




Abstract
Antimicrobial peptides (AMPs) are a unique and diverse group of molecules that play a crucial role in a myriad of biological processes and cellular functions. AMP-related studies have become increasingly popular in recent years due to antimicrobial resistance, which is becoming an emerging global concern. Systematic experimental identification of AMPs faces many difficulties due to the limitations of current methods. Given its significance, more than 30 computational methods have been developed for accurate prediction of AMPs. These approaches show high diversity in their data set size, data quality, core algorithms, feature extraction, feature selection techniques and evaluation strategies. Here, we provide a comprehensive survey on a variety of current approaches for AMP identification and point at the differences between these methods. In addition, we evaluate the predictive performance of the surveyed tools based on an independent test data set containing 1536 AMPs and 1536 non-AMPs. Furthermore, we construct six validation data sets based on six different common AMP databases and compare different computational methods based on these data sets. The results indicate that amPEPpy achieves the best predictive performance and outperforms the other compared methods. As the predictive performances are affected by the different data sets used by different methods, we additionally perform the 5-fold cross-validation test to benchmark different traditional machine learning methods on the same data set. These cross-validation results indicate that random forest, support vector machine and eXtreme Gradient Boosting achieve comparatively better performances than other machine learning methods and are often the algorithms of choice of multiple AMP prediction tools.
Introduction
Antimicrobial peptides (AMPs) are a unique and diverse group of gene-coded peptide antibiotics, which have been found in various species ranging from prokaryotes to eukaryotes, including mammals, amphibians, insects and plants [1]. AMPs are essential components of the immune system in a wide range of organisms, representing the first line of defense against a variety of pathogens [2]. AMPs display a broad spectrum of antimicrobial activities mainly against bacteria and also against fungi, viruses and cancer cells. They typically have a short length (6–100 amino acid residues) and fast and efficient action against microbes. In recent years, with resistance to antibiotics becoming an increasing global concern, AMPs have attracted significant interest as a potential substitute for conventional antibiotics [3]. The low toxicity to mammals and minimal development of resistance in target microorganisms make them potential candidates as peptide drugs [1]. AMPs’ targeting and mechanisms of action mean that they may have evolved to function in specific physiological and anatomical environments to minimize their potential damage to the host [4, 5]. Some AMPs can bind to protein receptors, whereas others appear to act directly against cell membranes [6], suggesting the existence of multiple modes of action. Besides, some AMPs are multifunctional effector molecules. They play a direct role in killing microorganisms and amplifying leukocytes’ antibacterial mechanism, which may bridge the innate and adaptive immunity [4, 5]. Compared with traditional antibacterials, AMPs have a more flexible mechanism, fewer side effects and are less prone to drug resistance.
Several models of antibacterial activity have been proposed; some features, such as helicity, flexibility and a cationic nature, have proven to be necessary for antibiotic activity [7–12]. Further, due to the functional significance of AMPs, several curated public resources have been established, providing comprehensive, experimentally verified annotations of AMPs [6, 13, 14]. In addition, the mode of action and activity-specific databases, such as AntiTbPdb [15], have also been made publicly available.
Based on the identification of AMPs, many efforts have been dedicated to the investigation of potential cellular mechanisms [16, 17]. Advances in AMP research have driven continued efforts for developing computational methods for accurate prediction of AMPs, aimed at significantly reducing the time and effort involved in experimental identification. Indeed, compared to labor-intensive and time-consuming experimental characterization of AMPs, computational prediction of AMPs provides a useful and complementary approach by shortlisting likely AMP candidates for subsequent experimental validation.
Thus far, a number of computational approaches have been developed and published for this purpose. At present, these tools can be classified into two major categories in terms of the adopted methodologies. The first category comprises conventional machine learning-based predictors, such as Collection of Anti-Microbial Peptides (CAMP) [3, 18], iAMP-2L [19] and iAMPpred [20]. These tools apply machine learning algorithms to features extracted from the peptide sequences to identify AMPs. Among the machine learning-based predictors, support vector machine (SVM) [21] and random forest (RF) [22] are the two most commonly used algorithms. The second category contains all deep learning-based methods. Deep learning is a hot direction in recent years, which is widely used in bioinformatics, especially in biological sequences [23, 24]. Such tools of the second category frequently use encodings of the original sequences, such as one-hot encoding, as the input, and in some cases, add the extracted sequence information from sequences by some third-party tools and then further extract features and output the classification labels through a neural network structure. Encodings of the original sequences as the inputs are seldomly used for training machine learning methods.
A number of attempts have been made to provide benchmark tests of prediction tools [25]; however, each study had certain limitations and drawbacks: either the study authors did not include a performance evaluation of all reviewed tools; several state-of-the-art prediction tools were not considered and benchmarked, or the studies did not provide a detailed algorithm description for each of the reviewed tools. In this article, we first provide a comprehensive survey on current machine learning-based computational methods for AMP prediction to the best of our knowledge to overcome these issues. We discuss a wide range of aspects including but not limited to the data set used, core algorithms selected for individual methods, feature selection techniques employed and performance evaluation strategies. Furthermore, we construct an independent data set and multiple validation data sets of different databases and compare different AMP prediction methods.
Materials and Methods
Construction of the independent test data set
In order to objectively evaluate the predictive performance of the existing available tools, an independent test data set must be constructed. For the positive data set, we integrated all AMPs from different comprehensive AMP public databases, including ADAM [26], ADAPTABLE [27], APD3 [17], CAMP [3, 18], dbAMP [28], DRAMP [29, 30], LAMP [14, 31], MilkAMP [32] and YADAMP [6]. Peptide sequences with length greater than 100 or less than 10 were not considered, and sequences with non-standard residues such as ‘B’, ‘J’, ‘O’, ‘U’, ‘X’ or ‘Z’ were eliminated since these peptides are rare and cannot be predicted by some tools [33]. Furthermore, to reduce homology bias and redundancy, the CD-HIT tool [34, 35] was used to filter out the sequences sharing the 40% sequence identity with any other in the same subset. And then, the sequences used as training data sets in the reviewed approaches were removed. Finally, 1536 AMP positive samples were obtained after all the steps mentioned above and were used as our training data set. The major steps for constructing the independent positive data set are illustrated in Supplementary Figure S1 available online at http://bib.oxfordjournals.org/.
For generating a negative data set, the following steps were taken: (1) peptide sequences were downloaded from UniProt (http://www.uniprot.org), and all entries containing the keyword ‘antimicrobial’ and related keywords (e.g. ‘antibacterial’, ‘antifungal’, ‘anticancer’, ‘antiviral’, ‘antiparasitic’, ‘antibiotic’, ‘antibiofilm’, ‘effector’ or ‘excreted’) were removed; (2) sequences with the length greater than 100 or less than 10 amino acid residues and with non-standard residues ‘B’, ‘J’, ‘O’, ‘U’, ‘X’ or ‘Z’ were eliminated, just as was done for the construction of the positive data set and (3) in order to reduce homology bias and redundancy, the CD-HIT program [34, 35] was employed to remove sequences that had 40% pairwise sequence identity to samples from the positive data set or were sharing 40% sequence identity to any other sequences in the same subset. Finally, 3062 negative samples were obtained. To balance the number between the positive and negative samples, we randomly selected 1536 samples from these 3062 samples, constituting our final negative data set.
Construction of the validation data sets
In order to compare the performance of different tools on different AMP databases, we constructed six data sets for additional performance evaluation based on six commonly used AMP public databases (APD3 [17], CAMP [3, 18], dbAMP [28], DRAMP [29, 30], LAMP [14, 31], YADAMP [6]).
We again removed sequences with the length less than 10 or greater than 100 and with non-standard residues to construct the positive data sets, and we used CD-HIT [34, 35] to filter out sequences sharing 40% sequence identity. To construct the negative data sets, we used the same approach as described in the previous section. Finally, 494 positive samples and 494 negative samples were obtained for the APD3 test, 203 positive samples and 203 negative samples were obtained for the CAMP test, 522 positive samples and 522 negative samples were obtained for the dbAMP test, 1408 positive samples and 1408 negative samples were obtained for the DRAMP test, 1054 positive samples and 1054 negative samples were obtained for the LAMP test and 324 positive samples and 324 negative samples were obtained for the YADAMP test, respectively. The major steps of constructing the six validation positive data sets are shown in Supplementary Figure S1 available online at http://bib.oxfordjournals.org/.
State-of-the-art computational approaches for AMP prediction
More than 30 computational methods have been developed for AMP identification to date. These methods differ in a variety of key aspects, including algorithms employed, adopted feature selection techniques and more. Table 1 summarizes 34 computational approaches currently available for AMP prediction according to the algorithm selected, features calculated, feature selection methods employed, data set size, performance evaluation strategy, web server availability, max data uploaded, emails of results and software availability.
