





Abstract
Intracellular myeloperoxidase (MPO) plays a specific role in the innate immune response; however, upon release into the extracellular space in the setting of inflammation, drives oxidative tissue injury. Extracellular MPO has recently been shown to be abundant in unstable atheroma and causally linked to plaque destabilization, erosion, and rupture, identifying it as a potential target for the surveillance and treatment of vulnerable atherosclerosis. Through the compartmentalization of MPO’s protective and deleterious effects, extracellular MPO can be selectively detected using non-invasive molecular imaging and targeted by burgeoning pharmacotherapies. Given its causal relationship to plaque destabilization coupled with an ability to preserve its beneficial properties, MPO is potentially a superior translational inflammatory target compared with other immunomodulatory therapies and imaging biomarkers utilized to date. This review explores the role of MPO in plaque destabilization and provides insights into how it can be harnessed in the management of patients with vulnerable atherosclerotic plaque.
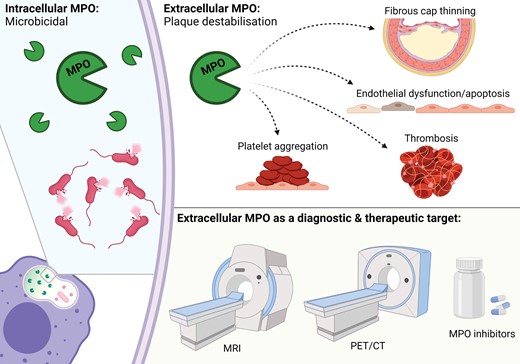
The advantage of MPO to other inflammatory mediators linked to vulnerable, inflammatory atherosclerosis is in the ability of therapeutic and diagnostic approaches to selectively target the damaging extracellular activity while preserving its beneficial cellular effects.
1. Introduction
The destabilization of atherosclerotic plaque and resultant cardiovascular events remain the leading cause of morbidity and mortality worldwide,1 with acute myocardial infarction (AMI) and stroke the most common and devastating pathological consequences. Atherosclerosis is a progressive disease of large arteries characterized by the accumulation of cholesterol and inflammatory cells. The disease typically culminates in plaque destabilization through rupture, fissuring, or endothelial cell erosion, and can result in thrombosis, embolization, and abrupt arterial occlusion leading to precipitous ischaemia or infarction.2 Such acute events often occur as the first disease manifestation in otherwise asymptomatic individuals, predominantly because culprit lesions are frequently not flow-limiting prior to an indexed event and symptoms of ischaemia do not manifest until significant stenoses ensue.3 Rather than being necessarily stenotic, thrombosis-prone plaques can be broadly divided into two histological phenotypes: appearing either as destabilized thin-capped fibroatheromas (TCFA) or having undergone endothelial cell erosion. Destabilized TCFA can be enriched with inflammatory cells, are lipid-laden and have thin fibrous caps leaving them susceptible to rupture due to reduced tensile strength, whereas erosive plaques undergo endothelial cell erosion resulting in metabolic and immune disturbances that promote a localized prothrombotic state.4 Plaque rupture accounts for 60–70% of fatal AMI,5 with endothelial erosion the culprit process in the remaining third of cases and found in ∼25% of patients presenting with ST-elevation myocardial infarction who undergo in vivo optical coherence tomography.6
Despite evidence that plaque structure and inflammatory activity are the most accurate determinants of acute events,2 escalation of therapy, surveillance and intervention is often determined by the degree of luminal stenosis—a parameter that poorly predicts future cardiovascular disease (CVD) events.7 This predilection toward stenotic grade has largely persisted due to an inability to determine the composition and pathological activity of atheroma reliably and pragmatically. Presently, there are no widely utilized non-invasive imaging modalities or specific therapies to assess and treat vulnerable atherosclerosis, distinguishing this area of cardiovascular medicine as an important unmet need.
This review focuses on the role of extracellular myeloperoxidase (MPO) in plaque destabilization, as well as its emerging clinical potential in the detection, monitoring, and treatment of vulnerable atherosclerotic plaque.
