR188,193 | Potent, non-selective | Less potent, more selective than spironolactone | Potent, non-selective | Potent, selective | Potent, selective |
| Binding mode 188,193 | Partial agonist | Partial agonist | Partial agonist | Bulky antagonist and inverse agonist | Full antagonist with large side-chain rearrangements |
| Tissue distribution (kidney/heart)a189,196 | Kidney > heart | Kidney > heart | NA | Equal distribution | Equal distribution |
| Pharmacokineticsb181,183,184,185,190,194 | Prodrug with multiple active metabolites with long half-lives (>12 h) | No active metabolites; half-life 4–6 h | Prodrug with multiple active metabolitesc with long half-lives (>12 h) | No active metabolites and short half-life (2–3 h) | No active metabolites and long half-life (>18 h) |
| Clinical trials | RALES83, TOPCAT,99 ALBATROSS,124 SPIRIT-HF (NCT04727073, ongoing clinical trials) | EPHESUS,84 EMPHASIS-HF,85 REMINDER123 | Di Pasquale et al.186 | FIDELIO-DKD,197 FIGARO-DKD,198 FINEARTS-HF, (NCT04435626, ongoing clinical trials) | ESAX-DN,199 ESAX-HTN200 |
MRAs, mineralocorticoid receptor antagonists; NA, not available.
Evaluated in rats.
Half-life in healthy volunteers.
Canrenone is a common metabolite of spironolactone and potassium canrenoate.
5. Conclusion
Aldosterone binds to the MR which activates two pathways: genomic and non-genomic. Extensive evidence supports the concept that dysregulation of either aldosterone secretion and/or MR levels or activation contributes to CV pathologic states. Several recent reviews strongly support a prominent role for this dysregulation in arguably the largest CV condition—hypertension.9 This review assesses the likelihood of that association extending to six other CV conditions based on published data during the past decade. Data from both clinical, including RCTs and pre-clinical studies strongly support an association between HF and dysregulated aldosterone/MR. This association is not surprising given aldosterone/MR’s prominent effect in regulating sodium/volume homeostasis. AF and MI also associate with dysregulated aldosterone/MR. The pre-clinical data are particularly supportive. However, the clinical data are not as supportive, specifically because of limited data from RCTs. For the most part, the available clinical data are insufficient to determine whether or not a relationship exists between atherosclerosis, stroke, or thrombosis and aldosterone/MR dysregulation. Although the pre-clinical data are more supportive of these relationships, they are still limited.
Thus, this review clearly documented an expanding role for dysregulation in aldosterone/MR in CV diseases beyond hypertension. How expansive these relationships might be are limited by the currently available data. It is anticipated that with an increased focus on aldosterone/MR’s role in these other CV diseases and the development and utilization of non-steroidal-based MRAs, additional clinical and pre-clinical studies will further clarify these relationships, thereby opening approaches to use modulators of aldosterone/MR action to more precisely treat CV diseases.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contribution
W.W.P. and G.H.W. established the survey plan, independently screened the individual publications and then, by consensus, identified the publications to be assessed given our inclusion/exclusion criteria. W.W.P. and G.H.W. wrote the manuscript. G.H.W. created the concept of the manuscript and provided the final editing of the manuscript.
Acknowledgements
We thank the students, fellows, and faculty of the Cardiovascular Endocrinology Section, Division of Endocrinology, Diabetes, and Hypertension, Brigham and Women’s Hospital, for inspiring and encouraging us to complete this project. We appreciate the administrative and editorial assistance of Haris Lefteri.
Funding
This work was supported in part by the Division of Endocrinology, Diabetes and Hypertension, Brigham and Women’s Hospital, Boston, MA, USA, as well as by a fellowship to W.W.P. from King Chulalongkorn Memorial Hospital, Thai Red Cross Society and Faculty of Medicine, Chulalongkorn University (Bangkok, Thailand).
Data availability
No new data were generated or analysed in support of this research.
References
Author notes
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}