

 Musculoskeletal Imaging Volume 2: Metabolic, Infectious, and Congenital Diseases; Internal Derangement of the Joints; and Arthrography and Ultrasound
Musculoskeletal Imaging Volume 2: Metabolic, Infectious, and Congenital Diseases; Internal Derangement of the Joints; and Arthrography and Ultrasound

Cite
Introduction
Lysosomal storage diseases (LSDs) are caused by the accumulation of proteins, polysaccharides, and lipids in the lysosomes of cells because of a genetic failure of enzymes needed for their breakdown. The progressive accumulation of substrates frequently affects the skeleton. The pathophysiology is not completely understood and likely involves a complicated interplay of substrate storage, tissue inflammation, and epigenetic factors. LSDs can be categorized as sphingolipidoses and mucolipidoses, and mucopolysaccharidoses (discussed in Chapter 84, “Mucopolysaccharidoses”). Table 83.1 summarizes the incidence and musculoskeletal findings of the sphingolipidoses and mucolipidoses, which are groups of diseases that share musculoskeletal imaging findings. Diagnosis relies primarily on peripheral blood lysosomal enzyme activity, but DNA testing is the most reliable technique for definitive diagnosis and detecting disease transmission.
| Condition | Incidence | Musculoskeletal Findings |
|---|---|---|
SPHINGOLIPIDOSES | ||
Fabry disease | 1:40,000–60,000 (males) | Short stature, crises of burning and aching pain in hands and feet, arthralgia, exercise intolerance, osteonecrosis with or without secondary osteoarthritis osteopenia/osteoporosis |
Gaucher disease type I (nonneuropathic type) | 1:40,000–60,000 | Short stature, chronic bone pain and/or acute bone crises, osteonecrosis with or without secondary osteoarthritis, osteopenia/osteoporosis, recurrent fractures, Erlenmeyer flask deformities |
Niemann-Pick disease (types A-C) | A-B: 1:250,000; C: 1:150,000 | Erlenmeyer flask deformities, osteonecrosis |
MUCOLIPIDOSES | ||
Mucolipidosis (ML) I (sialidosis syndrome) | 1:5,000,000 | Dysostosis multiplex (Table 83.2), myoclonic epilepsy |
ML II (I-cell disease) | 1:640,000 | Disproportional short stature, joint stiffness/contractures, hip dysplasia, genu valgum, carpal tunnel syndrome, trigger fingers, dysostosis multiplex |
ML III (pseudo-Hurler polydystrophy) | 1:315,000 | Disproportional short stature, joint stiffness/contractures, scoliosis, carpal tunnel syndrome, trigger fingers, dysostosis multiplex |
α-Mannosidosis | 1-1.6:500,000 | Dysostosis multiplex |
| Condition | Incidence | Musculoskeletal Findings |
|---|---|---|
SPHINGOLIPIDOSES | ||
Fabry disease | 1:40,000–60,000 (males) | Short stature, crises of burning and aching pain in hands and feet, arthralgia, exercise intolerance, osteonecrosis with or without secondary osteoarthritis osteopenia/osteoporosis |
Gaucher disease type I (nonneuropathic type) | 1:40,000–60,000 | Short stature, chronic bone pain and/or acute bone crises, osteonecrosis with or without secondary osteoarthritis, osteopenia/osteoporosis, recurrent fractures, Erlenmeyer flask deformities |
Niemann-Pick disease (types A-C) | A-B: 1:250,000; C: 1:150,000 | Erlenmeyer flask deformities, osteonecrosis |
MUCOLIPIDOSES | ||
Mucolipidosis (ML) I (sialidosis syndrome) | 1:5,000,000 | Dysostosis multiplex (Table 83.2), myoclonic epilepsy |
ML II (I-cell disease) | 1:640,000 | Disproportional short stature, joint stiffness/contractures, hip dysplasia, genu valgum, carpal tunnel syndrome, trigger fingers, dysostosis multiplex |
ML III (pseudo-Hurler polydystrophy) | 1:315,000 | Disproportional short stature, joint stiffness/contractures, scoliosis, carpal tunnel syndrome, trigger fingers, dysostosis multiplex |
α-Mannosidosis | 1-1.6:500,000 | Dysostosis multiplex |
Gaucher Disease
Pathophysiology and Clinical Findings
Gaucher disease (GD) is an autosomal recessive disorder caused by deficient β-glucocerebrosidase activity and accumulation of the lipid glucosylceramide. Macrophages, when filled with glucosylceramide, are referred to as Gaucher cells, which have a characteristic crumpled silk appearance of the cytoplasm. GD is the most common LSD and is classically subdivided into 3 different phenotypes. Type I GD is by far the most frequent with a particularly high frequency in the Ashkenazi Jewish population (1 in 850). There is no central nervous system involvement, although Parkinson disease has been described in these patients. Type 1 GD has a highly variable presentation, even among twins, which most often involves the bone (80%), but also results in cytopenia and hepatosplenomegaly. Types 2 and 3 GD are much less frequent and have either acute (type 2) or chronic (type 3) neurologic disease. Patients with type 2 GD die in infancy, and patients with type 3 GD present in childhood with a less severe phenotype.
