





Abstract
The chemokine receptor CCR5 and its inflammatory ligands have been linked to atherosclerosis, an accelerated form of which occurs in saphenous vein graft disease. We investigated the function of vascular smooth muscle CCR5 in human coronary artery and saphenous vein, vascular tissues susceptible to atherosclerosis, and vasospasm.
CCR5 ligands were vasoconstrictors in saphenous vein and coronary artery. In vein, constrictor responses to CCL4 were completely blocked by CCR5 antagonists, including maraviroc. CCR5 antagonists prevented the development of a neointima after 14 days in cultured saphenous vein. CCR5 and its ligands were expressed in normal and diseased coronary artery and saphenous vein and localized to medial and intimal smooth muscle, endothelial, and inflammatory cells. [125I]-CCL4 bound to venous smooth muscle with KD = 1.15 ± 0.26 nmol/L and density of 22 ± 9 fmol mg−1 protein.
Our data support a potential role for CCR5 in vasoconstriction and neointimal formation in vitro and imply that CCR5 chemokines may contribute to vascular remodelling and augmented vascular tone in human coronary artery and vein graft disease. The repurposing of maraviroc for the treatment of cardiovascular disease warrants further investigation.
1. Introduction
The G-protein-coupled receptor, CCR5,1–3 is activated by the inflammatory chemokines CCL3, CCL4, and CCL5, but only CCL4 exhibits selectivity for CCR5 over other CC chemokine receptors.4 CCR5 is a major co-receptor for the HIV-1 virus;5,6 however, in humans, a 32-bp deletion results in a non-functional receptor protein that confers resistance to HIV-1 infection.7,8 This observation gave impetus to the development of small molecular weight antagonists and led to the approval of maraviroc,9 the first of a new class of virus entry inhibitors, for use in drug-resistant CCR5-tropic HIV-1 infection in 2007.
As patient life-expectancy increases, HIV-infected individuals are at increased risk of cardiovascular events10,11 and poorer long-term outcomes following coronary artery bypass grafting.10 This may be a direct effect of HIV infection or the actions of some anti-viral protease inhibitors.12 However, CCR5 and its ligands have been increasingly linked to human atherosclerosis through studies on plasma chemokine levels13,14 and genetic polymorphisms,15 with individual homozygous for the delta-32 mutation exhibiting an inverse association with early onset of coronary artery disease.16 Hypertension is a major risk for cardiovascular disease, and while a direct correlation between blood pressure and CCR5 genotype is debatable,17,18 prolonged treatment with antiviral therapy is related to increased systolic blood pressure19,20 and pulmonary hypertension associated with vascular remodelling is increased in HIV-positive individuals.21,22
We were particularly interested in the dose-limiting postural hypotension reported for maraviroc23 that suggested to us a potential vasodilator action of this drug via blockade of endogenous CCR5 tone. Maraviroc is a highly selective CCR5 antagonist9 and therefore, we hypothesized that CCR5 receptors localized to vascular smooth muscle cells contribute to increased vascular tone that may be of relevance in coronary atherosclerosis in which expression of CCR5 ligands is increased. Additionally, we wished to determine whether the CCR5 receptor contributed to intimal hyperplasia in vein graft disease leading to graft failure that is exacerbated in individuals positive for HIV-1.
We now report for the first time that smooth muscle CCR5 mediates vasoconstriction in isolated saphenous vein and coronary artery in response to CCL4 and CCL5 that can be abolished by CCR5 antagonists, including maraviroc. We also find that neointimal development was completely inhibited by CCR5 antagonists in a human saphenous vein model of accelerated intimal hyperplasia. These data suggest that, in addition to roles proposed for CCR5 in endothelial dysfunction and atherosclerosis, increased levels of CCR5 chemokines in disease may contribute to vascular remodelling and augmented vascular tone or acute vasospasm in human coronary artery and vein graft disease.
2. Methods
An expanded method section is available in Supplementary material online.
2.1 Tissue samples
Human tissues were obtained with informed consent from the Papworth Hospital Research Tissue Bank (REC reference 08/H0304/56) and experiments carried out with local ethical approval (REC 05/Q0104/142). Saphenous vein and mammary artery were from 104 patients receiving coronary artery bypass grafts. Other cardiovascular tissues were from 79 patients undergoing cardiac or lung transplantation or nephrectomy. The study conformed to the principles outlined in the Declaration of Helsinki.