A comprehensive summary of the reviewed approaches for AMP prediction
| Type | Tool | Year | Algorithm | Feature selection | Evaluation strategy | Web server availability | Max data upload | File upload availability | Email of result | Software availability |
|---|---|---|---|---|---|---|---|---|---|---|
| Machine learning-based methods | AMPer | 2007 | HMMs | None | 10-fold CV | No | N.A. | N.A. | N.A. | Yes |
| CAMP | 2010, 2016 | SVM, RF, ANN, DA | RFE (RF Gini) | 10-fold CV, independent test | Yes | N.A. | Yes | No | No | |
| Porto et al. | 2010 | SVM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Song et al. | 2011 | k-NN, BLASTP | mRMR, IFS | Jack-knife validation, independent test | No | N.A. | N.A. | N.A. | No | |
| Torrent et al. | 2011 | ANN | None | Independent test | No | N.A. | N.A. | N.A. | No | |
| Fernandes et al. | 2012 | ANFIS | ANFIS | Independent test | No | N.A. | N.A. | N.A. | No | |
| ClassAMP | 2012 | RF, SVM | RFE (RF Gini) | 10-fold CV, independent test | Yes | N.A. | Yes | No | No | |
| CS-AMPPred | 2012 | SVM | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | Yes | |
| C-PAmP | 2013 | SVM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| iAMP-2L | 2013 | FKNN | None | Jack-knife validation, independent test | Yes | 500 | No | No | No | |
| Paola et al. | 2013 | SVM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Randou et al. | 2013 | LR | None | Independent test | No | N.A. | N.A. | N.A. | No | |
| dbaasp | 2014 | z-Score | None | Independent test | No | N.A. | N.A. | N.A. | No | |
| ADAM | 2015 | SVM, profile HMMs | None | N.A. | Yes | N.A. | No | NO | No | |
| Camacho et al. | 2015 | NB, RF, SVM | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Ng et al. | 2015 | SVM, BLASTP | None | Jack-knife validation, independent test | No | N.A. | N.A. | N.A. | No | |
| MLAMP | 2016 | RF | None | Jack-knife validation, independent test | Yes | 5 | Yes | Yes | No | |
| iAMPpred | 2017 | SVM | None | 10-fold CV, independent test | Yes | N.A. | No | No | No | |
| AmPEP | 2018 | RF | None | 10-fold CV, independent test | Yes | N.A. | Yes | Yes | Yes | |
| CLN-MLEM2 | 2018 | MLEM2, IRIM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| MOEA-FW | 2018 | RF, k-NN, SVM, ANN | None | 10-fold CV | No | N.A. | N.A. | N.A. | No | |
| AMAP | 2019 | SVM, XGBoost, one-versus-rest classifier fusion | None | LOCO, 5-fold CV, independent test | Yes | N.A. | No | No | No | |
| MAMPs-Pred | 2019 | RF, LC-RF, PS-RF | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| dbAMP | 2019 | RF | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| AMPfun | 2019 | DT, RF, SVM | FS | 10-fold CV, independent test | Yes | N.A. | No | No | No | |
| Ampir | 2020 | SVM | RFE | independent test | No | N.A. | N.A. | N.A. | Yes | |
| Chung et al. | 2020 | RF | OneR, FS | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Fu et al. | 2020 | ADA | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| AmpGram | 2020 | RF | None | 5-fold CV, benchmark test | Yes | 50 | Yes | No | Yes | |
| IAMPE | 2020 | RF, SVM, XGBoost, k-NN, NB | None | 10-fold CV, independent test | Yes | N.A. | Yes | No | No | |
| Deep learning-based methods | AMP Scanner V2 | 2018 | LSTM | None | 10-fold CV, independent test | Yes | 50 000 | Yes | No | No |
| APIN | 2019 | CNN | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | Yes | |
| Deep-AmPEP30 | 2020 | CNN | None | 10-fold CV, independent test | Yes | N.A. | Yes | Yes | No | |
| AMPlify | 2020 | Bi-LSTM, attention mechanism | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | Yes |
| Type | Tool | Year | Algorithm | Feature selection | Evaluation strategy | Web server availability | Max data upload | File upload availability | Email of result | Software availability |
|---|---|---|---|---|---|---|---|---|---|---|
| Machine learning-based methods | AMPer | 2007 | HMMs | None | 10-fold CV | No | N.A. | N.A. | N.A. | Yes |
| CAMP | 2010, 2016 | SVM, RF, ANN, DA | RFE (RF Gini) | 10-fold CV, independent test | Yes | N.A. | Yes | No | No | |
| Porto et al. | 2010 | SVM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Song et al. | 2011 | k-NN, BLASTP | mRMR, IFS | Jack-knife validation, independent test | No | N.A. | N.A. | N.A. | No | |
| Torrent et al. | 2011 | ANN | None | Independent test | No | N.A. | N.A. | N.A. | No | |
| Fernandes et al. | 2012 | ANFIS | ANFIS | Independent test | No | N.A. | N.A. | N.A. | No | |
| ClassAMP | 2012 | RF, SVM | RFE (RF Gini) | 10-fold CV, independent test | Yes | N.A. | Yes | No | No | |
| CS-AMPPred | 2012 | SVM | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | Yes | |
| C-PAmP | 2013 | SVM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| iAMP-2L | 2013 | FKNN | None | Jack-knife validation, independent test | Yes | 500 | No | No | No | |
| Paola et al. | 2013 | SVM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Randou et al. | 2013 | LR | None | Independent test | No | N.A. | N.A. | N.A. | No | |
| dbaasp | 2014 | z-Score | None | Independent test | No | N.A. | N.A. | N.A. | No | |
| ADAM | 2015 | SVM, profile HMMs | None | N.A. | Yes | N.A. | No | NO | No | |
| Camacho et al. | 2015 | NB, RF, SVM | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Ng et al. | 2015 | SVM, BLASTP | None | Jack-knife validation, independent test | No | N.A. | N.A. | N.A. | No | |
| MLAMP | 2016 | RF | None | Jack-knife validation, independent test | Yes | 5 | Yes | Yes | No | |
| iAMPpred | 2017 | SVM | None | 10-fold CV, independent test | Yes | N.A. | No | No | No | |
| AmPEP | 2018 | RF | None | 10-fold CV, independent test | Yes | N.A. | Yes | Yes | Yes | |
| CLN-MLEM2 | 2018 | MLEM2, IRIM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| MOEA-FW | 2018 | RF, k-NN, SVM, ANN | None | 10-fold CV | No | N.A. | N.A. | N.A. | No | |
| AMAP | 2019 | SVM, XGBoost, one-versus-rest classifier fusion | None | LOCO, 5-fold CV, independent test | Yes | N.A. | No | No | No | |
| MAMPs-Pred | 2019 | RF, LC-RF, PS-RF | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| dbAMP | 2019 | RF | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| AMPfun | 2019 | DT, RF, SVM | FS | 10-fold CV, independent test | Yes | N.A. | No | No | No | |
| Ampir | 2020 | SVM | RFE | independent test | No | N.A. | N.A. | N.A. | Yes | |
| Chung et al. | 2020 | RF | OneR, FS | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Fu et al. | 2020 | ADA | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| AmpGram | 2020 | RF | None | 5-fold CV, benchmark test | Yes | 50 | Yes | No | Yes | |
| IAMPE | 2020 | RF, SVM, XGBoost, k-NN, NB | None | 10-fold CV, independent test | Yes | N.A. | Yes | No | No | |
| Deep learning-based methods | AMP Scanner V2 | 2018 | LSTM | None | 10-fold CV, independent test | Yes | 50 000 | Yes | No | No |
| APIN | 2019 | CNN | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | Yes | |
| Deep-AmPEP30 | 2020 | CNN | None | 10-fold CV, independent test | Yes | N.A. | Yes | Yes | No | |
| AMPlify | 2020 | Bi-LSTM, attention mechanism | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | Yes |
Full names of the algorithms: N.A., not available; HMMs, hidden Markov models; SVM, support vector machine; RF, random forest; ANN, artificial neural networks; DA, discriminant analysis; DT, decision tree; LR, logistic regression; k-NN, k-nearest neighbor; BLASTP, basic local alignment search tool (protein); ANFIS, adaptive neuro-fuzzy inference system; NB, naive Bayes; FKNN, fuzzy k-nearest neighbor; profile HMMs, profile hidden Markov models; MLEM2, modified learning from examples module; IRIM, interesting rule induction module; XGBoost, extreme gradient boosting; PS-RF, pruned sets-random forests; LC-RF, label combination-random forests; ADA, adaboost; LSTM, long short-term memory; CNN, convolutional neural networks; Bi-LSTM, bidirectional LSTM.
A comprehensive summary of the reviewed approaches for AMP prediction
| Type | Tool | Year | Algorithm | Feature selection | Evaluation strategy | Web server availability | Max data upload | File upload availability | Email of result | Software availability |
|---|---|---|---|---|---|---|---|---|---|---|
| Machine learning-based methods | AMPer | 2007 | HMMs | None | 10-fold CV | No | N.A. | N.A. | N.A. | Yes |
| CAMP | 2010, 2016 | SVM, RF, ANN, DA | RFE (RF Gini) | 10-fold CV, independent test | Yes | N.A. | Yes | No | No | |
| Porto et al. | 2010 | SVM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Song et al. | 2011 | k-NN, BLASTP | mRMR, IFS | Jack-knife validation, independent test | No | N.A. | N.A. | N.A. | No | |
| Torrent et al. | 2011 | ANN | None | Independent test | No | N.A. | N.A. | N.A. | No | |
| Fernandes et al. | 2012 | ANFIS | ANFIS | Independent test | No | N.A. | N.A. | N.A. | No | |