Bone complications can be a first symptom of disease, presenting as bone crises, which are episodes of excruciating pain, possibly necessitating hospitalization for pain management. Patients also suffer from a chronic inflammatory state that is exacerbated during these crises, which are often accompanied by localized warmth and swelling, leukocytosis, raised ESR and fever, similar in presentation to juvenile idiopathic arthritis (JIA) and RA. Up to 43% of patients have a history of ON with up to 22% undergoing joint replacement. Osteoporosis is common in patients with GD, possibly related to chronic inflammation, in a similar fashion to the osteoporosis of JIA and RA. Both longer disease duration and history of splenectomy are associated with ON and osteoporosis.
Imaging Strategy
MRI is the imaging modality of choice. Sagittal T1W and STIR sequences of the lumbar spine, coronal T1W and STIR imaging of the pelvis, and coronal T1W and STIR sequences of both femora are optimal to evaluate disease status. In younger patients, T1W and STIR images of the tibias are useful, because the presence of red marrow in the femurs can make the detection of involvement difficult. Patients should have follow-up imaging every 12-24 months and/or at times of enzyme replacement therapy or substrate removal therapy (ERT and SRT, respectively). At the same sitting, axial T1W and T2W images are acquired through the upper abdomen to assess severity of spleen and liver involvement. DXA scanning is generally recommended for the evaluation and follow-up of bone density at 1-2 year intervals. The use of radiography is limited to evaluation of acute pain (eg, exclude fractures). Nuclear medicine, especially technetium 99m sestamibi (99mTc-sestamibi), is a useful alternative in evaluating disease status in patients with GD who are unable to undergo MRI or when MRI is unavailable.
Imaging Findings
Radiography
Radiography is insensitive to marrow infiltration:
Detects changes in 30-40% of patients with GD
Characteristic features include Erlenmeyer flask deformity of the femora, osteopenia, and sclerosis:
Osteopenia is nearly universal in both children and adults:
Localized or diffuse
Associated with an increased risk of fragility fracture and reported in up to 28% of patients with GD
Areas of sclerosis caused by ON may be detected, especially involving the spine, pelvis, and proximal long bones (Figure 83.2):
Femoral and humeral epiphyseal involvement can be complicated by articular collapse and secondary OA.
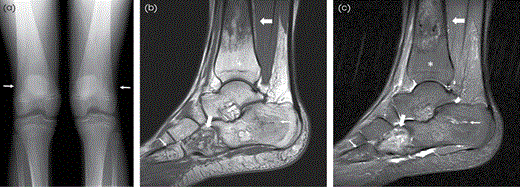
GD in a 13-year-old patient. (A) AP radiograph of the knees demonstrates the characteristic Erlenmeyer flask deformity of both distal femurs (thin arrows) with thin cortices consistent with osteopenia. (B) T1W and (C) STIR sagittal MR images of the ankle demonstrate areas of heterogeneous signal in the distal tibia and cuboid bone consistent with areas of ON (thick arrows). Heterogeneous signal within the calcaneus and proximal fifth metatarsal is abnormal (thin arrows), which may represent ON. Areas of normal fatty marrow (asterisk) caused by ERT are present.