2.2 Human in vitro pharmacology
Endothelium-denuded saphenous vein and coronary artery were set up for isometric force recordings, as described.24 Cumulative concentration response curves were constructed to CCL4, CCL5 (0.1 pmol/L–110 nmol/L), angiotensin-II (10 pmol/L–100 nmol/L), endothelin-1 (ET-1, 0.1–300 nmol/L), and phenylephrine (1 nmol/L–100 μmol/L). It should be noted that a limitation of these experiments was that the maximum possible concentration achievable in the organ bath for CCL4 and CCL5 was 110 nmol/L. In the vein, CCL4 responses were determined using ±300 nmol/L of maraviroc to verify involvement of CCR5 and confirmed using 10 and 100 nmol/L of the chemically distinct CCR5 antagonist PF-232796.25 For dilator studies, the vein was pre-constricted with 10 nmol/L of ET-1 and CCL4 (10 pmol/L–100 nmol/L) was added cumulatively. Data were analysed using a four parameter logistic equation (GraphPad Prism 5) to give values of pD2 (−log10 of the concentration that produces 50% of the fitted maximum response) and maximum response (EMAX). Wire myography was performed using the aorta from C57Bl/6 mice to determine whether CCL4 contracted a blood vessel susceptible to atherosclerosis in another species.
2.3 Human saphenous vein organ culture
Saphenous vein organ culture was performed as described.26–28 Using consecutive vein segments (1 cm), one segment (Day 0 control) was immediately formalin-fixed and remaining segments cultured with ±1 μmol/L of maraviroc, PF-232796, or vehicle (0.1% dimethyl sulfoxide) for 14 days. Conditioned culture medium was frozen (−70°C) until required. Formalin-fixed transverse segments were stained with haematoxylin and eosin, Alcian blue, Miller's elastin, and van Gieson's stains28 to determine the cell number and neointimal area, expressed as a percentage of total intimal + medial area.
2.4 Chemokine multiplex immunoassay
A custom 4-plex immunoassay for CCL2, CCL3, CCL4, and CCL5 was used to determine chemokine concentrations in vein culture supernatant.
2.5 Reverse transcription polymerase chain reaction assays
RNA was extracted and reverse transcribed from human tissues. PCR was carried out using primers specific for Gsα, CCR5,29 CCL3, CCL4, and CCL5.
2.6 Quantitative reverse transcription polymerase chain reaction
Total RNA was extracted and expression of CCR5, CCL3, CCL4, CCL5, and GAPDH determined using cDNA-specific TaqMan Gene Expression Inventoried Assays. Gene expression was quantified using the comparative (ΔΔCT) method. All samples were screened for Gsα to confirm cDNA integrity and the lack of gDNA contamination.
2.7 Western blot
Western blotting was performed on protein lysates of saphenous vein with rabbit anti-CCR5.
2.8 Immunohistochemistry
Immunohistochemistry was performed as described.30 Sections of saphenous vein and vein graft were used for additional colorimetric histology. Cultured vein sections were processed for the analysis of proliferation (phosphorylated-histone H3 and Ki67) and apoptotic (cleaved caspase-3) markers, in addition to TUNEL staining.
2.9 Dual labelled fluorescent confocal microscopy
Sections of human frozen tissues were processed30 and stained with rabbit anti-human CCR5, CCL3, CCL4, or CCL5 and markers; mouse anti-human von Willebrand factor (vWF), smooth muscle α-actin (SMαA), CD68 to identify macrophages, or CD3 to detect T-lymphocytes. Secondary antibodies Alexa Fluor 488-conjugated goat anti-rabbit IgG and Alexa Fluor 568-conjugated goat anti-mouse IgG were incubated for 1 h (22°C). Slides were mounted in ProLong Gold reagent, cured for 24 h, and viewed with a confocal laser scanning microscope.
2.10 Tissue profiling using receptor autoradiography
Receptor autoradiography31 was carried out using cryostat-cut sections of human tissues and 0.1 nmol/L of [125I]-CCL4. Non-specific binding (NSB) was defined using 100 nmol/L of CCL4.
2.11 Saturation and competition analysis
Saphenous vein (10 μm sections) was incubated for 2 h (22°C) with [125I]-CCL4 (2 pmol/L–2 nmol/L). NSB was defined by 1 µmol/L of CCL4. In competition experiments, sections were incubated with 0.1 nmol/L of [125I]-CCL4 and increasing concentrations of CCL4 or maraviroc (10 pmol/L–2 umol/L). Sections were opposed, with standards, for 5 days to radiation-sensitive film. Autoradiograms were analysed using computer-assisted densitometry.31
2.12 Statistical analysis
N-values are the number of patients from which tissues were obtained. For in vitro pharmacology data, EMAX and pD2 values were compared using Student's two-tailed t-test or one-way analysis of variance, followed by Bonferroni's multiple comparison tests. Receptor autoradiography was analysed by one-way analysis of variance followed by Tukey's or Bonferroni's multiple comparison tests. Where there was evidence of non-normality non-parametric statistical analysis was performed with data expressed as median (range). For quantitative reverse transcription polymerase chain reaction, analysis of different genes within the same tissues was done by the Friedman test followed by Dunn's multiple comparison test. Comparison between different tissues used the Kruskall–Wallis test followed by the Mann–Whitney U-test with Bonferroni correction applied. For vein culture, comparisons between paired data were by the Wilcoxon signed-rank test or the Friedman test for related samples followed by Dunn's multiple comparison test. A P-value of <0.05 was considered significant.