| ClassAMP | 2012 | RF, SVM | RFE (RF Gini) | 10-fold CV, independent test | Yes | N.A. | Yes | No | No | |
| CS-AMPPred | 2012 | SVM | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | Yes | |
| C-PAmP | 2013 | SVM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| iAMP-2L | 2013 | FKNN | None | Jack-knife validation, independent test | Yes | 500 | No | No | No | |
| Paola et al. | 2013 | SVM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Randou et al. | 2013 | LR | None | Independent test | No | N.A. | N.A. | N.A. | No | |
| dbaasp | 2014 | z-Score | None | Independent test | No | N.A. | N.A. | N.A. | No | |
| ADAM | 2015 | SVM, profile HMMs | None | N.A. | Yes | N.A. | No | NO | No | |
| Camacho et al. | 2015 | NB, RF, SVM | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Ng et al. | 2015 | SVM, BLASTP | None | Jack-knife validation, independent test | No | N.A. | N.A. | N.A. | No | |
| MLAMP | 2016 | RF | None | Jack-knife validation, independent test | Yes | 5 | Yes | Yes | No | |
| iAMPpred | 2017 | SVM | None | 10-fold CV, independent test | Yes | N.A. | No | No | No | |
| AmPEP | 2018 | RF | None | 10-fold CV, independent test | Yes | N.A. | Yes | Yes | Yes | |
| CLN-MLEM2 | 2018 | MLEM2, IRIM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| MOEA-FW | 2018 | RF, k-NN, SVM, ANN | None | 10-fold CV | No | N.A. | N.A. | N.A. | No | |
| AMAP | 2019 | SVM, XGBoost, one-versus-rest classifier fusion | None | LOCO, 5-fold CV, independent test | Yes | N.A. | No | No | No | |
| MAMPs-Pred | 2019 | RF, LC-RF, PS-RF | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| dbAMP | 2019 | RF | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| AMPfun | 2019 | DT, RF, SVM | FS | 10-fold CV, independent test | Yes | N.A. | No | No | No | |
| Ampir | 2020 | SVM | RFE | independent test | No | N.A. | N.A. | N.A. | Yes | |
| Chung et al. | 2020 | RF | OneR, FS | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Fu et al. | 2020 | ADA | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| AmpGram | 2020 | RF | None | 5-fold CV, benchmark test | Yes | 50 | Yes | No | Yes | |
| IAMPE | 2020 | RF, SVM, XGBoost, k-NN, NB | None | 10-fold CV, independent test | Yes | N.A. | Yes | No | No | |
| Deep learning-based methods | AMP Scanner V2 | 2018 | LSTM | None | 10-fold CV, independent test | Yes | 50 000 | Yes | No | No |
| APIN | 2019 | CNN | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | Yes | |
| Deep-AmPEP30 | 2020 | CNN | None | 10-fold CV, independent test | Yes | N.A. | Yes | Yes | No | |
| AMPlify | 2020 | Bi-LSTM, attention mechanism | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | Yes |
| Type | Tool | Year | Algorithm | Feature selection | Evaluation strategy | Web server availability | Max data upload | File upload availability | Email of result | Software availability |
|---|---|---|---|---|---|---|---|---|---|---|
| Machine learning-based methods | AMPer | 2007 | HMMs | None | 10-fold CV | No | N.A. | N.A. | N.A. | Yes |
| CAMP | 2010, 2016 | SVM, RF, ANN, DA | RFE (RF Gini) | 10-fold CV, independent test | Yes | N.A. | Yes | No | No | |
| Porto et al. | 2010 | SVM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Song et al. | 2011 | k-NN, BLASTP | mRMR, IFS | Jack-knife validation, independent test | No | N.A. | N.A. | N.A. | No | |
| Torrent et al. | 2011 | ANN | None | Independent test | No | N.A. | N.A. | N.A. | No | |
| Fernandes et al. | 2012 | ANFIS | ANFIS | Independent test | No | N.A. | N.A. | N.A. | No | |
| ClassAMP | 2012 | RF, SVM | RFE (RF Gini) | 10-fold CV, independent test | Yes | N.A. | Yes | No | No | |
| CS-AMPPred | 2012 | SVM | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | Yes | |
| C-PAmP | 2013 | SVM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| iAMP-2L | 2013 | FKNN | None | Jack-knife validation, independent test | Yes | 500 | No | No | No | |
| Paola et al. | 2013 | SVM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Randou et al. | 2013 | LR | None | Independent test | No | N.A. | N.A. | N.A. | No | |
| dbaasp | 2014 | z-Score | None | Independent test | No | N.A. | N.A. | N.A. | No | |
| ADAM | 2015 | SVM, profile HMMs | None | N.A. | Yes | N.A. | No | NO | No | |
| Camacho et al. | 2015 | NB, RF, SVM | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Ng et al. | 2015 | SVM, BLASTP | None | Jack-knife validation, independent test | No | N.A. | N.A. | N.A. | No | |
| MLAMP | 2016 | RF | None | Jack-knife validation, independent test | Yes | 5 | Yes | Yes | No | |
| iAMPpred | 2017 | SVM | None | 10-fold CV, independent test | Yes | N.A. | No | No | No | |
| AmPEP | 2018 | RF | None | 10-fold CV, independent test | Yes | N.A. | Yes | Yes | Yes | |
| CLN-MLEM2 | 2018 | MLEM2, IRIM | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| MOEA-FW | 2018 | RF, k-NN, SVM, ANN | None | 10-fold CV | No | N.A. | N.A. | N.A. | No | |
| AMAP | 2019 | SVM, XGBoost, one-versus-rest classifier fusion | None | LOCO, 5-fold CV, independent test | Yes | N.A. | No | No | No | |
| MAMPs-Pred | 2019 | RF, LC-RF, PS-RF | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| dbAMP | 2019 | RF | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| AMPfun | 2019 | DT, RF, SVM | FS | 10-fold CV, independent test | Yes | N.A. | No | No | No | |
| Ampir | 2020 | SVM | RFE | independent test | No | N.A. | N.A. | N.A. | Yes | |
| Chung et al. | 2020 | RF | OneR, FS | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| Fu et al. | 2020 | ADA | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | No | |
| AmpGram | 2020 | RF | None | 5-fold CV, benchmark test | Yes | 50 | Yes | No | Yes | |
| IAMPE | 2020 | RF, SVM, XGBoost, k-NN, NB | None | 10-fold CV, independent test | Yes | N.A. | Yes | No | No | |
| Deep learning-based methods | AMP Scanner V2 | 2018 | LSTM | None | 10-fold CV, independent test | Yes | 50 000 | Yes | No | No |
| APIN | 2019 | CNN | None | 10-fold CV, independent test | No | N.A. | N.A. | N.A. | Yes | |
| Deep-AmPEP30 | 2020 | CNN | None | 10-fold CV, independent test | Yes | N.A. | Yes | Yes | No | |
| AMPlify | 2020 | Bi-LSTM, attention mechanism | None | 5-fold CV, independent test | No | N.A. | N.A. | N.A. | Yes |
Full names of the algorithms: N.A., not available; HMMs, hidden Markov models; SVM, support vector machine; RF, random forest; ANN, artificial neural networks; DA, discriminant analysis; DT, decision tree; LR, logistic regression; k-NN, k-nearest neighbor; BLASTP, basic local alignment search tool (protein); ANFIS, adaptive neuro-fuzzy inference system; NB, naive Bayes; FKNN, fuzzy k-nearest neighbor; profile HMMs, profile hidden Markov models; MLEM2, modified learning from examples module; IRIM, interesting rule induction module; XGBoost, extreme gradient boosting; PS-RF, pruned sets-random forests; LC-RF, label combination-random forests; ADA, adaboost; LSTM, long short-term memory; CNN, convolutional neural networks; Bi-LSTM, bidirectional LSTM.
The AMP identification task can be generally classified into two categories. The first category formulates the AMP identification problem as a binary classification task. The most common classification task is to determine whether a peptide is an AMP or not, and the majority of methods listed in Table 1 belong to this category. The second category treats the AMP identification problem as a multi-classification task, with the goal to determine the specific functional types of AMPs, such as AMPs targeting Gram-positive bacteria, or AMPs targeting Gram-negative bacteria or other AMPs. In this work, we mainly focus on the review, survey and benchmarking of the first category and simply discuss the second category. For AMP identification, we classify approaches into two groups based on the adopted computational methodology. The first group is based on traditional machine learning algorithms that use sequence-derived features to train the models. According to our survey, most tools listed in Table 1 employ the first category of approaches to build their prediction models. In recent years, deep learning has become wildly popular due to its strong ability to learn and extract informative features. Accordingly, the second group of approaches is developed based on deep learning. A timeline and a general flow chart of computational methods for AMP prediction are shown in Figures 1 and 2, respectively.
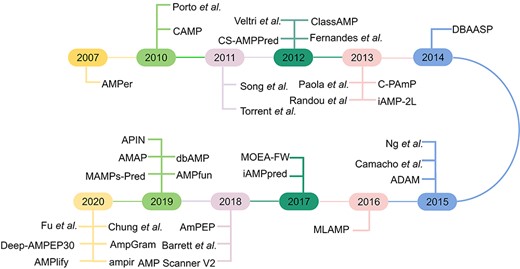
Timeline of current computational approaches for AMP prediction.
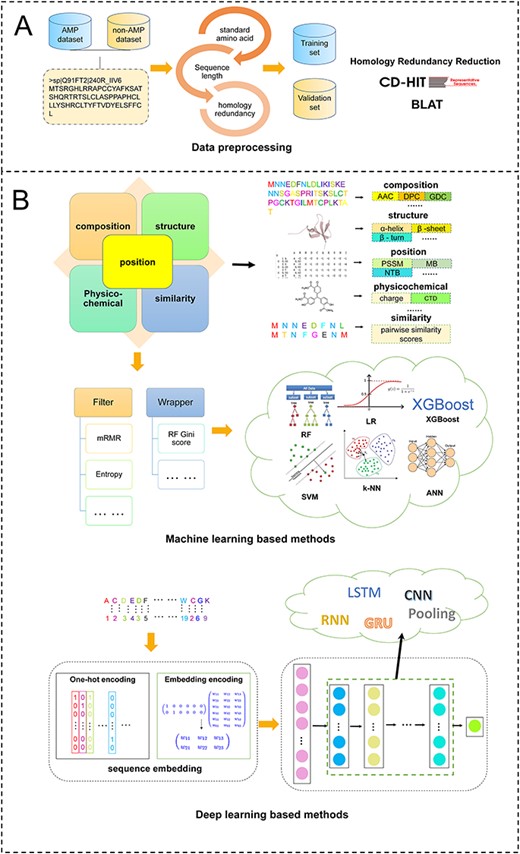
Overview of current computational approaches for AMP prediction. (A) pre-processing to build data set, (B) structure of the CNNs used in this study.
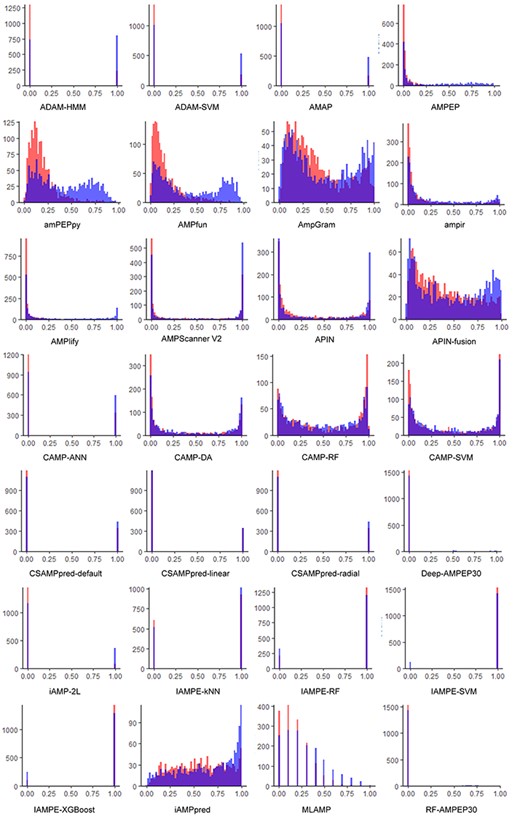
Prediction distributions of tests of reviewed computational approaches using the independent test data set. Red: non-AMPs; Blue: AMPs.
