





Abstract
Cardiovascular (CV) stiffening represents a complex series of events evolving from pathological changes in individual cells of the vasculature and heart which leads to overt tissue fibrosis. While vascular stiffening occurs naturally with ageing it is accelerated in states of insulin (INS) resistance, such as obesity and type 2 diabetes. CV stiffening is clinically manifested as increased arterial pulse wave velocity and myocardial fibrosis-induced diastolic dysfunction. A key question that remains is how are these events mechanistically linked. In this regard, heightened activation of vascular mineralocorticoid receptors (MR) and hyperinsulinaemia occur in obesity and INS resistance states. Further, a downstream mediator of MR and INS receptor activation, the endothelial cell Na+ channel (EnNaC), has recently been identified as a key molecular determinant of endothelial dysfunction and CV fibrosis and stiffening. Increased activity of the EnNaC results in a number of negative consequences including stiffening of the cortical actin cytoskeleton in endothelial cells, impaired endothelial NO release, increased oxidative stress-meditated NO destruction, increased vascular permeability, and stimulation of an inflammatory environment. Such endothelial alterations impact vascular function and stiffening through regulation of vascular tone and stimulation of tissue remodelling including fibrosis. In the case of the heart, obesity and INS resistance are associated with coronary vascular endothelial stiffening and associated reductions in bioavailable NO leading to heart failure with preserved systolic function (HFpEF). After a brief discussion on mechanisms leading to vascular stiffness per se, this review then focuses on recent findings regarding the role of INS and aldosterone to enhance EnNaC activity and associated CV stiffness in obesity/INS resistance states. Finally, we discuss how coronary artery-mediated EnNaC activation may lead to cardiac fibrosis and HFpEF, a condition that is especially pronounced in obese and diabetic females.
1. Introduction
Increased arterial stiffness has been shown to be predictive of cardiovascular disease (CVD) events and all-cause mortality.1–3 Arterial stiffening is known to increase with age and is commonly associated with isolated systolic hypertension after the age of 50 years.4 The process is accelerated in insulin (INS) resistant states, such as obesity and type 2 diabetes typically occurs at earlier ages.5–7 Further, arterial stiffening is especially accelerated in obese and diabetic females who tend to lose the normal protection afforded by female sex hormones against vascular disease and show an increase in CVD events relative to lean and non-diabetic premenopausal women.6,8,9
The current standard for clinically assessing vascular stiffness is measurement of carotid-femoral pulse wave velocity (PWV).10,11 PWV increases with vascular stiffening and is a risk predictor of future CVD events.12 In addition to its predictive value, increased PWV/arterial stiffening has pathophysiological consequences including an earlier propagation of the reflected wave from the periphery arriving at the ascending aorta during systole, as opposed to diastole, with a resulting augmentation of aortic systolic pressure and pulse pressure and a decrease in diastolic coronary perfusion pressure.13 An additional consequence of arterial stiffening is exposure of the microcirculation to higher pressures which has been implicated in organ damage, particularly in the kidney and brain.14–16
1.1 Components of CV stiffness
Stiffness of the arterial wall is a complex parameter dependent on the material properties of the vascular wall and its radial geometry.17–19 In turn, the material properties of the wall are dependent on non-cellular components, such as extracellular matrix (ECM) and endothelial cells (ECs), and vascular smooth muscle cells (VSMCs), as well as interactions between these elements, such as occurs through ECM–integrin–VSMC linkages.19–22 The relative contributions of ECM components to stiffness can be affected by a number of processes including increased deposition and decreased turnover of collagen, as well as collagen cross-linking and elastin fragmentation. Further, medial calcification which tends to increase with ageing, obesity, and diabetes adds to the passive stiffness of arteries.23 While earlier studies have tended to focus on the passive elements of the vessel wall as determinants of increased arterial stiffness, for example as shown by a relative decrease in the ratio of elastin to collagen, more recent emphasis has been placed on the additional contributions of specific cells of the vascular wall.24,25 VSMCs have been implicated in affecting wall stiffness through changes in contractile tone and through changes in their inherent cellular stiffness.20,22 Thus, during contractile activation VSMCs both remodel their cortical actin cytoskeleton and their adhesion to ECM via focal adhesion sites.20 Age and diabetes-induced alterations to ECM proteins, such as results from advanced non-enzymatic glycation, may impact these matricellular interactions.26