3. Results
3.1 CCR5 mediates constriction of human saphenous vein and coronary artery
In pre-constricted saphenous vein, CCL4 had no direct dilator actions (n = 4) (see Supplementary material online, Figure S1A). CCL4 contracted tissue from 13 of 19 veins tested. In seven of these responsive veins, a maximum response was achieved at 110 nmol/L CCL4; however, for the remaining tissues, a maximum response was not obtained, although there were sufficient data for a curve fit to derive both maximum response and EC50. Therefore, our values of potency and maximum response for CCL4 in saphenous vein should be regarded as estimates. With this caveat, the order of potency of the four agonists tested was angiotensin-II (pD2 = 8.80 ± 0.23, n = 10) > ET-1 (pD2 = 7.92 ± 0.17, n = 6) ≥ CCL4 (pD2 = 7.67 ± 0.19, n = 13) > phenylephrine (pD2 = 6.31 ± 0.21, n = 10). Comparing the maximum constrictor responses of the four agonists, the order of efficacy was ET-1 (EMAX = 98 ± 7% KCl) > phenylephrine (EMAX = 65 ± 8% KCl) > angiotensin-II (EMAX = 40 ± 3% KCl) = CCL4 (EMAX = 39 ± 8% KCl) (Figure 1A). CCL5 contracted saphenous vein with pD2 comparable with CCL4 (CCL5 pD2 = 7.56 ± 0.37, EMAX = 17 ± 8% KCl, n = 5).
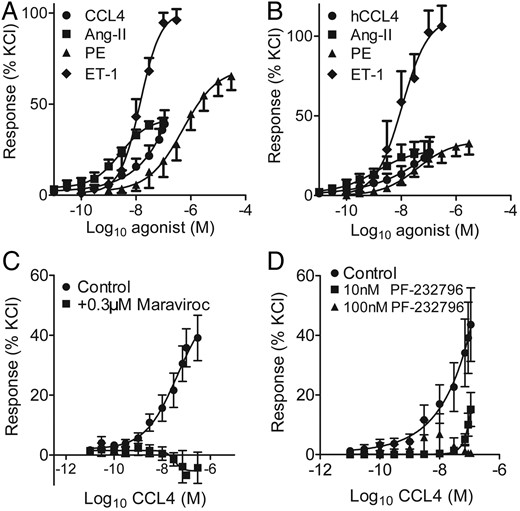
Vasoconstrictor responses to CCL4 (filled circle), angiotensin-II (Ang-II, filled square), phenylephrine (PE, filled triangle), and endothelin-1 (ET-1, filled diamond) in human endothelium-denuded (A) saphenous vein (n = 6–13) and (B) coronary artery (n = 4–9). Antagonism of (C) CCL4 (filled circle) by 300 nmol/L maraviroc (filled square) (n = 4) and by (D) 10 nmol/L (filled square) and 100 nmol/L (filled triangle) PF-232796 (n = 4) in saphenous vein.
In coronary artery vasoconstrictor responses to CCL4 were obtained in all the five arteries tested and a maximum response was achieved to CCL4 for 4/5 of these. The order of agonist potency was as for saphenous vein; angiotensin-II (pD2 = 9.20± 0.41, n = 6) > ET-1 (pD2 = 8.28 ± 0.18, n = 6) ≥ CCL4 (pD2 = 8.07 ± 0.42, n = 5) > phenylephrine (pD2 7.43 ± 0.14, n = 9/16) (Figure 1B). The maximum responses to CCL4 (EMAX 26 ± 7% KCl); angiotensin-II (EMAX 27 ± 9% KCl), and phenylephrine (EMAX 33 ± 8% KCl) were comparable, but all were significantly lower than that to ET-1 (106 ± 13% KCl) (P < 0.05). In the presence of 300 nmol/L maraviroc, CCL4 constriction was abolished (Figure 1C). PF-232796 (10 nmol/L) produced a rightward shift of the CCL4 concentration response curve with abolition at 100 nmol/L PF-232796 (Figure 1D). Maraviroc (300 nmol/L) had no effect on responses to phenylephrine or ET-1 in saphenous vein (not shown). CCL4 contracted mouse aorta with pD2 = 9.79 ± 0.23 (n = 10) (see Supplementary material online, Figure S1C).