Features calculated and extracted for model construction
Extracting feature information from the sequence is crucial for building a computational model, and there is a lot of work on feature construction [11, 12, 24, 36–42]. In order to construct robust and accurate approaches for AMP prediction, various features have been designed and extracted for encoding the peptide sequences. According to these features’ characteristics, we identified five major types of features in current computational approaches for AMP prediction (Table 2). These feature types are: (1) composition features, (2) position features, (3) structure features, (4) physicochemical properties and (5) similarity features. As part of our survey, we collected the most representative features for each type. Amino acid composition (AAC) and pseudo amino acid composition (PseAAC) are the most commonly used features, although some structure features are also often used. Composition features can be calculated based on the peptide sequence and are easy to obtain, while some features require third-party software. For instance, structure features can be calculated using the software Tango [43]. After feature encoding, the initial feature set sometimes has a high dimensionality, which may result in biased model training. Therefore, feature selection needs to be performed to reduce the dimensionality of the initial feature set before construction of the computational models.
Different types of features employed by the reviewed approaches for AMP prediction
| Feature type | Feature | Reference |
|---|---|---|
| Composition features | AAC | [20, 28, 64, 82, 83, 98, 103, 104, 112, 113] |
| Normalized amino acid composition (NAAC) | [20] | |
| AAPC | [105] | |
| DPC | [3, 18, 64, 71, 112, 122] | |
| TPC | [3, 18, 64] | |
| Peptide length | [49, 89] | |
| N-gram composition found by counting (NCC) | [103] | |
| N-gram composition found by t-test (NTC) | [103] | |
| Motifs composition (MC) | [103] | |
| Position features | N-gram binary profiling of position found by counting (NCB) | [103] |
| N-gram binary profiling of position found by t-test (NTB) | [103] | |
| Motifs binary profiling of position (MB) | [103] | |
| PSSM profile | [108] | |
| CMV | [71] | |
| Structure features | α-Helix | [20, 49–51, 64, 65, 85, 89, 98] |
| β-Sheet | [20, 49–51, 64, 65, 85, 89, 98] | |
| β-Turn | [20, 49–51, 64, 85, 89, 98] | |
| Loop formation | [65] | |
| Random coil | [51, 85] | |
| Physicochemical properties | Isoelectric point | [20, 49, 81, 85] |
| Charge | [3, 18, 20, 51, 65, 81, 98] | |
| Molecular mass | [50] | |
| Atom count | [50] | |
| Size | [81] | |
| Amino acid acidity and basicity | [81] | |
| Aromaticity | [81] | |
| Sulfur | [81] | |
| Oxygen, nitrogen, hydrogen and carbon atom contents | [81] | |
| Net charge at the physiological pH | [63] | |
| μH | [63] | |
| Aliphatic index | [3, 18, 81] | |
| Amphipathicity | [51, 65] | |
| Ratio between hydrophobic and charged residues | [63] | |
| Hydrophilicity | [3, 18] | |
| Hydrophobic moment | [63, 65] | |
| Hydrophobicity | [3, 18, 20, 51, 63, 65, 89, 98] | |
| Instability index | [3, 18] | |
| Disordering | [51] | |
| Solvent accessibility | [98] | |
| Surface tension | [98] | |
| Normalized van der Waals volume | [64, 98] | |
| Conformational similarity | [64] | |
| Polarity | [64, 81, 98] | |
| Polarizability | [64, 98] | |
| Flexibility | [65] | |
| Normalized MoreauBroto autocorrelation (NMBroto) | [71] | |
| Moran autocorrelation (Moran) | [71] | |
| Geary autocorrelation (Geary) | [71] | |
| CNMR | [81] | |
| Composition/transition/distribution (CTD) | [3, 18, 64, 71, 92, 95, 98, 103, 104] | |
| PseAAC | [19, 20, 66, 71, 80, 83, 96, 98, 103, 113] | |
| PseKRAAC | [113] | |
| 3-mer composition | [82] | |
| Sequence order coupling number | [71] | |
| Quasi sequence order | [71] | |
| Similarity features | BLOSUM-50 | [3, 18, 64] |
| LZ complexity pairwise similarity scores | [74] | |
| In vivo propensity | In vivo aggregation propensity | [85, 89] |
| In vivo stability | [64] |
| Feature type | Feature | Reference |
|---|---|---|
| Composition features | AAC | [20, 28, 64, 82, 83, 98, 103, 104, 112, 113] |
| Normalized amino acid composition (NAAC) | [20] | |
| AAPC | [105] | |
| DPC | [3, 18, 64, 71, 112, 122] | |
| TPC | [3, 18, 64] | |
| Peptide length | [49, 89] | |
| N-gram composition found by counting (NCC) | [103] | |
| N-gram composition found by t-test (NTC) | [103] | |
| Motifs composition (MC) | [103] | |
| Position features | N-gram binary profiling of position found by counting (NCB) | [103] |
| N-gram binary profiling of position found by t-test (NTB) | [103] | |
| Motifs binary profiling of position (MB) | [103] | |
| PSSM profile | [108] | |
| CMV | [71] | |
| Structure features | α-Helix | [20, 49–51, 64, 65, 85, 89, 98] |
| β-Sheet | [20, 49–51, 64, 65, 85, 89, 98] | |
| β-Turn | [20, 49–51, 64, 85, 89, 98] | |
| Loop formation | [65] | |
| Random coil | [51, 85] | |
| Physicochemical properties | Isoelectric point | [20, 49, 81, 85] |
| Charge | [3, 18, 20, 51, 65, 81, 98] | |
| Molecular mass | [50] | |
| Atom count | [50] | |
| Size | [81] | |
| Amino acid acidity and basicity | [81] | |
| Aromaticity | [81] | |
| Sulfur | [81] | |
| Oxygen, nitrogen, hydrogen and carbon atom contents | [81] | |
| Net charge at the physiological pH | [63] | |
| μH | [63] | |
| Aliphatic index | [3, 18, 81] | |
| Amphipathicity | [51, 65] | |
| Ratio between hydrophobic and charged residues | [63] | |
| Hydrophilicity | [3, 18] | |
| Hydrophobic moment | [63, 65] | |
| Hydrophobicity | [3, 18, 20, 51, 63, 65, 89, 98] | |
| Instability index | [3, 18] | |
| Disordering | [51] | |
| Solvent accessibility | [98] | |
| Surface tension | [98] | |
| Normalized van der Waals volume | [64, 98] | |
| Conformational similarity | [64] | |
| Polarity | [64, 81, 98] | |
| Polarizability | [64, 98] | |
| Flexibility | [65] | |
| Normalized MoreauBroto autocorrelation (NMBroto) | [71] | |
| Moran autocorrelation (Moran) | [71] | |
| Geary autocorrelation (Geary) | [71] | |
| CNMR | [81] | |
| Composition/transition/distribution (CTD) | [3, 18, 64, 71, 92, 95, 98, 103, 104] | |
| PseAAC | [19, 20, 66, 71, 80, 83, 96, 98, 103, 113] | |
| PseKRAAC | [113] | |
| 3-mer composition | [82] | |
| Sequence order coupling number | [71] | |
| Quasi sequence order | [71] | |
| Similarity features | BLOSUM-50 | [3, 18, 64] |
| LZ complexity pairwise similarity scores | [74] | |
| In vivo propensity | In vivo aggregation propensity | [85, 89] |
| In vivo stability | [64] |
Different types of features employed by the reviewed approaches for AMP prediction
| Feature type | Feature | Reference |
|---|---|---|
| Composition features | AAC | [20, 28, 64, 82, 83, 98, 103, 104, 112, 113] |
| Normalized amino acid composition (NAAC) | [20] | |
| AAPC | [105] | |
| DPC | [3, 18, 64, 71, 112, 122] | |
| TPC | [3, 18, 64] | |
| Peptide length | [49, 89] | |
| N-gram composition found by counting (NCC) | [103] | |
| N-gram composition found by t-test (NTC) | [103] | |
| Motifs composition (MC) | [103] | |
| Position features | N-gram binary profiling of position found by counting (NCB) | [103] |
| N-gram binary profiling of position found by t-test (NTB) | [103] | |
| Motifs binary profiling of position (MB) | [103] | |
| PSSM profile | [108] | |
| CMV | [71] | |
| Structure features | α-Helix | [20, 49–51, 64, 65, 85, 89, 98] |
| β-Sheet | [20, 49–51, 64, 65, 85, 89, 98] | |
| β-Turn | [20, 49–51, 64, 85, 89, 98] | |
| Loop formation | [65] | |
| Random coil | [51, 85] | |
| Physicochemical properties | Isoelectric point | [20, 49, 81, 85] |
| Charge | [3, 18, 20, 51, 65, 81, 98] | |
| Molecular mass | [50] | |
| Atom count | [50] | |
| Size | [81] | |
| Amino acid acidity and basicity | [81] | |
| Aromaticity | [81] | |
| Sulfur | [81] | |
| Oxygen, nitrogen, hydrogen and carbon atom contents | [81] | |
| Net charge at the physiological pH | [63] | |
| μH | [63] | |
| Aliphatic index | [3, 18, 81] | |
| Amphipathicity | [51, 65] | |
| Ratio between hydrophobic and charged residues | [63] | |
| Hydrophilicity | [3, 18] | |
| Hydrophobic moment | [63, 65] | |
| Hydrophobicity | [3, 18, 20, 51, 63, 65, 89, 98] | |
| Instability index | [3, 18] | |
| Disordering | [51] | |
| Solvent accessibility | [98] | |
| Surface tension | [98] | |
| Normalized van der Waals volume | [64, 98] | |
| Conformational similarity | [64] | |
| Polarity | [64, 81, 98] | |
| Polarizability | [64, 98] | |
| Flexibility | [65] | |
| Normalized MoreauBroto autocorrelation (NMBroto) | [71] | |
| Moran autocorrelation (Moran) | [71] | |
| Geary autocorrelation (Geary) | [71] | |
| CNMR | [81] | |
| Composition/transition/distribution (CTD) | [3, 18, 64, 71, 92, 95, 98, 103, 104] | |
| PseAAC | [19, 20, 66, 71, 80, 83, 96, 98, 103, 113] | |
| PseKRAAC | [113] | |
| 3-mer composition | [82] | |
| Sequence order coupling number | [71] | |
| Quasi sequence order | [71] | |
| Similarity features | BLOSUM-50 | [3, 18, 64] |
| LZ complexity pairwise similarity scores | [74] | |
| In vivo propensity | In vivo aggregation propensity | [85, 89] |
| In vivo stability | [64] |
| Feature type | Feature | Reference |
|---|---|---|
| Composition features | AAC | [20, 28, 64, 82, 83, 98, 103, 104, 112, 113] |
| Normalized amino acid composition (NAAC) | [20] | |
| AAPC | [105] | |
| DPC | [3, 18, 64, 71, 112, 122] | |
| TPC | [3, 18, 64] | |
| Peptide length | [49, 89] | |
| N-gram composition found by counting (NCC) | [103] | |
| N-gram composition found by t-test (NTC) | [103] | |
| Motifs composition (MC) | [103] | |
| Position features | N-gram binary profiling of position found by counting (NCB) | [103] |
| N-gram binary profiling of position found by t-test (NTB) | [103] | |
| Motifs binary profiling of position (MB) | [103] | |
| PSSM profile | [108] | |
| CMV | [71] | |
| Structure features | α-Helix | [20, 49–51, 64, 65, 85, 89, 98] |
| β-Sheet | [20, 49–51, 64, 65, 85, 89, 98] | |