2. Myeloperoxidase and its role in plaque destabilization
MPO is a haem-containing enzyme stored in azurophilic granules of neutrophils, monocytes, and certain tissue macrophages that performs a unique and specific role within the innate immune system. Following phagocytosis of a microbe as part of an immune response, granular MPO fuses with and is released into the phagolysosome where it serves the primary purpose of oxidizing chloride to hypochlorous acid (HOCl, or bleach) through activation of hydrogen peroxide. HOCl is highly reactive and contributes to the microbicidal activity of phagocytes (Figure 1). Separate to this role of phagolysosomal MPO, up to 30% of cellular MPO can be released into the extracellular space by degranulation and apoptosis, migration from the intravascular space or by association with neutrophil extracellular traps (NETs).8 Once in the extracellular compartment, MPO can cause oxidative tissue injury which contributes to plaque destabilization (Figure 2)9 as well as other cardiac and inflammatory disorders.10

Intracellular MPO. Membrane-bound NADPH oxidase (NOX2) generates superoxide radical anion (O2•–) from molecular oxygen (O2). Superoxide dismutase (SOD) then converts O2•– to H2O2 which reacts with MPO to oxidize chloride (Cl–) to hypochlorous acid (HOCl). HOCl contributes to microbicidal activity intracellularly or oxidative tissue injury extracellularly.
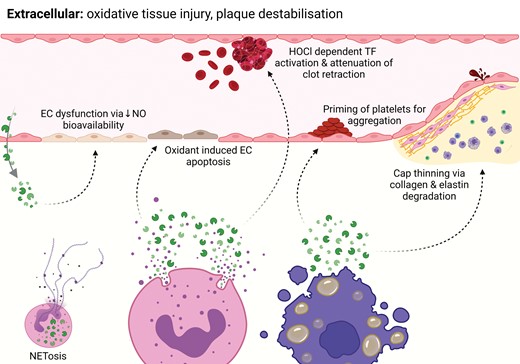
Extracellular MPO. MPO is released into the extracellular space via neutrophil degranulation, migration from the intravascular space and granulocyte apoptosis. This liberation of MPO is responsible for plaque destabilization through various specific mechanisms. In addition, MPO plays a causal role in the formation of NETs, which are implicated in plaque destabilization.
Like other inflammatory mediators linked to atherosclerotic plaque destabilization, it is within this dichotomy of immune protection and deleterious host effects that MPO treads a line between necessity and antipathy. Unlike other inflammatory mediators, however, MPO offers a potential clinical advantage through the compartmentalization of its protective intracellular activity and damaging extracellular effects, whereby extracellular MPO can become a distinct target for diagnostic and therapeutic purposes (Graphical Abstract).
The involvement of MPO in human atherosclerosis is well established with evidence of its association dating back to the 1990s. Enzymatically active MPO11 and HOCl-modified proteins12 are present in atheromatous lesions. More recently, however, the focus on the role of MPO and MPO-derived oxidants has shifted from atherogenesis to the key determinants of plaque vulnerability including plaque destabilization, rupture, and erosion.
2.1 MPO in plaque rupture
The thickness of a plaque’s fibrous cap is the main determinant of its stability and likelihood of rupture. In a mouse model of plaque instability characterized by fibrous cap thinning and rupture,13 MPO activity has been demonstrated to be abundantly higher in unstable plaque compared with adjacent stable plaques9 and there are ongoing clinical studies substantiating this correlation. Moreover, multiple murine models have identified that genetic or pharmacological blockade of MPO prevents unstable plaque formation and increases histological features associated with plaque stability, thus identifying a causal relationship between MPO activity, plaque destabilization, and the susceptibility to rupture.9,14
At a mechanistic level, MPO contributes to cap thinning through activation of matrix metalloproteinases (MMPs) and inactivation of their inhibitors, resulting in degradation of the cap matrix. Activated human neutrophils simultaneously release MPO and active MMP-9.15 In addition, low concentrations of HOCl directly activate MMP-7 in vitro,16 and MPO-generated oxidants inactivate tissue inhibition of metalloproteinase-1 that counteracts the action of MMP, thereby weakening the extracellular matrix.17 In addition, micromolar concentrations of reagent HOCl have been demonstrated to cause dysfunction and death of cultured human coronary smooth muscle cells, which are a vital component of fibrous cap architecture and plaque stability.18
MPO is also involved in plaque destabilization because it is required for the formation of NETs, i.e. mesh-like DNA fibres cast out by neutrophils in a process known as NETosis which mechanistically cannot occur in the absence of cellular MPO.19 NETosis activates endothelial cells, antigen-presenting cells, and platelets, resulting in a pro-inflammatory environment. NETs are present in plaques and thrombi, and they contribute to triggering arterial thrombosis.20
2.2 MPO in plaque erosion
Eroded plaques typically have denuded endothelium, exposing the intima. Although these lesions can demonstrate macrophage and T-cell infiltration, they are more consistently laden with neutrophils and infiltrated smooth muscle cells.21 MPO is directly involved in the development of eroded plaques through endothelial dysfunction and endothelial cell apoptosis, and indirectly involved via the effects of NETs.