3.2 CCR5 antagonists inhibit intimal thickening in a model of vein graft disease
By Day 14, vein segments had developed a neointima containing smooth muscle cells and extracellular matrix (Figure 2A and B, and see Supplementary material online, Figure S2A, B, E, and Supplementary Data). Medial and neointimal layers expressed CCR5 mRNA (n = 5) (see Supplementary material online, Figure S2J) and protein (n = 4 pooled, Figure 2C–E) localized to smooth muscle cells (see Supplementary material online, Figure S2K). CCL3, CCL4, and CCL5 mRNA (n = 5, Figure 2F) and protein (see Supplementary material online, Figure S3A–C) were also identified, localized to neointimal smooth muscle cells (see Supplementary material online, Figure S4), and all the three ligands were detected in culture medium (n = 7–8, Figure 2G). Total CCL2 release over the culture period, measured for comparison, was significantly greater than CCL3 or CCL5 release (n = 9, P < 0.01 and <0.001) and CCL4 release was significantly greater than CCL5 (n = 9, P < 0.05, see Supplementary material online, Figure S3D).
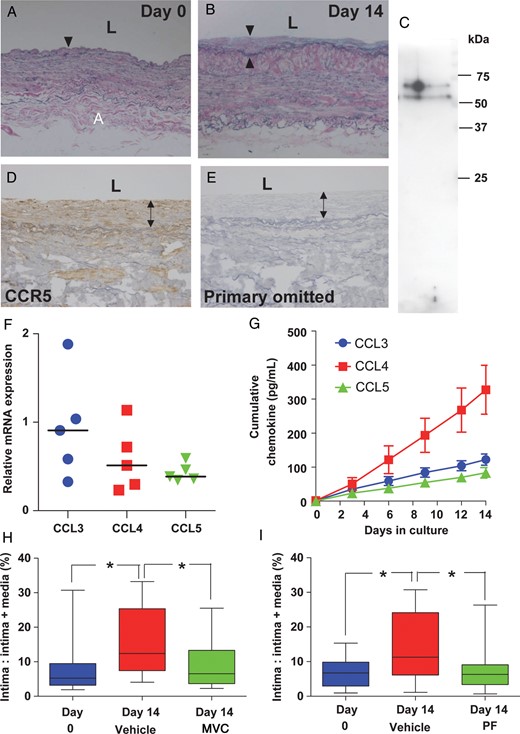
Contribution of CCR5 to intimal hyperplasia in cultured saphenous vein. Cross-section through saphenous vein segment after (A) 0 and (B) 14 days in culture showing the development of a neointima (arrow heads) by Day 14. (C) CCR5 protein detected by western blotting (n = 4, pooled). (D) CCR5 immunoreactivity localized to medial and neointimal layers of cultured vein with staining absent (E) when the primary antibody was omitted. (F) CCL3, CCL4, and CCL5 mRNA (n = 5) were detected in cultured vein and in culture medium (G) during culture (n = 7–8). Co-culture with CCR5 antagonists (H) maraviroc (MVC, n = 10) and (I) PF-232796 (PF, n = 9) inhibited the development of intimal thickening (P < 0.05, Friedman test followed by Dunn's multiple comparison test). Values are median (range).
In vein segments, co-culture with maraviroc (Figure 2H) and PF-232796 (Figure 2I) inhibited the development of intimal thickening (P < 0.05). Little staining for cleaved caspase-3 was observed in veins cultured without or with maraviroc or PF-232796 (see Supplementary material online, Figure S5A–F). There was no difference in cell density in segments cultured with vehicle or CCR5 antagonists (see Supplementary material online, Figure S5G and H). A significant increase in CCL5 release was seen with maraviroc (P < 0.05), although no difference was observed for CCL3, CCL4, or CCL2 (n = 7). Co-culture with PF-232796 did not lead to significant alterations in chemokine release (n = 6–7) (see Supplementary material online, Figure S6). There was no evidence for alterations in the extent of apoptosis or proliferation in veins cultured with antagonists compared with vehicle (see Supplementary material online, Figures S7–9).
3.3 CCR5 identified in human cardiovascular tissues
Full-length CCR5 transcripts were identified in cardiomyocytes, media of aorta, pulmonary and coronary artery, and saphenous vein (Figure 3A). Infrequently, a smaller band was identified consistent with individuals being homozygous or heterozygous for the CCR5 delta 32-deletion polymorphism (e.g. Figure 3A, lane 4). Western blotting confirmed a single band at ∼75 kDa in the saphenous vein (Figure 3B). Specific CCR5 immunoreactivity was detected in the normal saphenous vein and coronary artery (Figure 3C and D). Using specific markers, CCR5 immunoreactivity co-localized to endothelial and medial smooth muscle cells in the saphenous vein and coronary artery (see Supplementary material online, Figure S10) and to smooth muscle of small intramyocardial vessels and surrounding cardiomyocytes in the heart (Figure 3G).