More recently, attention has also turned towards the role of ECs in regulating stiffness of the arterial wall, with studies showing that ECs can impact vascular stiffness by a variety of mechanisms including through reduced nitric oxide (NO)-mediated alterations of vascular tone, changes in vascular permeability, secretion of vasoactive and inflammatory factors and the recruitment of monocytes and macrophages.7 Indeed, stiffening of ECs appears to be associated with cortical actin polymerization which leads to impaired ability to release NO which then contributes to a number of events leading to stiffening of the artery wall.7 These mechanical properties of the EC and associated reductions in bioavailable NO increase vascular stiffness independently of a direct physical contribution of endothelial stiffening to the overall material stiffness of the wall. An additional consideration is that stiffness of arterial vessels varies throughout the arterial tree due to differences in relative cellular content, extracellular matrix composition, and three-dimensional architecture.27,28
1.2 Myocardial stiffness
Similar to the arterial wall, cardiac tissue is subject to compressive, tensile, and shear forces and exhibits material properties, such as stiffness which are, in turn, related to their constituent cells (predominantly cardiomyocytes) and ECM. In regard to extracellular matrix proteins, deposition and cross-linking of collagen fibrils during fibrosis impacts myocardial stiffening, while intracellularly the phosphorylation state of the protein, titin, can directly impact cardiomyocyte stiffness. In states of impaired INS metabolic signalling, it is of interest that cardiomyocyte passive tension is increased by decreased phosphorylation of titin via a protein kinase B (Akt)/eNOS/NO-dependent mechanism.29 Thus, reduced coronary artery and cardiomyocyte release of NO and oxidative stress-mediated (including via reactive oxygen species and peroxynitrite) increases in its destruction can promote cardiomyocyte stiffening. To this point, increased cardiac tissue, and specifically increased cardiomyocyte stiffness is associated with heart failure with preserved systolic function (HFpEF) as determined by impaired diastolic relaxation.5,30–32
Recent studies support the concept that stiffness of the myocardium and consequent decreases in its ability to undergo normal diastolic relaxation can be impacted by stiffening of ECs of the coronary microcirculation which leads to impaired generation of NO.5,33,34 Thus, coronary endothelial abnormalities including decreases in NO production, increases in NO destruction, increases in coronary arterial immune cell adhesion, increased permeability, and immune cell transmigration and recruitment, generation of reactive oxygen species (ROS) and inflammatory factors creates a cardiac environment that is profibrotic (e.g. leading to increased collagen deposition) and promotes cardiac stiffening, thereby impairing diastolic relaxation.5 Further, these vascular mechanisms likely interact with additional factors, such as cardiomyocyte-dependent mechanisms for NO production (mediated by several NOS isoforms) and the mechanical state of the cardiac tissue.35–37 This provides a mechanistic framework for the development of HFpEF, an entity that is increasing in our ageing population and individuals with obesity and type 2 diabetes, especially in females.
This review highlights the contribution of ECs to pathological increases in vascular stiffness particularly as relates to CV dysfunction associated with INS resistance as exists in obesity and type 2 diabetes. For more general reviews on vascular stiffness and the roles played by the ECM and VSMCs, the reader is referred to a number of comprehensive recent publications.17–19 More specifically, we summarize recent studies demonstrating key roles for signalling through activated EC mineralocorticoid receptors (ECMRs) and EC INS receptors (IRs) and their target the endothelial Na+ channel (EnNAC) as determinants of vascular (Figures 1 and 2) and cardiac stiffness and dysfunction. In addition, important interactions, and points of convergence, between MR and IR signalling are highlighted due to their utilization of common signalling molecules (e.g. mTOR2/SGK-1) for EnNaC activation and the fact that states of heightened MR activation are associated with impaired INS metabolic signalling.38 Importantly, INS resistance is known to be clinically associated with increased arterial stiffness.39 These shared signalling mechanisms are highlighted through published studies and specifically for INS, additional preliminary data of EnNaC activation in ECs (Figure 2). These contemporary studies in ECs extend work over the past several decades demonstrating that INS and mineralocorticoids increase ENaC in renal distal tubule cells. Finally, we provide a conceptual framework for how altered endothelial function and stiffness, in the context of both enhanced ECMR and impaired INS metabolic signalling/hyperinsulinaemia, leads to coronary artery stiffening, cardiac fibrosis, and diastolic dysfunction (Figures 3 and 4).