{kind=link}
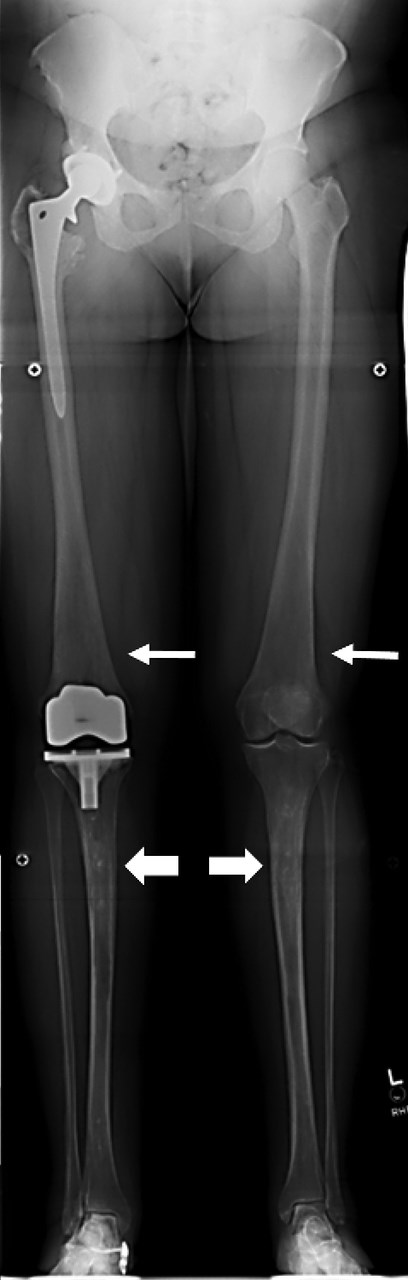
GD in a 42-year-old patient. A stitched leg length series demonstrates Erlenmeyer flask deformities (thin arrows) and patchy osteosclerosis of the tibial shafts caused by ON (thick arrows). Right hip and knee joint replacements were caused by secondary OA from ON.
{kind=link}
Marrow infiltration disorders
Gaucher disease
Niemann-Pick disease
Thalassemia
Lead poisoning
Craniotubular bone dysplasias
Frontometaphyseal dysplasia
Otopalatodigital syndrome
Craniometaphyseal dysplasia
Pyle disease
Engelmann disease (diaphyseal dysplasia)
Osteopetrosis
Dual-Energy X-ray Absorptiometry
Bone marrow density (BMD) in GD is significantly lower than expected for age and sex:
BMD is decreased centrally (lumbar spine, femoral neck) and peripherally (distal radius).
Magnetic Resonance Imaging
Imaging shows infiltrated bone marrow in GD (relative to subcutaneous fat):
Decreased bone marrow signal on T1 and T2W sequences without fat saturation and relatively hyperintense fluid sensitive sequences is related to infiltration by Gaucher cells.
MRI may show a salt-and-pepper pattern because of scattered involvement.
Diffusely homogeneous or heterogenous patterns are seen.
Abnormal signal may persist after treatment.
Degree of bone involvement is unrelated to visceral involvement.
Treatment with ERT or SRT may lead to rapid recurrence of bright T1 fatty marrow signal within 1 year.
ON with characteristic signal changes may be present (Figure 83.1B,C).
Extraosseous extension of Gaucher cells is rare and resembles malignancy, usually requiring biopsy.
Assessment of GD burden is primarily qualitative (descriptive), but other methods are used at specialty centers:
Bone marrow burden (BMB) scores severity of disease by degree of signal intensity loss relative to subcutaneous fat on T1W and T2W imaging and by how diffuse replacement is (eg, basivertebral fat replacement, epiphyseal involvement) (Figure 83.3).
Stabilizes 5 years after ERT treatment
Quantitation of GD burden using T1 relaxation, chemical shift imaging and spectroscopy is used primarily in research.
Marrow signal changes of ON in GD is similar to other causes (see Chapter 115, “Osteonecroses and Osteochondroses”).
ON is detected in GD even without a history of bone crisis.
Focal areas of increased T2 signal may represent bone crisis and represent a precursor to ON.
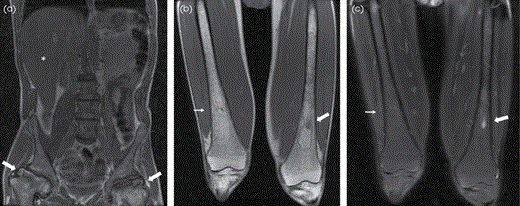
MRI of GD in a 45-year-old man. (A) Large field-of-view (FOV) T1W image demonstrates diffusely abnormal marrow signal with femoral head ON with articular surface collapse (thick arrows) and hepatomegaly (asterisk). Coronal T1W (B) and T2W with FS (C) images demonstrate diffuse marrow heterogeneity from ON (thick arrow) with asymmetric Erlenmeyer flask deformity on the right (thin arrow).