| β-Turn | [20, 49–51, 64, 85, 89, 98] | |
| Loop formation | [65] | |
| Random coil | [51, 85] | |
| Physicochemical properties | Isoelectric point | [20, 49, 81, 85] |
| Charge | [3, 18, 20, 51, 65, 81, 98] | |
| Molecular mass | [50] | |
| Atom count | [50] | |
| Size | [81] | |
| Amino acid acidity and basicity | [81] | |
| Aromaticity | [81] | |
| Sulfur | [81] | |
| Oxygen, nitrogen, hydrogen and carbon atom contents | [81] | |
| Net charge at the physiological pH | [63] | |
| μH | [63] | |
| Aliphatic index | [3, 18, 81] | |
| Amphipathicity | [51, 65] | |
| Ratio between hydrophobic and charged residues | [63] | |
| Hydrophilicity | [3, 18] | |
| Hydrophobic moment | [63, 65] | |
| Hydrophobicity | [3, 18, 20, 51, 63, 65, 89, 98] | |
| Instability index | [3, 18] | |
| Disordering | [51] | |
| Solvent accessibility | [98] | |
| Surface tension | [98] | |
| Normalized van der Waals volume | [64, 98] | |
| Conformational similarity | [64] | |
| Polarity | [64, 81, 98] | |
| Polarizability | [64, 98] | |
| Flexibility | [65] | |
| Normalized MoreauBroto autocorrelation (NMBroto) | [71] | |
| Moran autocorrelation (Moran) | [71] | |
| Geary autocorrelation (Geary) | [71] | |
| CNMR | [81] | |
| Composition/transition/distribution (CTD) | [3, 18, 64, 71, 92, 95, 98, 103, 104] | |
| PseAAC | [19, 20, 66, 71, 80, 83, 96, 98, 103, 113] | |
| PseKRAAC | [113] | |
| 3-mer composition | [82] | |
| Sequence order coupling number | [71] | |
| Quasi sequence order | [71] | |
| Similarity features | BLOSUM-50 | [3, 18, 64] |
| LZ complexity pairwise similarity scores | [74] | |
| In vivo propensity | In vivo aggregation propensity | [85, 89] |
| In vivo stability | [64] |
Feature selection strategy
As previously mentioned, before model construction, feature selection is a nontrivial step that measures the importance of all the features and eliminates the less-informative ones. Six of the 34 surveyed predictors in Table 1 adopted the feature selection procedure. The commonly used feature selection algorithms include the incremental feature selection (IFS) [44, 45] method based on maximum relevance minimum redundancy (mRMR) [46], fast correlation-based filter (FCBF) method based on entropy [47], rigorous recursive feature elimination (RFE) method based on RF Gini score [48] and forward selection method based on the one-rule attribute evaluation (OneR) method. There are also studies focusing on new feature selection methods for AMP prediction, such as Fernandes et al. [49] and MOEA-FW [50].
Machine learning-based AMP predictors
As listed in Table 1, in addition to dbaasp [51] which optimizes the features by z-score for predicting linear cationic AMPs, and most computational approaches for AMP identification are developed based on well-established machine learning algorithms. These algorithms include the hidden Markov model (HMM) [52], SVM [21], RF [22], eXtreme Gradient Boosting (XGBoost) [53], discriminant analysis (DA) [54], decision tree (DT) [55], Bayesian network (BN) [56], fuzzy k-nearest neighbor (fuzzy k-NN) [57], artificial neural network (ANN) [58], logistic regression (LR) [58] and AdaBoost (ADA) [59]. Based on our survey, SVM and RF stand out as the two most commonly used machine learning algorithms (Table 1). Next, we will briefly describe these tools.
HMM-based predictors
AMPer [52] is a database and an automated discovery tool for AMPs, which is based on HMM models. First, an initial AMP set was constructed by performing a pairwise comparison between the AMPs in its reference data set and the peptides from the Uniprot database (Swiss-Prot and TrEMBL) using Basic Local Alignment Search Tool (BLAST) [60]. Clusters of similar peptides were constructed based on pairwise alignments using BLAST by setting a threshold value, and multiple alignments were created for each cluster using ClustalW [61]. With these clusters, HMMs were created using the HMM software (hmmer) [62]. Through these HMMs, new AMPs can be identified and added to these clusters according to the matching scores, and the HMMs are updated.
SVM-based predictors
This model only considered the order information of the amino acids in peptide sequences without any physicochemical information.
ADAM [26] is another public AMP database built to systematically establish comprehensive associations between AMP sequences and structures and to provide easy access to view their relationships. It also provides two computational tools based on SVM and HMM to predict the AMPs.
Of course, the issue of unbalanced data sets could also have an impact on AMP prediction. To address this, Camacho et al. extracted 10 groups of features from peptide sequences [71], 9 groups of physicochemical properties using Propy [72] plus the composition moment vector (CMV) [73]. Using these 10 feature groups, they constructed 10 data sets for training, built models based on SVM, RF and NB algorithms with unbalanced data sets and compared their AMP identification performances.
Ng et al. [74] proposed an AMP prediction approach, which, as a first step, classifies a peptide by comparing the maximum of high-scoring segment pairs (HSP) scores with BLASTP. To classify those peptides that cannot be classified by BLASTP [60], Ng et al. instead employed the SVM-LZ complexity pairwise algorithm [75–77]. Due to the fixed number of letters in sequences, LZ complexity is suitable for calculating the distance between AMPs.
iAMPpred [20] is a tool for predicting antibacterial, antiviral and antifungal peptides using three different categories of features, including compositional, structural and physicochemical properties. It is developed based on three SVM models with the RBF kernel. In order to identify the importance of each feature for predicting antibacterial, antiviral or antifungal peptides, the information gain was computed for all the features. The differences in the predictive performance among different feature combinations were discussed as well.
Additionally, Ampir [80] is an R package that employed two SVM with the RBF kernel classifiers for AMP prediction including two built-in models, one trained on peptide data (10–50 amino acids) and another trained on full-length precursor protein sequences data. And IAMPE [81] is a recent AMP predictive web server which utilized the clusters of CNMR spectral of amino acids and several physicochemical properties for identifying AMPs based on SVM, k-NN, NB, RF and XGBoost.
In addition, there are some approaches that can not only identify whether a peptide is an AMP or non-AMP but can also identify which functional type it belongs to. AMAP [82] is such an AMP prediction tool which has two prediction levels. The first level used SVM to predict AMPs, and the second level used one-versus-rest classifier fusion and SVM to predict the type of the biological activity of a peptide and the effect of mutations on its biological activity.
k-NN-based predictors
Song et al.’ s work employed BLASTP [60] and the k-NN algorithm for AMP prediction [83]. This approach consisted of two stages. First, BLASTP was employed to identify AMPs with HSP scores. However, BLASTP was unable to deal with certain sequences that had no overlap with the training sequences. For those sequences, Song et al. used the k-NN algorithm. To this end, 270 features from each sequence were calculated and the mRMR feature selection algorithm [46] was employed. The value of k was set to 1 so that unknown samples were assigned to the class of their nearest neighbor.
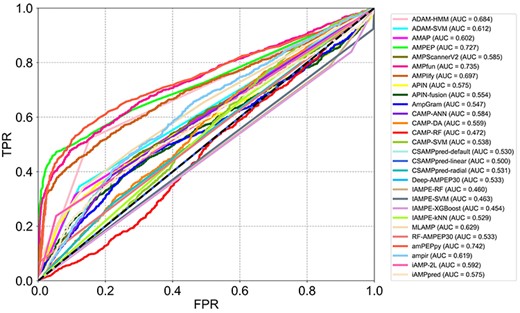
ROC curves and the corresponding AUC values of tests of reviewed computational approaches on the independent test data set.
ANN-based predictors
Torrent et al. constructed an ANN structure to classify AMPs [85]. In this work, secondary structure prediction features and physicochemical features were calculated by the TANGO software [43], AGGRESCAN [86], the Expasy reference values [87] and the GRAVY scale [88]. For AMP prediction, an ANN structure containing a two-layer feed-forward network with sigmoid function and 50 nodes in the hidden layer were used.
LR-based predictors
Considering that thousands of sequence features are irrelevant and redundant and that some might even mislead the correct classification of the peptide activity, some studies instead focus on feature analysis and feature selection. Randou et al. [89] used TANGO [43], AGGRESCAN [86], ExPASy [87] and the GRAVY scale [88] to obtain eight features for prediction. The statistical significance of each of these features was measured by two-sample randomization tests, upon which seven of them were chosen. Features were further selected if their estimated coefficient was significantly different from 0 at α = 0.5 in the LR model. After that, the Akaike Information Criterion (AIC) [90] measure was employed to choose the best model, and the Brier score [91] was used to measure the overall prediction accuracy.
RF-based predictors
In addition to the approaches described above, there are also some approaches employing RF algorithms. AmPEP [92] is an AMP prediction approach based on the RF algorithm with 100 trees, which uses numerical descriptors to characterize different properties of peptides converted from their amino acid sequences [93]. AmPEP employed the synthetic minority over-sampling technique (SMOTE) [94] to rebalance the proportion of positive and negative samples to improve prediction performance. The descriptors were additionally split into three subsets based on the Pearson correlation coefficient (PCC) analysis of AMP/non-AMP distributions to further compare their individual importance in the prediction. dbAMP [28] is an AMP database that contains a tool for AMP prediction. This tool employed the RF algorithm with AAC and physicochemical properties to identify AMPs. And amPEPpy [95] is the latest publicly available Python toolkit using a RF classifier and the same features as [92] for predicting AMPs.