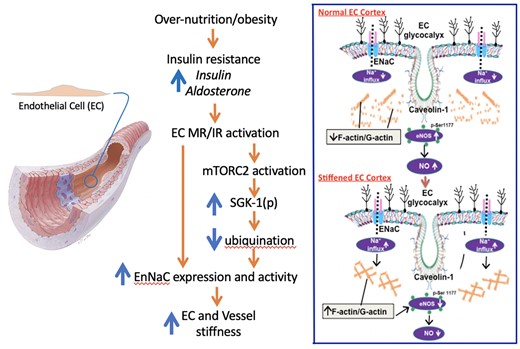
Schematic diagram illustrating an overview of the hypothesized pathway by which western diet/INS resistance together with enhanced ALDO and INS-mediated signalling promotes abnormally increased vascular stiffness (left panel). Both INS receptor (IR) and MR activation are proposed to mediate stiffening through mTOR and SGK-1-dependent mechanisms that lead to both increased Na+ channel (EnNaC) expression and activity. As described in the text, EnNaC activity is increased through decreased ubiquination and internalization. The right-hand panel contrasts at the EC level the ‘normal’ EC cortical flexibility (upper) to the situation where the cell cortex is ‘stiffened’ leading to impaired eNOS activity and decreased NO bioavailability (lower panel). Specifically, enhanced Na+ entry occurs as a result of enhanced EnNaC activity. Increased Na+ influx promotes polymerization of the cortical actin cytoskeleton leading to decreased eNOS activation and diminished production of NO. As further described in the text decreased NO bioavailability leads to increased vascular fibrosis and stiffening.
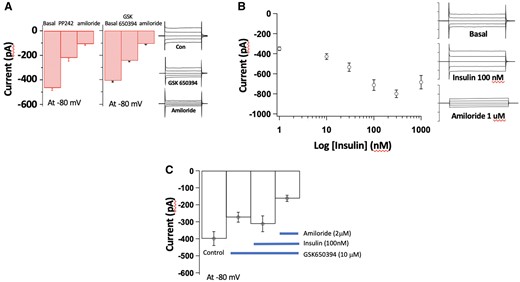
Consistent with data from epithelial cells of the distal renal tubule40–44 ECs exhibit amiloride-sensitive Na+ currents that are modulated by mTORC2, SGK-1, and INS signalling. To illustrate this, preliminary studies have been undertaken in mouse pulmonary ECs isolated by a dual antibody-linked magnetic bead purification method described in our recent publications.7,30,45,46 Electrophysiological recordings were conducted using conventional whole cell patch clamp. Protocols were designed to examine elements of the signalling pathways outlined in Figure 1. (A) mTORC2 (PP242 10 μM; n = 10–14 cells) and SGK-1 (GSK 20 μM; n = 12–17 cells) inhibitors acutely attenuate amiloride-sensitive Na+ currents (EnNaC-mediated) in vascular ECs. Example patch clamp tracings are shown to the right of the panel. (B) INS acutely increases amiloride-sensitive Na+ currents in a concentration-dependent manner over the range 10–300nM (n = 4–11 cells). Example patch clamp tracings are shown to the right of the panel. (C) In vascular ECs, INS (100 nM)-induced increases in Na+ current are inhibited by pre-treatment with the SGK-1 inhibitor, GSK650394 (10 µM) (n = 6–8 cells). Results are shown as mean ± SEM.
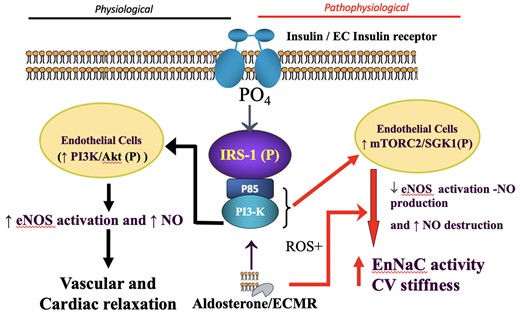
Schematic diagram proposing that INS and ALDO act both independently and additively through their respective receptors. Under physiological levels (left-hand side) of these hormones appropriate vascular and cardiac relaxation is favoured, via NO-dependent mechanisms. In contrast, when excessive levels of INS and ALDO/MR activation (right-hand side) result from nutrient excess/obesity (Figure 1), this relationship shifts to promote cardiovascular stiffening and dysfunction by pathways involving increased activity of the endothelial Na+ channel, EnNaC, and decreased NO bioavailability (see text). In excess, INS and ALDO synergize to promote mTORC2/SGK1 activation of EnNaC and consequent CV fibrosis, stiffening and impaired function. Akt, Protein kinase B; CV, cardiovascular; EC MR, endothelial cell mineralocorticoid receptor; EnNaC, endothelial cell epithelial Na+ channel; eNOS, endothelial nitric oxide synthase; IRS-1, insulin receptor substrate 1; mTORC2, rapamycin-insensitive mTOR protein complex; NO, nitric oxide; PI3K, phosphoinositide 3 kinase; ROS, reactive oxygen species; SGK1, serum and glucocorticoid regulated kinase.