{kind=link}
Nuclear Medicine
In GD, 99mTc-sestamibi accumulates in areas of infiltration and is direct evidence of disease burden:
This is particularly useful in children in differentiating residual normal red marrow from pathologically infiltrated marrow.
99mTc-sulfur colloid is taken up by normal bone marrow with reduced or abnormally distributed tracer uptake seen with GD infiltration.
The main disadvantages are poor spatial resolution and high radiation dose.
Treatment Options
ERT with purified glucocerebrosidase is the most effective treatment for GD, which can prevent bone disease, reduce organomegaly, and improve anemia.
Oral SRT with miglustat can give some benefit in mild- to moderately affected patients with GD with improvement in bone pain, improvement in BMD, and stabilization of MRI findings.
Treatment for osteoporosis, such as bisphosphonates, calcium, and vitamin D, is given.
Arthroplasty is recommended for treatment of ON-related OA.
Fabry Disease
Pathophysiology and Clinical Findings
Fabry disease (FD) is an X-linked recessive disease resulting in deficient galactosidase A. Together, GD and FD account for 20% of LSDs with a combined prevalence of 1 in 38,400 (live) births. Attacks of pain (Fabry crisis) in the extremities, similar to the bone crisis of GD, begin during adulthood or adolescence, decreasing in frequency and severity with age. Like GD, the Fabry crisis may be accompanied by systemic inflammation resulting in a misdiagnosis of inflammatory arthritis (eg, JIA, RA). The mean delay in diagnosis is approximately 14 years for male patients and 16 years in female heterozygotes from symptom onset. Similar to GD, osteopenia and osteoporosis are reported in patients with FD. More specific signs of FD include skin papules (angiokeratoma) and cornea color changes (cornea verticillata) with hypohidrosis or anhidrosis. Patients also experience gastrointestinal symptoms. Renal, cardiac, and cerebrovascular complications result in major morbidity if untreated.
Imaging Strategy
Although not as well-defined as for GD, a similar approach to evaluate the severity of disease involvement and response to treatment using MRI is practical. Nuclear medicine techniques are not well described for FD.
Imaging Findings
Radiography
Loss of bone mass with osteopenia or osteoporosis is nearly universal in both children and adults with FD.
ON may be detected, especially involving the spine, pelvis, and proximal long bones.
As in GD, this can be complicated by articular collapse and secondary OA.
Magnetic Resonance Imaging
Assessment of marrow infiltration in FD has not been well described.
Marrow signal changes of ON in FD is similar to other causes.
Treatment Options
ERT is used for FD, but with a less consistent clinical response than GD.
Bisphosphonates, calcium, and vitamin D are recommended for osteoporosis.
Arthroplasty is used for severe joint disease.
Niemann-Pick Disease
Pathophysiology and Clinical Findings
NPD is an autosomal recessive disorder caused by deficiency of acid sphingomyelinase composed of 3 subtypes (A-C). The infiltration of bone marrow and spleen by lipid-engorged cells with a soap bubble appearance results in anemia and thrombocytopenia (also seen in GD). Neurologic involvement is common.
Imaging Strategy
A similar approach to evaluate the severity of musculoskeletal disease involvement and response to treatment using MRI is practical. Nuclear medicine techniques are not well described for NPD.
Imaging Findings
Radiography
Erlenmeyer flask deformity is seen on imaging, which is neither sensitive or specific (Box 83.1).
ON may be detected, which may be complicated by secondary OA.
Magnetic Resonance Imaging
Assessment of marrow infiltration in NPD has not been well described.
ON is similar in appearance to other causes.
Treatment Options
Oral SRT with miglustat for NPD (type C)
Bisphosphonates, calcium, and vitamin D for osteoporosis
Arthroplasty
Mucolipidoses
Pathophysiology and Clinical Findings
Mucolipidosis (ML)—ML I, ML II, ML III, ML IV—and α-mannosidosis are rare, autosomal recessive genetic disorders caused by a deficiency in enzymes that metabolize glycoproteins. ML I-III result in musculoskeletal deformities resembling mucopolysaccharidosis (MPS) I. ML IV is not associated with skeletal disease. ML II rarely presents in adults because of death during childhood. There is a similar presentation of ML and MPS patients, described in Chapter 84, “Mucopolysaccharidoses.” The treatment options are limited to treating symptoms as there are no medical treatments. These include surgery for cervical stenosis, kyphoscoliosis and hip dysplasia.