By using the numerical value of the five physicochemical properties for each of the 20 amino acids in the same manner as shown in [19], a 30-dimensional feature vector representing a peptide was obtained. For the first predictive level, the RF algorithm was employed for predicting whether a peptide is an AMP or not. For the second level, a multi-label classifier based on the RF algorithm was used. Considering unbalanced functional types of AMPs, ML-SMOTE, a novel oversampling model, was used.
MAMPs-Pred [98] is also a two-level model that predicts AMPs and their functional types based on the RF algorithm. For AMP prediction, 188 features were used based on eight types of physicochemical and AAC properties and were calculated using SVM-Prot [99, 100]. Random under-sampling method [101] and weighted random sampling [102] were used for data balancing.
AMPfun [103] is a recent useful identification web server for AMPs and their functional activities. It is a two-stage framework, and each stage consists of three steps: feature calculation, feature selection and application of classification algorithms. The sequential forward selection algorithm was employed for feature selection, and RF was used as the prediction method.
Instead of predicting an AMP’s functional activities, Chung et al. proposed a method for predicting AMPs from seven categories of organisms, including amphibians, humans, fish, insects, plants, bacteria and mammals [104]. AAC, amino acid pair composition (AAPC) [105] and seven physicochemical properties were used as features representing each peptide. After feature selection based on the OneR [106], RF, SVM, k-NN and DT models were constructed, out of which, the RF model performed best and was selected.
AmpGram [107] is a recent AMP predictor that employed two RF models using n-grams to represent the information hidden in amino acid sequences. First, each sequence in the training data set was scanned with a sliding window of 10 amino acids and was divided into overlapping 10 amino acid long subsequences (10-mers). All 10-mers from the AMPs were then considered as positive samples, whereas all 10-mers from the non-AMPs were labeled as negative samples. N-grams, which are continuous or discontinuous sequences of n elements, were extracted from each 10-mer in the positive and negative data set as features. If an n-gram was present in the sequence, the feature representing the n-gram was 1, otherwise 0. With these features, an RF was trained to predict the antimicrobial property of 10-mers. In order to predict a peptide, several statistics for each peptide using prediction for its 10-mers were calculated. These statistics were used to train the second RF model with 500 trees to decide whether a peptide is an AMP or not.
ADA-based predictors
Deep learning-based AMP predictors
Deep learning architectures are derived from the simpler ANN. Convolutional neural networks (CNNs) [110] and recurrent neural networks (RNNs) [111] are deep learning frameworks that have been previously used to identify AMPs [33, 112–114]. Deep learning-based predictors frequently use original sequences as the input, which are seldom used to train ML-based methods, occasionally add the extracted sequence information by some third-party software, then extract features through several neural network layers and finally output classification labels. The recently developed methods based on deep learning architectures are provided in Table 1.
AMP Scanner v.2 [33], a web server based on a long short-term memory (LSTM) architecture [115], a kind of RNN, was the first to propose a deep neural network (DNN) model for AMP classification. First, peptide sequences were converted into zero-padded numerical vectors of length 200, and each of the 20 essential amino acids was assigned a number from 1 to 20. Then, such vectors were fed into an embedding layer to convert them into a vector representation of fixed size. After that, a CNN layer, a max pooling layer and an LSTM layer were used to further extract features from these vectors. Finally, the label prediction probability was obtained by a fully connected layer with the sigmoid function.
APIN [112] is an AMP identification model mainly based on multi-CNNs. At first, peptide sequences were converted into numerical vectors of fixed length through an embedding layer, in the same way as was done in [33]. Then, these vectors were fed into multiple convolutional layers of different filter lengths to extract several sequence features of different lengths. After two max-pooling layers, the most important feature vector was selected. Finally, the label prediction probability was obtained by a fully connected layer with the sigmoid function. Of note, APIN integrated two kinds of sequence features (i.e. AAC and DPC) to further improve the prediction performance.
Deep-AmPEP30 [113] is another DNN tool for short-length AMP prediction using a deep CNN framework consisting of two conventional layers, two max-pooling layers and a hidden fully connected layer. Different from [33, 113], Deep-AmPEP30 used AAC, composition/transition/distribution (CTD) [116], PseAAC [67–69] and pseudo K-tuple reduced amino acids composition (PseKRAAC) [117] as features fed into the DNN structure. Deep-AmPEP30 has been made publicly available as a web server. This web server also provides another prediction tool based on RF.
AMPlify [114] is a recently proposed DNN approach for AMP identification. Its architecture includes a bidirectional long short-term memory (Bi-LSTM) [118] layer, a multi-head scaled dot-product attention (MHSDPA) [118] layer and a context attention (CA) [119] layer. It uses the one-hot encoding as the input. According to our review, AMPlify appears to be the first DNN tool using the attention mechanism, which can help the deep learning model generate different weights to each part of the input, extract more critical and important information and make the model more accurate.
Performance evaluation strategies and metrics
Performance evaluation strategies including K-fold cross-validation test, jack-knife validation test and independent test are commonly used for performance evaluations of AMP predictors and parameter optimization. For the K-fold cross-validation test, the original data are randomly divided into K parts and, each time, (K − 1) parts are selected as the training set while the remaining part is used as the test set. Cross-validation is repeated K times, and the average of K accuracies is taken as performance metric of the model. Jack-knife validation test (also known as leave-one-out cross-validation test) is commonly used to evaluate models that are trained only on smaller data sets due to the lack of available data in those cases. If there are a total of N samples in the data set, then the predictor is trained on N − 1 training examples and tested on the remaining data point. For a data set with N samples, this procedure is repeated N times, and the final performance is the average of all N tests. In the independent test, the test data set is non-overlapping with the training data of the tools. As a result, an independent test is used as a uniform validation method to test the performance of different methods.
Software availability and usability
Alongside the publications of AMP predictors, user-friendly web servers and/or a locally executable software with proper documentation promote a broader research on AMPs and their application. As part of this survey, we studied the availability and usability of AMP predictors (Table 1) and found that 21 out of 34 approaches were made available as web servers and/or stand-alone software. However, five of the provided server links could not be opened. The general functionalities of the currently available tools are discussed below.
Web servers
For users, the most concerning issue is typically how to upload sequences to web servers. While all web servers allow users to predict the AMP status of multiple peptide sequences at a time, some of them have different limitations on the maximum number of sequences. For instance, the limit on the maximum number for MLAMP is 5, for AMPGram is 50, for iAMP-2L is 500 and for AMP Scanner Vr.2, it is 50 000.
In addition, only a few servers, including CAMP, ClassAMP, MLAMP, AmPEP, Deep-AmPEP30, RF-AmPEP30, AmpGram and AMP scanner Vr.2 provide a button to upload a sequence file in the FASTA format. Among these servers, AMP scanner Vr.2 allows the upload of a file of size ≤50 MB.
Another crucial issue of web server design is how to deliver the prediction results back to the user. Some web servers, including MLAMP, are able to send prediction results to users via an email address provided by the user. Another important functionality aspect is the possibility to revisit historical prediction results (based on the job ID). However, among the reviewed predictors, no web server provided this functionality. Moreover, all web servers allow checking of the prediction results online, but only the AMP scanner Vr.2 allows users to download the prediction results for follow-up analysis.
Stand-alone tools
There are also some stand-alone AMP prediction tools available for users to install and run locally. These tools include CS-AMPPred, AmPEP, ampir, APIN, AmpGram and AMPlify. Among them, ampir and AmpGram are the two user-friendly R packages that come with additional instructions.
Comparison based on the same training data set
Since different tools were trained with different feature descriptors, different feature selection methods and different training sets from different databases, it is objectively inaccurate to evaluate the prediction performances of different machine learning and deep learning methods based on independent data sets. Therefore, in order to further compare and evaluate the performance of different machine learning methods in AMP prediction, we trained different machine learning and deep learning methods based on the same data set.
Data sets
We integrated all AMPs from a set of comprehensive AMP databases and removed those peptide sequences with length greater than 100 or less than 10 and with non-standard residues ‘B’, ‘J’, ‘O’, ‘U’, ‘X’ or ‘Z’. After that, we used the program CD-HIT to filter out those sequences that share 70% or more sequence identity to existing samples, obtaining 10 019 positive samples in total. For the negative data set, we did the same as previously explained in the Construction of the independent test data set section, except that the CD-HIT parameter was set to 70%. We then randomly selected 10 019 negative samples.
Sequence feature generation and machine learning methods
Since peptide sequences cannot be used as inputs in machine learning classifiers, they need to be mapped onto numeric feature vectors. Recently, iFeature [120] and iLearn [121] were created, which are comprehensive Python-based toolkits that can calculate and analyze various features, construct machine learning models and evaluate their performances for classification problems involving deoxyribonucleic acid (DNA), ribonucleic acid (RNA) or protein sequences. Here, we used the iLearn platform to generate several peptide sequence features. These features are AAC, CKSAAP, DPC, TPC, PAAC, CTD, GDPC, GAAC, GTPC, CKSAAGP, Moran, Geary, NMBroto, CTriad, KSCTriad, SOCNumber, QSOrder, APAAC and PseKRAAC.
Considering the huge number of sequence features and the resulting computational complexity, we used the feature selection methods provided in iLearn, including CHI-Square feature selection, information gain feature selection, F-score, mutual information feature selection, recursive feature elimination based on RF (RFE-RF) and Max-Relevance, Min-Redundancy (mRMR), to reduce the number of features to 100.
Subsequently, 11 machine learning methods were used to build AMP predictors, and their performances were evaluated and compared. Specifically, SVM, RF, LR, k-NN, DT, ANN, NB, ADA and XGBoost—all algorithms that had been previously used in the tools reviewed here—were used to identify AMPs before. In addition, a gradient boosting tree (GDBT) model and an extremely randomized trees (ET) model were built and compared. The performances of all models were assessed using 5-fold cross-validation.