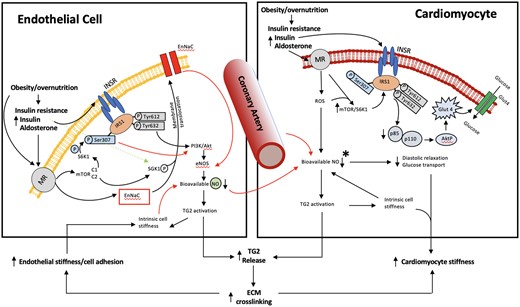
Schematic diagram illustrating cellular signalling mechanisms underlying MR-dependent cardiovascular stiffening and dysfunction in over nutrition, obesity, and insulin resistance. The model is based on both experimental data and the authors’ interpretation of the existing literature. Emphasis is placed on altered MR/INS signalling both independently and synergistically impacting tissue mechanical properties (fibrosis/stiffening) and function. The left-hand side illustrates MR/INS-mediated events occurring in endothelial cells and the right-hand side those occurring in cardiomyocytes. Collectively, coronary arterial endothelial cell and cardiomyocyte stiffening interact to impair cardiac relaxation which is manifested as diastolic dysfunction contributing to the clinical syndrome of HFpEF. HFpEF is thus viewed to result from several mechanisms which result from decreased NO bioavailability which manifest as increased afterload (vascular stiffness), altered myocardial structure (fibrosis), and impaired energetics (impaired insulin signalling). Black arrows indicate stimulatory pathways while red arrows depict inhibition. *Not shown are possible contributions of other cell types (e.g. cardiomyocytes per se) to NO availability or detail of downstream mechanisms involving ROS, peroxynitrate and soluble guanylate cyclase which have been implicated in HFpEF.
2. Mineralocorticoid receptors
Data from randomized controlled clinical trials strongly implicate elevated MR activation in increased risk for CVD and have shown that MR antagonists (MRAs; spironolactone and eplerenone) reduce CVD events.47 Thus, the RALES, EPHESUS, and EMPHASIS trials reported decreased morbidity and mortality in patients with heart failure treated with MRAs.48–51 Consistent with these clinical and recent basic research observations, MRAs also ameliorate cardiac diastolic dysfunction in obese INS resistant patients.43,52 As MRAs have also been shown to decrease risk for CVD events in chronic dialysis patients it can be surmised that their effects may, at least in part, be independent of renal mechanisms for Na+ and potassium balance54 and by direct effects on the vasculature and heart.
2.1 Vascular mineralocorticoid receptors
While MRs have classically been extensively characterized for their role in the renal aldosterone (ALDO)-sensitive distal tubular epithelium,55 studies over the last decade have demonstrated their existence and importance in ECs56,57 and VSMC.58,59 Activation of MRs at the level of the vasculature is now recognized to play a major role in the development of CV and renal diseases60,61 although precise roles played under physiological conditions are currently less clear.62 For example, the VSMC MR has been implicated in the age-dependent increases in arterial pressure and vascular fibrosis, in part, resulting from altered expression of voltage-gated Ca2+ channels.58,63 Recently, ECMR activation has been shown to negatively impact NO bioavailability while enhancing inflammatory responses and oxidative stress, particularly in the presence of known CVD risk factors, such as diet-induced obesity and associated INS resistance.58,64 In addition, MRs have now been identified in other cells associated with the vascular wall including macrophages,65,66 adipocytes,67 and fibroblasts.68 Activation of the MR in ECs and VSMCs occurs predominantly in response to ALDO, and not by glucocorticoids as both cell types express the enzyme 11-beta-hydroxysteroid dehydrogenase 2 (11β-HSD2) which converts glucocorticoids into inactive metabolites.69–71 Conceivably, this may not be the case in states of heightened glucocorticoid production, oxidative stress, or where 11β-HSD2 activity is diminished. To this point, evidence exists for MR activation to occur independently of ALDO including at the level of MR gene transcription and through Rac1 signalling mechanisms.72,73 Interestingly, these latter mechanisms may contribute to EC and VSMC MR activation-mediated vascular fibrosis and stiffening.74
2.2 Mineralocorticoid receptor activation and INS resistance/hyperinsulinaemia
Excessive MR activation, as occurs in states of INS resistance and associated hyperinsulinaemia, induces inflammation and associated CV abnormalities.30,61,64 These abnormalities may be mediated, in part, by the increased plasma ALDO levels in persons with obesity and INS resistance.75,76 Supporting this notion clinical trials have shown that MRAs ameliorate diastolic dysfunction in obese patients.52,53 Recent experimental studies indicate that specific activation of ECMRs initiates events which underlie vascular and cardiac fibrosis and associated increases in CV stiffness.7,30,61,64 Thus, in mice fed a diet high in refined carbohydrates and saturated fat (western diet), inhibition of MRs with a very low dose of spironolactone, not affecting blood pressure or Na+ excretion, or specific genetic deletion of the ECMR prevented increased vascular stiffness and cardiac fibrosis.77 These effects of ECMR activation were attributed, in part, to reduced EC-derived NO and resultant promotion of monocyte/macrophage recruitment. Importantly, these effects were particularly evident in obese INS resistant females.77