Imaging Strategy
The imaging strategy mirrors that of the MPS (see Chapter 84). The spectrum of findings seen in ML and MPS called dysostosis multiplex (Table 83.2) is evaluated using a baseline skeletal survey, typically acquired during childhood. Lateral flexion and extension radiographs of the cervical spine, a scoliosis series, and an AP pelvis radiograph should be obtained until skeletal maturity to evaluate for instability, gibbus deformity, and hip dysplasia. An annual cervical spine MRI may also be valuable given the frequency of cervical stenosis and its insidious onset.
Head | Macrocephaly; thickened skull; J-shaped sella turcica |
Thorax | Short, wide clavicles; wide oar-shaped ribs; |
Spine | Odontoid hypoplasia; anterior beaking of the lower thoracic and upper lumbar vertebral bodies; thoracolumbar kyphosis |
Legs/arms | Shortened long bones; wide diaphysis; irregular metaphyses; narrow epiphyses |
Hands/feet | Bullet-shaped phalanges; proximal pointing of metacarpals and metatarsals |
Hips | Flattened acetabula; flaring of iliac wings; coxa valga deformity; dysplastic femoral heads; secondary osteoarthritis |
Knees | Genu valgum; secondary subtalar varus |
Head | Macrocephaly; thickened skull; J-shaped sella turcica |
Thorax | Short, wide clavicles; wide oar-shaped ribs; |
Spine | Odontoid hypoplasia; anterior beaking of the lower thoracic and upper lumbar vertebral bodies; thoracolumbar kyphosis |
Legs/arms | Shortened long bones; wide diaphysis; irregular metaphyses; narrow epiphyses |
Hands/feet | Bullet-shaped phalanges; proximal pointing of metacarpals and metatarsals |
Hips | Flattened acetabula; flaring of iliac wings; coxa valga deformity; dysplastic femoral heads; secondary osteoarthritis |
Knees | Genu valgum; secondary subtalar varus |
Imaging Findings
Radiography
Dysostosis multiplex is detected on radiographs (Table 83.2).
Radiography is the primary technique to evaluate symptomatic patients.
Atlantoaxial instability is detected on flexion-extension radiographs.
Gibbus deformity with kyphosis is centered at the thoracolumbar junction:
Caused by L1 and L2 vertebral wedging deformity (see Chapter 84)
Magnetic Resonance Imaging
Craniocervical stenosis caused by odontoid dysplasia and glycoprotein deposition (see Chapter 84)
Treatment Options
Treatment is symptomatic.
GD is the most common LSD and lipidosis with bone marrow replacement resulting in ON and osteoporosis. The other sphingolipidoses, FD and NPD, demonstrate similar findings.
MRI of the spine, pelvis, and femurs is the primary imaging modality to evaluate disease and monitor treatment.
Erlenmeyer flask deformity of the femurs is characteristic of GD and NPD, but is neither sensitive nor specific for disease.
ML is a rare condition without effective treatments that results in bone changes similar to mucopolysaccharidoses.
ERT is the primary treatment for GD with frequent joint replacements secondary to ON.
Recommended Reading
Katz R, Booth T, Hargunani R, Wylie P, Holloway B.
References
1. Aldenhoven M, Sakkers RJ, Boelens J, de Koning TJ, Wulffraat NM.
2. Clarke LA, Hollak CE.
3. Fedida B, Touraine S, Stirnemann J, Belmatoug N, Laredo JD, Petrover D.
4. Katz R, Booth T, Hargunani R, Wylie P, Holloway B.
5. Mikosch P.
6. Parker EI, Xing M, Moreno-De-Luca A, Harmouche E, Terk MR.
7. Pastores GM.
| Month: | Total Views: |
|---|---|
| October 2022 | 3 |
| December 2022 | 3 |
| January 2023 | 2 |
| February 2023 | 4 |
| March 2023 | 4 |
| April 2023 | 2 |
| May 2023 | 1 |
| June 2023 | 5 |
| July 2023 | 2 |
| August 2023 | 2 |
| September 2023 | 2 |
| October 2023 | 2 |
| November 2023 | 2 |
| December 2023 | 2 |
| January 2024 | 1 |
| February 2024 | 1 |
| March 2024 | 2 |
| April 2024 | 5 |
| May 2024 | 1 |
| June 2024 | 3 |
| July 2024 | 3 |
| August 2024 | 1 |