Results and Discussion
Performance evaluation based on an independent data set
Considering that the training data sets of some reviewed tools are not available and several other tools have been updated with expanded training data sets since their first release, there might be some overlap between the data sets used for developing some tools and our independent data set. Therefore, we downloaded the training data sets of these tools and removed the peptides that are included in our validation data set to avoid any possible overlaps. Then we submitted the independent data set to these tools and collected the prediction results, which are shown in Figure 3. For this evaluation, the tools’ parameters were set to the recommended configurations in the corresponding publications, or to the default values if no recommendations were given. Six commonly used metrics, namely AUC, Sn, Sp, Pr, Acc and F-score, were previously utilized to assess the performance of the different tools. Consequently, we also used these metrics to evaluate the tools on our independent data set. In order to illustrate the prediction performance of each tool, ROC curves and other metrics are shown in Figure 4 and Supplementary Table S1 available online at http://bib.oxfordjournals.org/. The results show that amPEPpy achieved the best AUC value (74.2%) among all tools, while ADAM–HMM achieved the highest accuracy and the highest F-score. On one hand, amPEPpy used a range of AMP databases, including APD3 [17], CAMPR3 [18] and LAMP [14] for constructing the positive training data set, which was larger than those used by some other approaches. We conclude that use of abundant data is one of the reasons why amPEPpy predicted AMPs more accurately than other approaches. In addition, amPEPpy also randomly subsampled sequences in five different sequence length distributions to match the proportions of the AMP data to obtain a more balanced training set. All these steps for the training data set pre-processing contributed to the performance improvement. On the other hand, amPEPpy used the out-Of-bag (OOB) error to optimize the RF model parameters to achieve a better predictive performance. ADAM–HMM performs a kind of sequence alignment analysis and thus has a great advantage in identifying non-AMPs. This may explain the much higher Sn score compared to amPEPpy. In contrast, ADAM–HMM could not identify AMPs accurately compared with some other AMP predictors, such as amPEPpy, because ADAM–HMM achieved a lower Pr score than amPEPpy.
We collected AMPs from multiple AMP databases to construct the independent test data set while trying our best to remove those samples that might appear in the training sets of all approaches. However, most of the current approaches were trained based on only one or a few public AMP databases. Therefore, these approaches showed worse performance of AMP prediction on the independent data set constructed by us. Some approaches had high false-positive rates, such as IAMPE and AmpGram, while some approaches had high false-negative rates, such as ADAM–HMM.
Performance evaluation and comparison for AMPs with different functional activities
It is well known that AMPs have specific functional activities, and these AMPs with specific functions have attracted more interest and attention of biologists. On one hand, due to the limitation of experimental techniques, a large number of AMPs cannot be annotated with specific functions. On the other hand, from a computational perspective, the number of AMPs with different functions is highly unbalanced. Accordingly, it is a difficult and challenging task to predict the specific functional activities of AMPs.
Currently, most computational methods are developed to predict AMPs. Considering that AMPs with different functions differ from each other in sequences, secondary structure and physicochemical properties, these differences will result in the differences in predictive difficulty. We thus evaluated the predictive performances and compared the predictive differences of these approaches.
First, we counted the number of AMPs with different functions in the independent data set, and we have summarized the 12 common functional activities in Supplementary Table S2 available online at http://bib.oxfordjournals.org/. Based on the 12 functional groups, we calculated the Acc values of different computational approaches and generated bar plots, as shown in Figure 5). For AMPs with anti-Gram-positive, antibacterial, anticancer, antifungal, antitumor or antiviral functional activities, IAMPE–SVM was observed to be the most accurate approach that can predict whether such peptides are AMPs. For AMPs with anti-Gram-negative or antibiofilm activities, IAMPE–XGBoost achieved the best predictive performance. Meanwhile, IAMPE based on four different machine learning methods performed quite well for identifying AMPs with antiparasitic or antiproliferative functions. In addition, CAMP–DA, CAMP–SVM and CAMP–RF also performed as well as IAMPE when they were used to predict AMPs with antiproliferative activities. IAMPE–SVM and IAMPE–XGBoost also achieved the best performance for predicting AMPs with insecticidal activities. Additionally, AMPfun achieved the highest Acc score for AMPs with the antiangiogenic type.
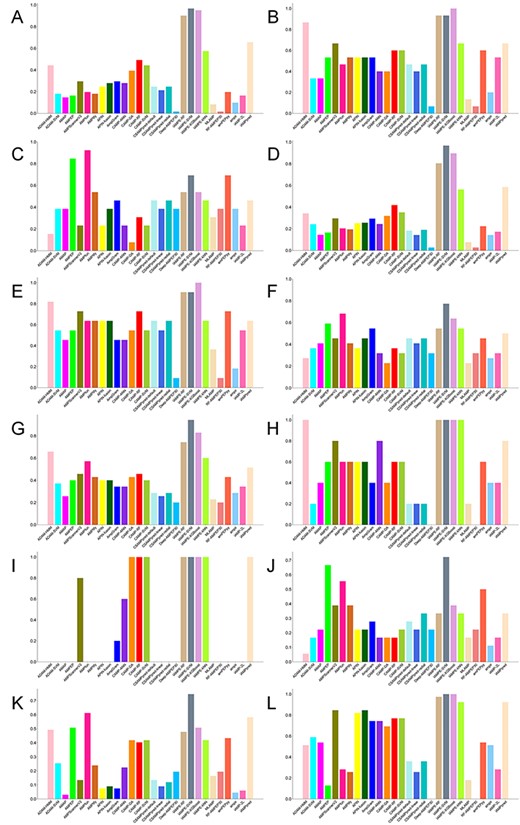
Bar plots comparing accuracy of reviewed computational approaches for AMP identification on 12 common functional types of the independent test data set. (A) anti-Gram-positive, (B) anti-Gram-negative, (C) antiangiogenic, (D) antibacterial, (E) antibiofilm, (F) anticancer, (G) antifungal, (H) antiparasitic, (I) antiproliferative, (J) antitumor, (K) antiviral and (L) insecticidal.
Furthermore, we evaluated those approaches that can be used to predict an AMP’s specific functional activity. Similarly, we calculated the accuracy values of these approaches with respect to different functional groups and have provided the results in Supplementary Table S3 available online at http://bib.oxfordjournals.org/. It can be seen that ClassAMP and iAMPpred performed better than the other compared approaches in this task. One reason is that ClassAMP and iAMPpred can often predict an AMP’s function based on the assumption that a peptide is an AMP. However, although the two approaches can predict functional activities accurately (Supplementary Table S3 available online at http://bib.oxfordjournals.org/), they cannot predict AMPs very well due to their high false-positive rates because these tools have low AUC and Acc values when testing on the independent data set constructed by us.
Performance evaluation based on six validation data sets of different databases
Since our independent test set was integrated from many AMP databases, prediction differences of individual methods across different databases could not be identified. In order to investigate the prediction differences, we tested these approaches on six data sets constructed from six commonly used AMP databases. We used ROC curves and AUC, Sn, Sp, Pr, Acc, MCC and F-score to measure the predictive performance of the different methods. The results are shown in Figure 6 and Supplementary Tables S4–S9 available online at http://bib.oxfordjournals.org/.
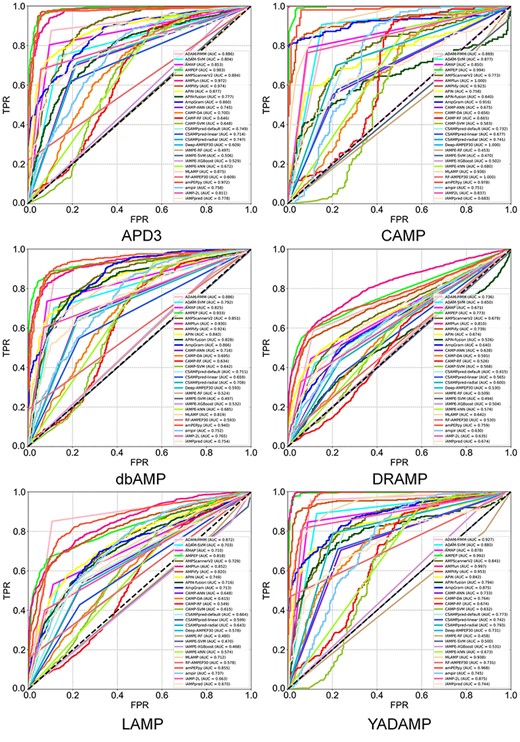
ROC curves and the corresponding AUC values of tests of reviewed computational approaches on six validation data sets.
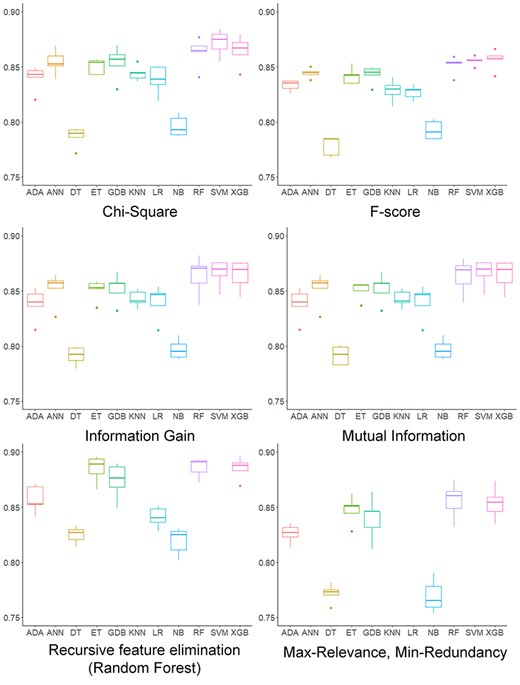
Boxplots comparing accuracy of 11 traditional machine learning methods based on 6 feature selection methods using 5-fold cross validation test.
When tested on the validation data set constructed from APD3, AMPEP achieved the largest AUC, while amPEPpy achieved the largest Acc. AMPfun and AMPlify also achieved high AUC values. When tested on the validation data set constructed from CAMP, both RF–AMPEP30 and Deep-AMPEP30 achieved the best performance. When tested on the validation data set constructed from dbAMP, amPEPpy outperformed all other methods, achieving the highest AUC value and Acc value. When tested on the validation data set built from DRAMP, AMPfun performed best with highest AUC value, and it is the only tool with an AUC value of more than 80%. When tested on the validation data set built from LAMP, ADAM–HMM was considered the best AMP predictor based on achieving the highest AUC value. Lastly, when tested on the validation data set built from YADAMP, AMPfun performed better than all other methods.
Performance evaluation for different machine learning methods based on 5-fold cross-validation
Because the methods are all trained on different training sets, the difference in predictions is partly caused by the training set. We first performed 5-fold cross-validation to compare the traditional machine learning methods on the same data set. The positive data set was collected from multiple AMP data sets, and the redundant information was identified and removed using CD-HIT. We then tested 11 machine learning methods using six different feature selection methods. Boxplots for accuracy values of these methods are shown in Figure 7, and boxplots for other metrics are shown in Supplementary Figures S2–S6 available online at http://bib.oxfordjournals.org/. Using feature selection based on CHI-Square and mutual information, SVM achieved the highest accuracy score. Upon using feature selection based on information gain, Max-Relevance, Min-Redundancy (mRMR) and using RFE-RF, RF got the highest accuracy score. When the F-score feature selection was used, XGBoost performed best in terms of accuracy. Despite the feature selection method used, RF and XGBoost usually achieved a competitive if not the best performance for AMP prediction.