An additional consequence of decreased NO bioavailability in diet-induced MR activation involves activation of the endothelial enzyme transglutaminase 2 (TG2).64,78 TG2 is a protein cross-linking enzyme that exerts both intracellular and extracellular actions contributing to cell stiffening, vascular remodelling, and CV tissue fibrosis.64,79–81 In studies of western diet-induced INS resistance, female mice displayed increased vascular stiffness, attenuated endothelial NO synthase (eNOS) activation, heightened inflammatory responses, increased expression of TG2, and tissue remodelling/fibrosis.7,30,64 These events were significantly attenuated in mice with ECMR depletion demonstrating a linkage to diet-induced ECMR activation in concert with INS resistance.75,76 Further, INS- and ALDO-mediated enhancement of EnNaC82 decreases NO production leading to TG2 activation and CV stiffening associated with obesity and accompanying INS resistance (Figure 4).64,78
As shown in Figures 1 and 3, available data suggest a model whereby INS and ALDO act both independently and additively through their respective EC receptors. At physiological hormone levels appropriate vascular and cardiac relaxation is mediated via NO-dependent mechanisms. In contrast, at excessive levels of INS and ALDO under conditions of nutrient excess/obesity and INS resistance, activation of their EC receptors shifts this relationship to promote CV stiffening and dysfunction through increased activity of an endothelial Na+ channel and decreased NO bioavailability (Figure 3) as described in the following sections.
3. Role of the endothelium Na+ channel (EnNaC) in CV fibrosis and stiffening
A well-recognized target of MR activation in the renal distal nephron tubules of the kidney is the amiloride-sensitive epithelial Na+ channel (ENaC).40–44 Activated MR binds the ENaC promoter element in epithelial cells and induces expression of the ion channel and its insertion into the plasma membrane. In addition to ALDO-mediated MR activation, angiotensin II exerts an effect on ENaC open probability (Po) through NADPH-mediated production of reactive oxygen species.83 Functionally, ENaC in the distal nephron then acts to fine tune the reabsorption of filtered Na+ and regulate extracellular fluid volume. ENaC has also been extensively studied in epithelial cells of the respiratory and gastrointestinal tracts initially and various glands, where the channel similarly regulates transepithelial cell Na+ movement.84–86 More recently the existence of ENaC has been demonstrated in the vasculature.42,87–89 Vascular ENaC have been implicated as a mechano-sensor, detecting changes in intraluminal pressure by VSMCs88,90,91 and shear stress by ECs.87,92,93 The mechano-sensitivity of ENaC extends beyond the vasculature and is evident in renal tubules where tubular flow rate affects Na+ and K+ reabsorption.94,95 Shear stress, through an effect on the extracellular domains of the Na+ channel proteins modulates open probability of the channel.96 Endothelial ENaC, termed EnNaC by Warnock et al.,97 has been linked to regulation of NO synthase activity and NO bioavailability7,30,64,98–100 possibly consistent with more generalized effects on vasomotor function. More recently, ENaC has been localized to perivascular immune cells (dendritic cells) providing another potential mechanism by which this channel could conceivably impact vascular function and stiffness.101 ENaC is also mechanically activated in nerve terminals baroreceptors providing a further mechanism by which it may impact the vasculature.102,103
ENaC is member of the ENaC/degenerin family of cation-selective channels with the functional trimeric channel generally being composed of three subunits termed α, β, and γ.104–106 In some species and tissues, an additional δ subunit has been cloned and characterized.107 Po of the channel is determined by subunit composition with the α-subunit being typically considered as the functional core of the channel.108 Additional conformations of the channel have been suggested to include δ,β,γ and β,γ in association with ASIC proteins (additional members of the degenerin family).88,109,110 The existence and functional significance of these alternate conformations in the vasculature are currently uncertain. The heteromeric channel is characterized by each subunit having two transmembrane domains and a large extracellular loop that regulates channel gating to a number of stimuli including proteases, extracellular Na+ and, as mentioned above, shear stress.40,92–94,96,110–112 In contrast to the large extracellular domain, ENaC has a relatively small intracellular domain with the C- and N-termini located intracellularly.40 The intracellular domains contain regulatory sites including for palmitoylation and phospholipids.40,113,114 The intracellular domain also binds the cortical actin cytoskeleton, or actin-binding proteins, such as filamin, likely contributing to the channel’s mechanosensory properties.115–117 Mechano-sensory properties of vascular ENaC may also be affected through colocalization with integrins and interactions with extracellular matrix proteins.118 Conceivably, the flow-sensitive properties of EnNaC are also affected by interactions between the Na+ channel activation and the glycocalyx disruption and shedding.92,119,120 (Figure 1). A process implicated in the regulation and effects of other ion channels.121 In addition to these mechano-sensory regulatory modes, vascular ENaC activity is modulated through a number of hormonal mechanisms (including ALDO, angiotensin II, and INS) and paracrine factors (including NO and ATP).40,122–124