MPO and MPO-derived oxidants contribute to endothelial dysfunction by limiting the bioavailability of nitric oxide and oxidation of its receptor, soluble guanylate cyclase.22 Indeed, individuals with high plasma MPO content are more likely to demonstrate endothelial dysfunction compared to those with low circulating MPO, even after adjustment for pharmacological therapies and traditional cardiovascular risk factors.23 Pharmacological or genetic blockade of MPO attenuates endothelial dysfunction in models of vascular inflammation by decreasing the oxidation of soluble guanylyl cyclase and therefore increasing nitric oxide-mediated signalling.24
There is in vitro evidence to suggest that MPO also provokes superficial plaque erosion and increases the propensity for thrombus formation.25 Exposure of human endothelial cells to MPO-expressing macrophages or pathophysiological concentrations of HOCl causes apoptosis and subsequent desquamation, whereas sub-lethal concentrations of HOCl induce tissue factor activity, and thereby generate a prothrombotic environment.25 Given its specific role in both plaque erosion and rupture, extracellular MPO stands out as a more selective and encompassing target for the full spectrum of atherosclerotic plaque destabilization compared to other inflammatory targets.
2.3 Putative mechanism of MPO in plaque rupture vs. endothelial erosion
Plaque rupture and erosion are two distinct modes of atherosclerotic plaque destabilization, and it appears that MPO plays distinct mechanistic roles in each process. Circulating MPO is increased in patients with eroded plaques compared to ruptured plaques, just as MPO+-granulocytes are abundant in the thrombi of eroded but not ruptured plaques.26 This infers a difference in not only the pathological process by which these distinct thrombotic states occur but also the disparate role of MPO in these plaque phenotypes.
We postulate that elevations in plasma MPO may be more pronounced in eroded plaques due to a luminal process driving the disease, where an acute endothelial insult results in an abrupt release of MPO from circulating neutrophils. In the setting of plaque rupture, the process appears more insidious, relying on intramural MPO from within mature inflamed atherosclerotic plaques. This hypothesis is supported by evidence demonstrating that eroded plaques are more likely to contain juvenile neutrophils and less likely to contain macrophages and T-cells, which are conventionally associated with sub-acute and chronic immune activation2,21
In addition, the relative dearth of macrophages seen in erosive plaques may provide further insight into the differences of these pathophysiological states. Blood contains monocytes rather than macrophages and only after migration from the endovascular space into tissue do monocytes differentiate into proficient phagocytes. The relative lower abundance of macrophages found in eroded compared with ruptured lesions may further substantiate an acute and predominantly luminal process underpinning erosion. This supposition is supported not only by the histological absence of macrophages in erosive plaques, but also large lipid pools, necrotic cores, calcific deposits, and positive remodelling, i.e. features of progressive, mature, and advanced lesions typical of ruptured and rupture-prone atheroma.2,27
2.4 MPO in coagulation and platelet aggregation
MPO is integral to thrombosis and platelet aggregation. Procoagulant activity is exerted via the generation of reactive oxygen species and associated lipid peroxidation.28 This leads to activation of tissue factor and inactivation of its inhibitor pathways.29 Being involved in NET formation, MPO also has the potential to affect coagulation and platelet aggregation indirectly, as NETs activate the contact pathway30 and increase thrombin generation.31
Prothrombotic effects relate to the ability of positively charged MPO to bind negatively charged glycosaminoglycans and proteoglycans, as well as the MPO-derived oxidants to degrade proteoglycans including heparan sulfate, thereby decreasing its established anticoagulant effects.32 HOCl has also been reported to concentration-dependently inhibit thrombus formation under flow, as well as clot retraction and fibrinolysis in whole blood.33 Platelet priming and aggregation are further effects of MPO and its oxidation products.34 MPO does not appear to be a direct platelet agonist but rather a mediator that binds to platelets, inducing reorganization of the cytoskeleton and increased platelet elasticity.35 This results in potentiation of store-operated calcium entry and agonist-induced platelet aggregation.