![CCR5 mRNA and protein expression in human cardiovascular tissues. (A) Transcripts for CCR5 (179 bp) in media of coronary artery (CA), aorta (Ao), pulmonary artery (PA), cardiomyocytes (cm), and saphenous vein (SV). Transcripts for Δ32 polymorphic (147 bp) CCR5 in PA (lane 4). Ladders are 100 bp. (B) Western blot confirmed a single band at ∼75 kDa in saphenous vein (n = 9, three pooled samples). CCR5 immunoreactivity localized to media (M) and endothelium (EC) of normal (C) saphenous vein and (D) coronary artery with staining abolished on omission of the primary antibody (E and F). CCR5 was co-localized to the smooth muscle of (G) small intramyocardial coronary vessels with SMαA and was present on surrounding cardiomyocytes with (H) staining abolished when primary antibodies were omitted. (I) Relative density of [125I]-CCL4 binding in human tissues (n = 3–9; * significantly different from coronary media; † significantly different from mammary artery; ‡ significantly different from radial artery; P < 0.05). (J) Representative autoradiograms showing total and NSB in sections of coronary artery (CA), saphenous vein (SV), mammary artery (MA), and radial artery (RA), scale bar = 2 mm.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/101/3/10.1093_cvr_cvt333/3/m_cvt33303.jpeg?Expires=1750282460&Signature=1xcfTNll2IZwfoow5yM93J~bmIiLV6ON6Tfotos6JNFF99PIdD4nwKfwpn6eNo8wVzkUlj3c~y6P9dSdABxt~68Dj5WadcHrrsZUXunpojP6HfH8llisJPUw2ADfg2rmOXDx04lGlRWR4DfX4A56rVC8sq6ocgPQFumZTo-2L62me9FI0KNvxvnEjScQv8SFJtYixEFCF~C5sMKyR8goqhR728uRnmUV-pr7gxafUIHYLcjRVM2Y7NgiNUYPu4l5gjODpK-Aw49n7yYrDLC9GGVWB4eGT2q8ox~cHYAxcdvj3EmKkJmhtWq02C1t6miWdTN21sAD5YPkStUy9Hf3dg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CCR5 mRNA and protein expression in human cardiovascular tissues. (A) Transcripts for CCR5 (179 bp) in media of coronary artery (CA), aorta (Ao), pulmonary artery (PA), cardiomyocytes (cm), and saphenous vein (SV). Transcripts for Δ32 polymorphic (147 bp) CCR5 in PA (lane 4). Ladders are 100 bp. (B) Western blot confirmed a single band at ∼75 kDa in saphenous vein (n = 9, three pooled samples). CCR5 immunoreactivity localized to media (M) and endothelium (EC) of normal (C) saphenous vein and (D) coronary artery with staining abolished on omission of the primary antibody (E and F). CCR5 was co-localized to the smooth muscle of (G) small intramyocardial coronary vessels with SMαA and was present on surrounding cardiomyocytes with (H) staining abolished when primary antibodies were omitted. (I) Relative density of [125I]-CCL4 binding in human tissues (n = 3–9; * significantly different from coronary media; † significantly different from mammary artery; ‡ significantly different from radial artery; P < 0.05). (J) Representative autoradiograms showing total and NSB in sections of coronary artery (CA), saphenous vein (SV), mammary artery (MA), and radial artery (RA), scale bar = 2 mm.
By autoradiography, we detected a significant difference in specific binding of [125I]-CCL4 across a panel of human tissues (P < 0.001, one-way ANOVA; Figure 3I and J, and see Supplementary material online, Figure S11A). Compared with the highest density in coronary artery media, levels of receptor expression were not different in the media of other arteries, heart, or kidney medulla, but were significantly lower (Tukey's multiple comparison test, P < 0.05) in saphenous vein, kidney cortex, and lung. Binding of [125I]-CCL4 was reduced by maraviroc, consistent with it acting as an allosteric modulator (see Supplementary material online, Figure S11C). Saturation analysis confirmed that [125I]-CCL4 bound to media of saphenous vein with a single nanomolar affinity (KD = 1.15 ± 0.26 nmol/L, n = 3). The Hill slope was 1.14 ± 0.06 with a receptor density of 22 ± 9 fmol mg−1 protein.
3.4 CCR5 ligands are expressed in human cardiovascular tissues
CCL3, CCL4, and CCL5 mRNA were detected in aorta, pulmonary, coronary and mammary artery, saphenous vein, and cardiomyocytes (Figure 4A). Immunoreactivity to all the three ligands was detected in the endothelium and media of epicardial and intramyocardial coronary artery, saphenous vein, and in cardiomyocytes (Figure 4B, and see Supplementary material online, Figure S12).