3.1 Increased EnNaC activity leads to EC cell stiffening, decreased NO bioavailability, and impaired vasodilator function
Studies from Oberleithner’s group in cultured human umbilical vein ECs demonstrated that ALDO promoted increased cell surface expression of the αEnNaC subunit as measured by quantum dot immunofluorescence and western blotting.125 The ALDO-mediated effect was prevented by spironolactone consistent with a role for MR activation.125 Atomic force microscopy (AFM) further showed that ALDO increased cell surface area, an assumed consequence of Na+ entry via ENaC as the effect was blocked by amiloride.125 This group further demonstrated that ALDO mediated Na+ entry led to stiffening of the EC cortical actin cytoskeleton as measured by indentation AFM which, in turn, impaired NO production through inhibition of eNOS activation. In addition, it was argued that the under physiological conditions the negative charge-rich cell surface proteoglycan layer, the glycocalyx (Figure 1), acts to limit Na+ access to EnNaC while under certain pathological conditions this protective barrier is disrupted favouring enhanced EnNaC activity.126 Collectively, under conditions of enhanced EnNaC activity this series of events was proposed to favour development of increased smooth muscle tone and a vasoconstricted state in an intact artery.
These cell-based studies provided, in part, impetus for in vivo mouse studies of MR activation, specifically via chronic infusion of ALDO (via osmotic mini pumps) or feeding (20 weeks) of an obesogenic western diet (high in refined carbohydrates and saturated fat) which is associated with increased INS resistance, elevated plasma ALDO and INS levels and increased CV MR expression.64 Mice fed the western diet showed increased endothelial and aortic stiffness, impaired endothelial-dependent vasorelaxation, aortic fibrosis, aortic oxidative stress, and increased vascular expression of EnNaC.127 Treatment of the mice with a low dose of amiloride (1 mg/kg/day, administered in the drinking water) prevented or significantly attenuated these diet-induced abnormalities.127 Further, in vivo evidence for a role for EnNaC was provided by studies of the global ENaC gain-of-function Liddle mice which showed increased stiffening of the cortical actin cytoskeleton in aortic ECs.128
To gain further insight into the vascular role played by EnNaC, we have characterized a mouse model with endothelial cell-specific deletion of the α, pore-forming, subunit of ENaC.7,64,87 As the deletion strategy involved the use of Cre recombinase expression by the Tie2 promoter studies in macrophages were performed to confirm that αEnNaC was not affected in haematopoetic cells. Further, blood pressure and renal Na+ handling were unaffected. Consistent with a role for EnNaC in sensing shear stress these mice were shown to have impaired flow-dependent dilation in small arteries of the intestinal mesentery.87 The lack of an effect of the channel protein deletion on systemic blood pressure under physiological conditions may reflect compensatory mechanisms due to the multiple systems capable of altering blood pressure. In contrast, we have observed that the ENaC deletion does reduce the increase in blood pressure that occurs in response to MR activation by subcutaneous deoxycorticosterone acetate (DOCA) administration and consumption of 1% NaCl (unpublished data). Interestingly, this has parallels with the ECMR model where a marked phentotype is not evident under basal conditions but can be unmasked by a stressor, such as diet-induced obesity or aldosterone infusion.7,64 Further demonstrating a role in pathophysiological states, it has been shown that in renal ischaemia/perfusion injury, a situation characterized by decreased NO bioavailability, deletion of αENaC prevents renal tubular injury, and renal dysfunction.129
As mentioned earlier and schematically illustrated in Figure 1, obesity and INS resistance in both human subjects and experimental dietary mouse models leads to increased ALDO and INS levels and increased vascular MR expression,130,131 resulting in increased EnNaC protein expression and its translocation to the cell membrane, an effect inhibited by MR antagonists in cultured umbilical vein ECs.98,132 To more directly demonstrate that enhanced EnNaC activation underlies increases in EC stiffness and decreased NO mediated vascular relaxation western diet feeding studies were conducted in αEnNaC−/− mouse models and their littermate controls.7,45 Sixteen weeks of the western diet resulted in increased EnNaC activation as shown by increased Na+ currents measured in isolated ECs.7,45 Increased endothelial stiffness was demonstrated in segments of aorta by an en face AFM indentation protocol and this increase in endothelial stiffness was associated with reduced bioavailable NO and impaired aortic ring endothelium-dependent relaxation.7,45 These western diet-induced abnormalities, along with vascular remodelling and fibrosis, were all significantly attenuated in the αEnNaC−/− mouse. From a mechanistic standpoint, these studies showed that western diet feeding resulted in a heightened inflammatory response that was associated with reduced eNOS phosphorylation/activation and NO production and bioavailability.7 These latter events likely emanated from increased EnNaC activity leading to polymerization of cortical actin fibres, subsequently reducing eNOS activity, and decreasing NO production leading to increased vascular stiffness7 (Figure 1).