Nevertheless, MPOs role in coagulation appears complex, with some in vitro studies implicating it in the inhibition of thrombus formation.33,36 One such study reported that acquired neutrophil MPO deficiency was associated with increased fibrinogen and prothrombotic tendency in leukaemia.36 Although haemostatic mechanisms are complex and MPO-derived oxidants may play agonistic and antagonistic roles, positive or negative effects may depend on the location of MPO activity. Specifically, we postulate that extracellular MPO provokes thrombosis,33 whereas intracellular MPO has antithrombotic properties as observed in studies employing MPO-depleted granulocytes.36 This further underscores the dichotomous effects of the enzyme in different physiological compartments. Intravital imaging of MPO activity in relation to platelets and neutrophils may clarify MPO’s in vivo function during thrombus formation and confirm this hypothesis.
3. Imaging of vulnerable plaque
Novel approaches toward the non-invasive assessment of plaque composition and disease activity may assist in differentiating stable from unstable atherosclerosis, thus complimenting established imaging techniques, and improving decision making around management. Fundamentally, imaging assessment of plaque phenotype can be broadly categorized into the anatomical assessment of plaque structure or the molecular assessment of disease activity, both of which help to differentiate stable from vulnerable plaque.37 An outline of the available non-invasive techniques for the imaging of vulnerable plaque and their targets are summarized in Table 1. Key differences in the vascular beds in which plaques are located makes imaging strategies heterogenous. As an example, coronary arteries are relatively small and subject to motion from respiration and the cardiac cycle, therefore, lend themselves to a modality like computed tomography (CT), which has excellent spatial resolution and rapid acquisition.38 Coronary artery assessment by CT can be coupled with single-photon or positron emission tomography (SPECT and PET) using molecular imaging to assess the biological activity of plaque, making up for the former’s poor contrast resolution. Conversely, extracardiac arteries such as the carotids are larger and less prone to motion, making their imaging more adaptable to magnetic resonance imaging (MRI), which has superior contrast resolution to CT and avoids exposure to ionizing radiation. MRI is particularly useful in examining plaque composition and adverse plaque features and has excellent concordance with histology.39,40 Molecular probes have similarly been integrated into MRI for the detection of pathological plaque activity.37
Non-invasive imaging techniques for vulnerable plaque and their targets
| Modality | Technique | Target |
|---|---|---|
| CT | Plaque quantification | Positive remodelling, necrotic core size, microcalcification |
| Perivascular fat attenuation | Inflammation/adipose tissue | |
| PET | 18F-Fluorodeoxyglucose (18F-FDG) | Inflammation/macrophages |
| 68Ga-DOTATATE | Inflammation/somatostatin receptor | |
| 18F-Sodiumfluoride | Microcalcification/mineral deposition | |
| 18F-galacto-RGD | Neoangiogenesis/integrin αvβ3 | |
| SPECT | 125Iodine-labelled oxidation specific antibodies | Oxidized low-density lipoproteins |
| 99mTc-labelled annexin-V | Inflammation/macrophage apoptosis | |
| 99mTc-MMP inhibitors | Fibrous cap degradation | |
| MRI | T1 and T2 weighted imaging | Necrotic core size, thrombosis, intraplaque haemorrhage |
| Gadolinium enhancement | Fibrous cap thickness | |
| Iron oxide nanoparticles | Macrophages |
| Modality | Technique | Target |
|---|---|---|
| CT | Plaque quantification | Positive remodelling, necrotic core size, microcalcification |
| Perivascular fat attenuation | Inflammation/adipose tissue | |
| PET | 18F-Fluorodeoxyglucose (18F-FDG) | Inflammation/macrophages |
| 68Ga-DOTATATE | Inflammation/somatostatin receptor | |
| 18F-Sodiumfluoride | Microcalcification/mineral deposition | |
| 18F-galacto-RGD | Neoangiogenesis/integrin αvβ3 | |
| SPECT | 125Iodine-labelled oxidation specific antibodies | Oxidized low-density lipoproteins |
| 99mTc-labelled annexin-V | Inflammation/macrophage apoptosis | |
| 99mTc-MMP inhibitors | Fibrous cap degradation | |
| MRI | T1 and T2 weighted imaging | Necrotic core size, thrombosis, intraplaque haemorrhage |
| Gadolinium enhancement | Fibrous cap thickness | |
| Iron oxide nanoparticles | Macrophages |
F, fluorine; Ga, gallium; Tc, technetium.