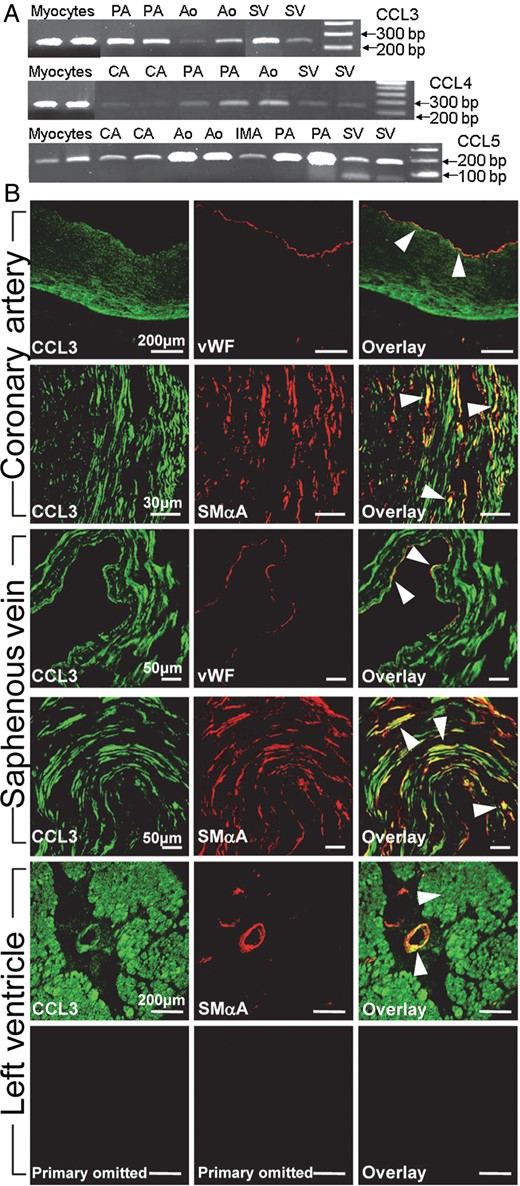
Expression of CCR5 ligands in human cardiovascular tissues. (A) Transcripts for CCL3, CCL4, and CCL5 in myocytes, aorta (Ao), pulmonary (PA), coronary (CA), and mammary (IMA) artery and saphenous vein (SV). Ladders are 100 bp. (B) Expression of CCL3 immunoreactivity (green) co-localized (orange/yellow in overlay) with vWF and SMαA (red) in coronary artery, saphenous vein and to a small intramyocardial vessel and surrounding myocardium in the left ventricle. CCR5 immunoreactivity was absent on omission of primary antibody.
3.5 CCR5 and ligand expression in coronary atherosclerosis, saphenous vein graft, and heart failure
In atherosclerotic plaque of diseased coronary artery (Figure 5A) and thickened intima of failed saphenous vein graft (Figure 5B), CCR5 immunoreactivity was expressed in smooth muscle cells, macrophages, and to lesser extent CD3-positive cells. Compared with saphenous vein and donor myocardium, there was no significant alteration in CCR5 mRNA in saphenous vein graft or in hearts from patients transplanted for dilated cardiomyopathy (DCM) or ischaemic heart disease (IHD), the two most common diagnoses for cardiac transplantation32 (Figure 5C and D). There was no difference in receptor density between coronary artery media and intimal layers and media of saphenous vein and vein graft (P > 0.05, one-way analysis of variance followed by Bonferroni's multiple comparison test; Figure 3I). A similar cellular localization was obtained for CCL3, CCL4, and CCL5 as for CCR5 in coronary artery plaque and intima of failed vein graft (Figure 5E, and see Supplementary material online, Figure S13) and whereas levels of mRNA encoding CCR5 ligands were not different in normal and diseased saphenous vein (Figure 5F) comparison in donor compared with DCM or IHD ventricle showed highest expression of ligands in the IHD group, with CCL5 significantly increased compared with donors (Figure 5G, P < 0.05).