3.2 EnNaC activation contributes to abnormal cardiac stiffening and diastolic dysfunction
Emerging evidence also suggests that coronary artery EnNaC activation plays a vital role in the pathogenesis of cardiac fibrosis leading to cardiac diastolic dysfunction46 providing a potential mechanistic link between coronary microvascular abnormalities and HFpEF as mentioned above in the context of ECMR activation5,30,61 (Figure 4). To explore this relationship, female mice were fed a western diet (16 weeks) with or without oral administration of a low dose of the EnNaC antagonist, amiloride (1 mg/kg/day).46 These studies centred on females as earlier work indicated that females fed a western diet exhibit earlier development of INS resistance, vascular stiffness, and cardiac fibrosis and diastolic dysfunction than in males.133–135 Amiloride significantly reduced diet-induced impairment of left ventricular initial filling rate and relaxation time as measured by magnetic resonance imaging.46 Impaired cardiac diastolic relaxation was associated with increases in coronary endothelium remodelling and permeability that paralleled diet-induced increases in F-actin and fibronectin, decreased expression of the tight junction molecules claudin-5 and occludin, and increased macrophage recruitment, M1 polarization, cardiac oxidative stress, fibrosis, and maladaptive remodelling.46 These data have been interpreted as supporting the hypothesis that coronary EnNaC activation leads to reduced eNOS activation and increased ROS-mediated destruction of NO, increased coronary endothelial monocyte adhesion, and endothelium permeability, thereby promoting macrophage infiltration and inflammation. Collectively, these events ultimately resulted in cardiac fibrosis and diastolic dysfunction in the MR-activated state of diet induced obesity and INS resistance. To provide further support for the proposed link between obesity-induced ECMR/ENaC signalling and cardiac dysfunction additional experiments were recently conducted using the genetic αEnNaC deletion model.30 Obesity-induced impairment in diastolic relaxation was attenuated in αEnNaC deletion mice which displayed functional impairment of EnNaC, confirmed by decreased Na+ currents in ECs.30 In addition, αEnNaC deletion decreased myocardial oxidative stress and diet-induced increases in stiffness of isolated cardiomyocytes.30 Collectively these studies suggest that ECMR activation in coronary vessels likely has a marked impact on initiating cardiomyocyte stiffness, cardiac fibrosis, and diastolic dysfunction. Further, these adverse effects of coronary ECMR activation likely occur via recently described interactions involving increased coronary EnNaC activity and decreased eNOS activation in coronary ECs.30 Future in vivo and in vitro studies will be required to confirm elements of this hypothesis by evaluating coronary flow reserve and conducting electrophysiological and AFM studies on coronary microvascular ECs to fully confirm the role of coronary EnNaC activation in the pathogenesis of cardiac fibrosis and diastolic dysfunction in states of INS resistance and accompanying hyperinsulinaemia with elevated plasma ALDO levels (Figure 4).
4. ALDO and INS interact to regulate EnNaC via mTORC2/SGK-1 signalling
As outlined above the epithelial cell literature has highlighted the complexity of ENaC regulation. In addition to post-translational modifications including protease activation, trafficking of subunits to the cell membrane leads to activation. Conversely, the channel is inactivated by, hormone-dependent, ENaC internalization, and ubiquitination/destruction pathways which act to regulate membrane levels of the functional channel.40 Central to this, considerable interest has developed regarding the role on mTOR signalling in in ENaC regulation (Figure 3). mTORC1 and C2 are multiprotein complexes that are involved in the regulation of multiple biological processes including metabolism, ion transport, and cellular growth/proliferation. For example, TORC1 is activated by MR activation and ROS to phosphorylate S6K1 which in turn phosphorylates IRS-1 on ser307 and other serine residues to inhibit IRS-1 tyrosine phosphorylation and downstream INS metabolic signalling (INS resistance) and NO production.5,34 This, in turn, leads to hyperinsulinaemia which likely promotes CV fibrosis and stiffness and decreased cardiac glucose utilization38 (Figures 3 and 4).