Non-invasive imaging techniques for vulnerable plaque and their targets
| Modality | Technique | Target |
|---|---|---|
| CT | Plaque quantification | Positive remodelling, necrotic core size, microcalcification |
| Perivascular fat attenuation | Inflammation/adipose tissue | |
| PET | 18F-Fluorodeoxyglucose (18F-FDG) | Inflammation/macrophages |
| 68Ga-DOTATATE | Inflammation/somatostatin receptor | |
| 18F-Sodiumfluoride | Microcalcification/mineral deposition | |
| 18F-galacto-RGD | Neoangiogenesis/integrin αvβ3 | |
| SPECT | 125Iodine-labelled oxidation specific antibodies | Oxidized low-density lipoproteins |
| 99mTc-labelled annexin-V | Inflammation/macrophage apoptosis | |
| 99mTc-MMP inhibitors | Fibrous cap degradation | |
| MRI | T1 and T2 weighted imaging | Necrotic core size, thrombosis, intraplaque haemorrhage |
| Gadolinium enhancement | Fibrous cap thickness | |
| Iron oxide nanoparticles | Macrophages |
| Modality | Technique | Target |
|---|---|---|
| CT | Plaque quantification | Positive remodelling, necrotic core size, microcalcification |
| Perivascular fat attenuation | Inflammation/adipose tissue | |
| PET | 18F-Fluorodeoxyglucose (18F-FDG) | Inflammation/macrophages |
| 68Ga-DOTATATE | Inflammation/somatostatin receptor | |
| 18F-Sodiumfluoride | Microcalcification/mineral deposition | |
| 18F-galacto-RGD | Neoangiogenesis/integrin αvβ3 | |
| SPECT | 125Iodine-labelled oxidation specific antibodies | Oxidized low-density lipoproteins |
| 99mTc-labelled annexin-V | Inflammation/macrophage apoptosis | |
| 99mTc-MMP inhibitors | Fibrous cap degradation | |
| MRI | T1 and T2 weighted imaging | Necrotic core size, thrombosis, intraplaque haemorrhage |
| Gadolinium enhancement | Fibrous cap thickness | |
| Iron oxide nanoparticles | Macrophages |
F, fluorine; Ga, gallium; Tc, technetium.
A recent position paper41 lays the foundations for how and why imaging of vulnerable atherosclerotic plaque may improve clinical assessment and outcomes. The paper also identifies the current limitations of such imaging strategies and the need to develop an understanding of how to implement them as well as determine which modalities are best suited for the identification of adverse plaques.41 Given the causal and specific role of extracellular MPO in plaque destabilization and vascular inflammation, the enzyme presents itself as a molecular target of interest in this emerging field, offering a specificity to the disease process beyond current probes that target inflammation more broadly.
Utilizing a probe that selectively targets extracellular MPO may allow for the selective detection of harmful inflammation and oxidative tissue injury. This is something that currently available probes generally cannot achieve. As an example, the widely studied FDG-PET detects the presence of macrophages within a given tissue and not M1 and M2 subsets which play vastly different roles in the inflammatory cascade. As such, the presence of FDG-PET avidity does not differentiate between oxidative tissue injury and the beneficial, reparative role of these phagocytes.
Despite the promise and exploration of non-invasive imaging for the detection of vulnerable plaque, there remains an absence of such methods in the clinical realm, highlighting the importance of ongoing exploration and discovery in this field.
4. MPO as an imaging target to identify vulnerable plaque
Several molecular imaging tracers with fluorescent, radioactive, or paramagnetic properties that specifically bind to or react with target molecular structures have been developed to selectively detect MPO activity. When coupled with non-invasive imaging platforms, these tracers allow for in vivo localization of MPO activity, which advances the non-specific assessment of circulating MPO protein by isolating enzymatic activity within affected tissues. Molecular MRI, PET, and SPECT probes with chemical moieties that specifically react with MPO-derived oxidants exist. Importantly, these probes assess MPO activity, and at least in the case of gadolinium (Gd)-based MRI probes, selectively target extracellular MPO, therefore, allowing for a determination of detrimental CVD activity rather than a quantification of the total enzyme within a given tissue.