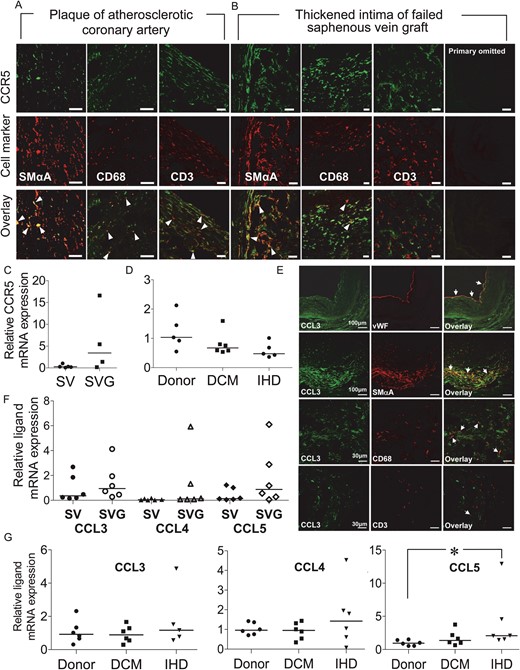
CCR5 receptor protein expression in (A) coronary atherosclerotic plaque and (B) thickened intima of failed saphenous vein graft. Co-localization (overlay: yellow/orange, arrow heads) of CCR5 immunoreactivity (green) with SMαA, CD68, and CD3-positive cells (red). Lack of fluorescence on omission of primary antibody shown for the vein graft. Scale bars: 30 μm. Relative levels of CCR5 mRNA in (C) saphenous vein (SV n = 5) and vein graft (SVG n = 4) and (D) donor (n = 5), DCM (n = 6) and ischaemic (IHD n = 5) myocardium. (E) Expression of CCL3 in saphenous vein graft: CCL3 immunoreactivity (green) co-localized in overlay (orange/yellow) with vWF, SMαA, CD68, and rarely CD3-positive cells (red). (F) Expression of CCL3, CCL4, and CCL5 mRNA was comparable in graft (SVG, n = 6) and normal vein (SV, n = 6). (G) In the left ventricle, CCL5 mRNA was significantly increased in IHD compared with donor (*P < 0.05, n = 6).
4. Discussion
We have examined whether CCR5 ligands have direct vasoactive actions on human blood vessels to understand the mechanism underlying the postural hypotension reported for maraviroc in healthy volunteers.23 We now report for the first time that, at least in vitro, the CCR5 receptor on human vascular smooth muscle mediates vasoconstriction in response to nanomolar concentrations of CCL4 and CCL5 and contributes to neointimal hyperplasia in a model of human vein graft disease. The involvement of CCR5 in these responses was confirmed by their blockade by maraviroc.
We next carried out a detailed analysis of the receptor pharmacology and expression pattern of CCR5 and its ligands in human cardiovascular tissues. In saphenous vein, CCR5 identified a single nanomolar affinity in agreement with that reported for human CCR5 expressed in HEK 293 cells.2 We obtained a CCR5 binding density of ∼20 fmol mg−1 protein, which is comparable with other vasoconstrictor peptide receptors including the endothelin ETA receptor (190 ± 23 fmol mg−1)33 and the thromboxane TP receptor (6 ± 2 fmol mg−1).34 Binding of [125I]-CCL4 was inhibited by the CCR5 antagonists, confirming the interaction of ligand with receptor rather than tethering of CCL4 to cell surface glycosaminoglycans. Using receptor autoradiography and immunocytochemistry, we have shown that CCR5 is expressed on vascular smooth muscle in a range of human arteries, including coronary artery. In contrast to previous reports,35 we have demonstrated CCR5 mRNA and protein on venous smooth muscle consistent with our receptor binding and observation that CCL4 and CCL5 are vasoconstrictors in the human saphenous vein. As far as we are aware this is the first report of vasoactivity of CC chemokines, although the CXCR4 ligand SDF-1α has been shown to contract human coronary artery microvessels.36 Importantly, we found that CCL4 was an equally effective constrictor of human epicardial coronary arteries in vitro.
A direct effect of CCR5 ligands on vascular smooth muscle has not previously been studied to any extent, although the HIV-1 virus has recently been shown to infect vascular smooth muscle cells resulting in release of the pro-inflammatory chemokine CCL2.37 This action is dependent on the expression of CD4 and a co-receptor such as CCR5. Activation of smooth muscle CCR5 by viral gp120 is linked to the release of tissue factor.38 Interestingly, CCL4 was reported to increase tissue factor activity in human cultured vascular smooth muscle cells;39 however, the consequence of activation of smooth muscle CCR5 by endogenous chemokine ligands has not been further investigated. One reason for this may be that individuals homozygous for the CCR5 delta 32 polymorphism do not exhibit a significant cardiovascular phenotype. Despite this, the mutation has been associated with decreased cardiovascular risk40 and circulating levels of CCL5 are increased in cardiovascular disease.41,42 In patients with unstable angina,41 CCL5 is detectable in the nanomolar range, and in a study of hypertensives, those individuals with the highest plasma levels of CCL4 had a higher risk of stroke and cardiovascular events with the authors concluding that CCL4 contributed to the development of atherosclerosis.43 The plasma levels of CCL4 detected in these hypertensives was 44–336 pg mL−1 which based on the affinity for CCL4 determined in our radioligand binding assay would produce a level of CCR5 receptor occupancy of between 84 and 97%. Our in vitro vasoconstrictor data suggest that these levels of plasma CCL4, at least in hypertensive patients, would occupy most of the available receptors with the potential to contribute to increased vascular tone. Plasma levels of chemokine/cytokine biomarkers, including CCL4 and CCL5, are also significantly increased in patients receiving coronary artery bypass grafts.44 Perioperative vein graft spasm is a serious complication contributing to graft failure45 and postoperative spasm, though rare, can be fatal.46 If CCR5 ligands are present locally at sufficient concentrations, our observations suggest that these mediators, acting through vascular CCR5, may also contribute to graft spasm at the time of operation.