Accumulating information supports the idea that mTORC2 and downstream serum and glucocorticoid regulated-kinase (SGK1) signalling constitutes a convergence point linking obesity, metabolic alterations, vascular stiffening, and hypertension. Studies conducted largely in renal tubular cells indicate that activation of mTORC2 causes phosphorylation of SGK-1 decreasing the ubiquitin ligase, Nedd 4-2, mediated unbiquitination and increasing plasma membrane localization and activation of ENaC.136–138 Additional studies have also suggested that SGK-1 mediates direct stimulatory effects of ALDO on ENaC production and membrane localization, although the exact mechanisms are uncertain.139,140 Recent studies have shown that ECMR activation following consumption of a western diet stimulates mTORC2 and SGK1 signalling pathways in ECs known to contribute to membrane localization and activation of EnNaC.64 Further, in preliminary unpublished studies to confirm the relevance of these pathways in vascular cells, we have found that mTORC2 and SGK1 inhibition attenuates amiloride-sensitive Na+ currents in isolated ECs (Figure 2). Also, consistent with this, in vitro treatment of ECs with ALDO and INS leads to mTORC2 activation and increased expression of the alpha subunit of EnNaC. These observations are consistent with studies conducted with renal tubule cells showing that INS also activates ENaC via the mTOR and SGK1 signalling pathway.123,124,141–143 Additionally, INS and ALDO have additive effects on activation of ENaC in these renal derived cells. Consistent with this, we have recently demonstrated that INS exerts EnNaC stimulatory effects in isolated ECs that is related to activation of the mTOR2/SGK1 signalling pathway (Figure 2). In addition, ECs cultured in the presence of ALDO (10 nM) for 48 h show enhanced Na+ current in response to acutely applied INS (100 nM) suggesting an additive action of these signalling pathways on ENaC activity (data not shown). While further studies are required to establish concentration-response and temporal relationships between these hormones, these preliminary observations in vascular ECs provide insight into integrative mechanisms by which INS resistance/hyperinsulinaemia, elevated ALDO and associated increases in ECMR and EC IR signalling activate EnNaC as an instigator for increased CV stiffness in INS resistant states, such as obesity and type 2 diabetes mellitus (Figures 3 and 4).
5. Conclusion
In conclusion, recent studies have uncovered an important role for the vascular Na+ channel activation in mediating normal and pathological CV structure and function. This research has revealed that activation of the endothelial Na+ channel by ALDO and INS leads to endothelial cortical stiffening, impaired NO production, and subsequent vascular fibrosis and stiffening. Additional recent observations also suggest that EnNaC activation in the coronary vasculature promotes myocardial fibrosis, myocardial stiffening, and impaired diastolic relaxation. Amplifying the effects mediated by coronary artery dysfunction, systemic artery stiffening (afterload) associated with EC IR and MR activation likely further contributes to cardiac stiffening/remodelling. This multilevel scenario (Figure 4) has potential translational relevance in the pathogenesis of HFpEF. To this point, diet-induced obesity is associated with increases in plasma ALDO and INS levels, and increased ECMR expression which leads to EnNaC activation and CV stiffness, especially in overweight females. These observations may explain why obese women and those with type 2 diabetes lose their normal premenopausal CVD protection and are especially prone to develop HFpEF. Finally, targeting EnNaC signalling presents a promising strategy for prevention and treatment of vascular stiffening and HFpEF.
Acknowledgements
The authors would like to acknowledge the important contributions of members of their laboratories to studies underling this review, including Guanghong Jia, PhD, Annayya Aroor, MD, PhD, Yan Yang, MD, Luke Sun, PhD, Benjamin Bonnard, PhD, and Liping Zhang, MS.
Conflict of interest: none declared.
Funding
Work of the authors described in this manuscript was supported by funding from Heart, Lung and Blood Institute, National Institutes of Health, USA for M.A.H. and J.R.S. and the Fight-HF Avenir Investment Program (ANR-15-RHUS-0004) and the Fondation de France (Cardio 00086498) for F.J.

{kind=link}
{kind=link}
{kind=link}
{kind=link}