MPO-Gd is a molecular imaging agent used for the non-invasive localization of MPO activity by MRI. The probe links the commonly used diethylenetriaminepentaacetic acid-Gd (DTPA-Gd) with two moieties of 5-hydroxytryptamine. MPO-mediated oxidation of the 5-hydroxytryptamine moiety leads to the formation of oligomers and protein cross-links.42 The resulting effect is the retention of Gd in tissues with MPO activity, giving rise to a >2-fold increase in T1-weighted MRI signal.43,44
MPO-Gd has successfully been utilized in vivo for the localization of MPO activity and inflammation in animal models of atherosclerosis, vasculitis, and myocardial infarction.43,44 In a mouse model, MPO-Gd demonstrated selective enhancement of unstable plaque on T1-weighted imaging when compared to stable plaque.9 MPO-Gd enhancement in this model was similarly attenuated by deleting the MPO gene or the use of a specific pharmacological inhibitor of MPO. Unlike the linear MPO-Gd, heMAMP45 is a macrocyclic probe with comparatively improved gadolinium stability and increased signal intensity, making it an appealing translational candidate. The efficacy of heMAMP has been confirmed in the same mouse model of plaque instability used to assess MPO-Gd,45 as well as in a currently unpublished rabbit model of both plaque erosion and rupture. As indicated, due to its chemical structure, Gd-based probes do not enter cells, so that the signal obtained from these probes selectively reflects extracellular MPO activity.
A fluorine-18-based PET imaging radioprobe (18F-MAPP) has been validated for the selective detection of MPO activity.46 Upon MPO-mediated oxidation, the probe is retained and hence identifies sites enriched with MPO activity in vivo using PET imaging. The specificity of 18F-MAPP has been validated in mouse models of myocardial infarction through genetic and pharmacological blockade of MPO. Moreover, 18F-MAPP can accurately report on the degree of MPO inhibition when an irreversible MPO inhibitor is used to decrease inflammation. Thus, this radioprobe presents itself as a potentially useful tool for drug development and treatment surveillance and is currently being considered for Investigation New Drug status.46 It should be noted that due to its lipophilicity, 18F-MAPP enters cells and therefore detects total MPO.
Although several MPO-specific SPECT probes have been developed, their relatively long radioactive half-lives have hampered their clinical translation. Therefore, it is likely the pursuit of available PET probes (combined with CT in the assessment of coronary atheroma) and MRI (for extracardiac plaque) will lead to clinical translation of MPO for the detection of vulnerable atherosclerosis.
5. Treatment of vulnerable plaque and the notion of residual risk
Currently, statins remain the best available therapy for plaque stabilization. Statins decrease circulating C-reactive protein (CRP), augment collagen content of atherosclerotic lesions, alter the endothelial function and decrease the inflammatory components of atheroma. Numerous clinical trials have highlighted the role of statins in promoting plaque stability, inducing plaque regression, and reducing fibro-fatty components.47 Statins have also been associated with reduced intraplaque angiogenesis.48 Unfortunately, however, the atherosclerotic disease progresses in up to one-third of patients,49 reinforcing the need to reduce the ‘residual risk; of CVD events through the development of new treatments.
Following aggressive medical therapy for cholesterol reduction, patients with persistent elevations in LDL (>70 mg/dL) or CRP (>2 mg/L) are at increased risk of acute CVD events.50 This cohort with residual risk is not optimally managed under current guideline-directed therapy and warrants additional treatment and surveillance measures. Reducing arterial inflammation and the inflammatory processes underlying atherogenesis and plaque destabilization could provide the breakthrough required to reduce residual risk.
Several agents have been trialled in addition to optimal medical therapy. Using a therapeutic monoclonal antibody targeting inhibition of interleukin-1β, a cytokine central to the inflammatory response, CANTOS demonstrated a net benefit in the reduction of hospitalization for unstable angina that led to a need for urgent revascularization, but this was associated with a higher incidence of fatal infections.51 The CIRT trial using low-dose methotrexate was stopped early due to an inability to demonstrate a benefit in CVD outcomes and demonstrable increases in adverse outcomes.52
Colchicine is another drug with postulated anti-inflammatory effects that has demonstrated promising reductions in stroke and AMI in certain patient populations. The LoDoCo and LoDoCo2 trials utilized low-dose colchicine as a secondary prevention in patients with established coronary artery disease and demonstrated a reduction in CVD endpoints without a significant increase in adverse events.53 Similarly, the COLCOT study, which enrolled patients following an AMI, found a reduction in a composite CVD outcome, the benefit of which was driven by a reduction in stroke rather than coronary events or mortality.54 Conversely, the smaller COPS trial looking at a similar cohort to COLCOT demonstrated no CVD benefit and a disproportionate increase in all-cause death and in particular non-CVD mortality in those treated with colchicine,55 leaving questions about the benefits of this therapy.