As reported by others,39,47 we observed that CCR5 and its ligands were expressed in atherosclerotic coronary artery and failed saphenous vein graft by intimal smooth muscle cells likely to be of a ‘synthetic’ phenotype,48 macrophages, and rarely by CD3+ T-cells. The presence of both receptor and ligands in the intima provides evidence for paracrine signalling during disease progression promoting neointimal thickening. In particular, CCL5/CCR5 is considered crucial to monocyte recruitment during the development of native vessel atherosclerosis.49,50 We observed expression of CCR5 by both contractile and proliferative vascular smooth muscle cells, comparable with the constrictor thromboxane,34 but this contrasts with our observation that receptors for ET-1 are downregulated in the intima of diseased coronary artery and saphenous vein.51,52
Having identified the CCR5 system in saphenous vein and failed vein graft, we next addressed the question of a functional role for CCR5 in graft disease. There is increasing evidence from animal models53 that chemokine receptors, including CCR5, contribute to intimal hyperplasia; however, maraviroc has little if any affinity for rodent CCR5 and therefore, we used a model of human intimal hyperplasia in which to interrogate the consequence of CCR5 antagonism. This is a static organ culture model, without flow conditions, that recapitulates some aspects of the in vivo condition and therefore, caution is required when interpreting the results. However, the advantage of this model is that it utilizes a clinically relevant human tissue. Following saphenous vein culture, development of a neointimal layer was evident, as previously reported.26,27 Similar to native vein graft, we detected CCR5 and ligands in cultured vein localized to intimal smooth muscle cells. CCL3, CCL4, CCL5, and CCL2 were released during culture, with intimal smooth muscle cells54,55 a likely source. Increased CCL5 release was observed on co-culture with maraviroc consistent with a feedback cycle between CCL5 and CCR5.56 The detection of CCL2 release is supported by a previously reported association between CCL2 expression and intimal thickening in rat vein graft.57 Importantly, in this model, antagonism of CCR5 by two structurally distinct antagonists abolished the development of intimal thickening, confirming a role for CCR5. This is unlikely to be due to toxicity as maraviroc was non-toxic with respect to cell proliferation in culture,9 and we found no evidence for increased cell death and no difference in cell density between segments cultured with vehicle or CCR5 antagonists.
Finally, we detected expression of CCR5 in cardiomyocytes, but any effect of CCR5 ligands on cardiac contractility remains to be investigated. SDF-1 reportedly inhibited cardiac contractility in isolated rodent myocytes and was blocked by the CXCR4-selective antagonist AMD3100.58 In human heart failure, we found no change in CCR5 receptor density but detected increased levels of CCR5 ligands in patients transplanted for IHD, the relevance of which is to be determined.
In summary, we report that CCR5 is widely expressed by human cardiovascular tissues and mediates potent vasoconstriction of human arteries and veins by its ligands CCL4 and CCL5. This response can be antagonized by maraviroc. We speculate that CCR5 antagonists, used as virus entry inhibitors, may have additional benefits to slow progression of virus- or drug- treatment-associated vascular complications in this patient group. In addition, CCR5 and its ligands are present in human saphenous vein and saphenous vein graft tissue, and antagonism of CCR5 inhibits the development of intimal thickening in vein in vitro. There are limitations to our studies in that they report only on the vasoconstrictor and proliferative potential of CCR5 ligands in human blood vessels in vitro. Therefore, extrapolation to the in vivo clinical setting requires further investigation to confirm whether blocking chemokine receptors, such as CCR5, may be a novel strategy for the treatment of vascular disease.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This work was supported by the British Heart Foundation (grant numbers PS/02/001 and PG/05/127/19872). This study was supported in part by the NIHR Cambridge Biomedical Research Centre and the Pulmonary Hypertension Association UK. K.J. was supported by a BBSRC Cooperative Awards in Science and Engineering studentship. Funding to pay the Open Access publication charges for this article was provided by the British Heart Foundation.
References
Author notes
Present address. University Hospital of Wales, Cardiff CF14 4XW, UK.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}