Nevertheless, the potential for targeted anti-inflammatory therapies to stabilize unstable plaque and reduce the residual risk of CVD events remains the subject of great interest, and here MPO holds promise as a drug target due to the spatial segregation of its beneficial and harmful effects.
6. MPO as a drug target for arterial inflammation and vulnerable plaque
The pervasive issue with anti-inflammatory and immunomodulatory medicines trialled to date is that they also downregulate the protective effects of their targets. Inhibition of the beneficial properties of the immune response may result in an unacceptable increase in non-CVD morbidity and mortality, typically through infection and malignancy. It is here where pharmacotherapy with MPO inhibitors may prove advantageous. MPO offers an advantage over other inflammatory mediators given the compartmentalization of its protective (cellular) and deleterious (extracellular) effects, the latter of which can be selectively targeted by currently available diagnostic and therapeutic strategies (Graphical Abstract). The thioxanthines AZD5904, AZD3241, and AZD4831, and the thiopyrimidinone PF06282999 have been clinically trialled as MPO inhibitors.56–60 These suicide inhibitors have the same mechanism of action whereby a thioether bridge is formed between MPO’s haem and the oxidized inhibitor, resulting in irreversible inhibition of MPO.56 Due to their relative polarity and differences in pharmacokinetic properties of cellular and extracellular MPO, these MPO inhibitors at select dosages specifically target extracellular MPO, thereby preserving the immune-protective role of cellular MPO.
MPO has long attracted the attention of medicinal chemists and the pharmaceutical industry who have endeavoured to develop an inhibitor for the treatment and prevention of inflammatory diseases in which MPO is implicated. Although currently not used clinically, these MPO inhibitors are in pre-clinical and early clinical trial stages for various inflammatory diseases including neurodegenerative disorders,61,62 vasculitis,63 cardiomyopathy,64 and atherosclerosis/myocardial infarction.9,65 A comprehensive review of MPO inhibitor-related patents indicates no adverse effects in human phase I and II trials with proven efficacy for plaque stabilization in pre-clinical models.56
In animal studies of vulnerable plaque, treatment with a thioxanthine-based MPO inhibitor has resulted in improved endothelial function, increased cap thickness and plaque stability as confirmed by both non-invasive imaging and histology.9,24 Effective plaque stabilization is demonstrated without a reduction in circulating immune cells or deleterious side effects. Despite the accumulating evidence of MPO’s role in many chronic diseases, especially atherosclerosis, and the activity of these compounds in inhibiting MPO in vivo without demonstrable adverse effects, no human trials exploring these compounds in atherosclerosis are presently underway.
Conclusion
If we are to improve the assessment and management of patients with CVD, a shift away from luminal stenosis and toward plaque morphology and biological activity is required. With advances in clinical diagnostic tools and treatments, extracellular MPO presents itself as a unique target that holds potential as an imaging biomarker and therapeutic target for vulnerable atherosclerosis. Future studies should look to translate pre-clinical data supporting the role and potential benefit of targeting extracellular MPO in the assessment and management of the atherosclerotic disease.
Acknowledgements
The authors thank Dr Freda Passam (Heart Research Institute) for her insight and critical reading of the manuscript.
Funding
J.N. is supported by scholarships from the National Health and Medical Research Council of Australia, the National Heart Foundation of Australia, and the University of New South Wales. This work was supported by an NSW Department of Health grant as well as National Health and Medical Research Council of Australia Program Grant 1052616 and Senior Principal Research Fellowship 1111632 to R.S.
References
Author notes
Conflict of interest: R.S. receives in-kind support from AstraZeneca in relation to the development of MPO inhibitors. The remaining authors have nothing to disclose.

{kind=link}
{kind=link}
{kind=link}