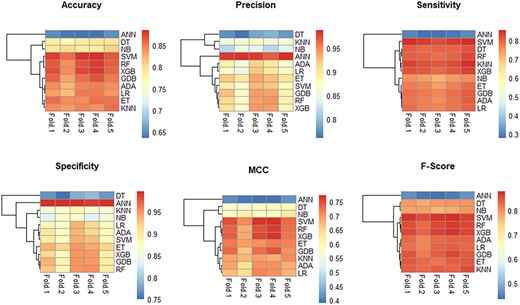
Heatmaps for different metrics of 11 traditional machine learning methods based on CHI-2 square feature selection methods. NB, naive Bayes; XGB, XGBoost; GDB: gradient boosting DT.
In addition, we clustered these machine learning methods based on accuracy, precision, sensitivity, specificity, MCC and F-score of the 5-fold cross-validation test to compare their similarity. The clustering results are shown in Figure 8 and Supplementary Figures S7–S11 available online at http://bib.oxfordjournals.org/. According to these heatmaps, prediction performances of RF and XGBoost are considered to be similar.
Performance evaluation for feature importance
According to the 5-fold cross-validation results of the previous section, we further compared the importance of different feature contributions. First of all, we compared the types of 100 features selected by different feature selection methods and the number of each type of feature. Then, because RF and XGBoost yielded the best performances, we compared the contributions of the selected features to these two methods. Results are shown in Figure 9. For feature selection based on CHI-Square, F-score, information gain and mutual information, PseKRAAC turned out to be the most informative feature as it contributes significantly to RF’s and XGBoost’s prediction. In terms of the number of selected features, for feature selection based on mRMR, CKSAAP features were most selected, while in terms of the feature importance for RF and XGBoost, the most informative and contributing feature type is PAAC. For RFE based on RF, CTDD is the most contributing feature for RF and XGBoost.
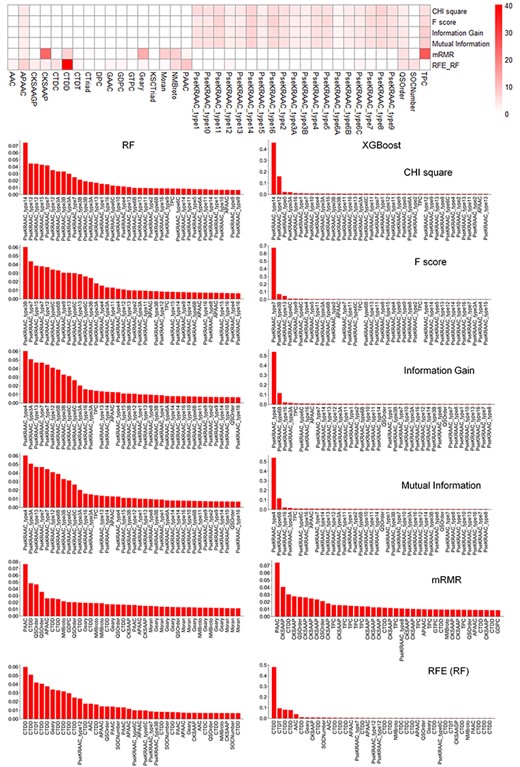
A heatmap for the contributions of features based on different feature selection methods and two contribution distributions of selected features based on RF and XGBoost methods.
Furthermore, we selected 10 AMPs from the Uniprot database which had been manually annotated in 2019 and 2020 and tested them with different tools (Supplementary Table S10 available online at http://bib.oxfordjournals.org/). As a result, IAMPE–RF, IAMPE–SVM, IAMPE–XGBoost and AmpGram predicted AMPs quite correctly with only one mistake. Notably, all predictions of ADAM–HMM were wrong, implying that ADAM–HMM has certain problems, especially for predicting previously unidentified AMPs.
How to develop effective feature extraction and enhance the representation of the learning samples remains a challenging but important question. In this regard, combining feature engineering and representation learning techniques might provide a potentially useful strategy for the AMP peptide representation and identification [11, 12, 24, 36, 39–42]. Based on some common features calculated from sequences, this paper preliminarily compared the prediction results of different traditional machine learning methods and feature selection methods and preliminarily evaluated the importance of different features. In the future, more in-depth feature extraction, construction and selection and model construction are needed for the prediction problem.
Future prospects
Due to their importance in the immune system, there has been a significant progress in the characterization of AMPs in recent years. Simultaneously, because of the increasing availability of AMPs annotated, a variety of computational methods for AMPs have emerged. These methods employ different traditional machine learning-based algorithms as well as the feature calculation and selection strategies. However, most current approaches still suffer from high false-positive rates; there is an urgency to develop new and improved methods to address this problem.
Several potentially useful strategies may be helpful for improving the predictive perfomance of machine learning-based methods. First, deep learning has recently emerged as a powerful machine learning technique and has gained popularity in the field of bioinformatics and computational biomedicine. Although several approaches used deep learning frameworks for AMP identification, their deep learning structures are simple. A more comprehensive and more accurate deep learning structure needs to be developed in future work. Second, the idea of ensemble machine learning has been seldom used in AMP identification. Integrating different machine learning methods can combine the advantages of different machine learning methods and may help to develop more powerful AMP predictors. In addition, predicting the functional activities of AMPs is also an important task. However, only a few approaches are currently available for predicting AMPs and their functional activities. These approaches can predict a limited number of activities with low accuracy. Therefore, new methods for improved prediction of AMPs’ functional activities are expected to be developed in the future.
Conclusions
AMPs, a vital part of the immune system of organisms, have generated considerable interest. Accordingly, tremendous research efforts have been put into developing new approaches to predict AMPs accurately. In this study, we provided a comprehensive review to these approaches in terms of a wide range of aspects, including algorithms selected, feature selection techniques employed, performance evaluation strategy and web server/tool availability. To obtain a more objective performance evaluation, we constructed an independent test data set to benchmark all tools. Based on the assessment results, amPEPpy achieved the highest predictive performance. We also constructed six validation test data sets based on commonly used AMP databases to further evaluate these approaches. Based on AUC metrics, AMPfun showed good performances for most validation data sets. In addition, we tested 11 traditional machine learning methods and 6 commonly used feature selection algorithms using 5-fold cross-validation. Based on several metrics, we concluded SVM, RF and XGBoost were remarkable machine learning methods for AMP prediction.
This study should provide useful guidance to researchers who are interested in developing innovative and even more powerful AMP prediction models. We expect that, in the near future, more accurate AMP prediction methods will be developed and made available to facilitate community-wide efforts for characterizing and discovering new AMPs.
We provide a comprehensive survey and assessment of tools for the prediction of AMPs.
We systematically review and benchmark 34 existing tools with respect to the algorithms employed, chosen feature selection methods, calculated features, performance evaluation strategies and web server/software functionality.
All predictors underwent a comprehensive performance assessment based on an independent data set and six validation data sets from six commonly used databases.
Eleven traditional machine learning methods based on six feature selection strategies that were assessed using 5-fold cross-validation test based on the same data set.
This paper provides a useful guidance to researchers who are interested in developing innovative and even more powerful AMP prediction models.
Data Availability
The data are available from the authors upon reasonable request.
Conflict of Interest
The authors declare that they have no competing interests.
Funding
National Health and Medical Research Council of Australia (NHMRC) (1144652, 1127948); the Young Scientists Fund of the National Natural Science Foundation of China (31701142); the Australian Research Council (ARC) (LP110200333, DP120104460); a Major Inter-Disciplinary Research (IDR) project awarded by the Monash University, the Collaborative Research Program of Institute for Chemical Research, Kyoto University (2019-32 and 2018-28); the National Natural Science Foundation of China (62072243, 61772273).
Jing Xu received her BSc and MSc degrees from the Nankai University, China. She is currently a PhD student in the Department of Biochemistry and Molecular Biology and Biomedicine Discovery Institute, Monash University, Australia. Her research interests are bioinformatics, computational oncology, machine learning and pattern recognition.
Fuyi Li is currently a research fellow in the Department of Microbiology and Immunology, the Peter Doherty Institute for Infection and Immunity, the University of Melbourne, Australia. His research interests are bioinformatics, computational biology, machine learning and data mining.
André Leier is currently an assistant professor in the Department of Genetics, UAB School of Medicine, USA. He is also an associate scientist in the UAB’s O’Neal Comprehensive Cancer Center and the Gregory Fleming James Cystic Fibrosis Research Center. His research interests are in computational biomedicine, bioengineering, bioinformatics and machine learning.
Dongxu Xiang received his BEng from the Northwest A&F University, China. He is currently a research assistant in the Department of Biochemistry and Molecular Biology and Biomedicine Discovery Institute, Monash University, Australia. His research interests are bioinformatics, computational biology, machine learning and data mining.
Hsin-Hui Shen received her PhD in physical chemistry from the Oxford University, UK. She is an NHMRC Career Development Fellow and group leader in the Department of Biochemistry & Molecular Biology and Department of Materials Science & Engineering, Monash University. Her research interests are biophysical chemistry, chemical biology, antibiotics and biotechnology.
Tatiana T. Marquez Lago is currently an associate professor in the Departments of Genetics and Microbiology, UAB School of Medicine, USA. She is also a scientist in the UAB Gregory Fleming James Cystic Fibrosis Research Center. Her research interests are in host–microbiota interactions, evolution of antibiotic resistance, computational systems biology, bioengineering and artificial intelligence.
Jian Li is a professor and group leader in the Monash Biomedicine Discovery Institute and Department of Microbiology, Monash University, Australia. He is a Web of Science 2015–2017 Highly Cited Researcher in Pharmacology & Toxicology. He is currently an NHMRC Principal Research Fellow. His research interests include the pharmacology of polymyxins and the discovery of novel, safer polymyxins.
Dong-Jun Yu received his PhD degree from the Nanjing University of Science and Technology on the subject of pattern recognition and intelligence systems in 2003. He is currently a full professor in the School of Computer Science and Engineering, Nanjing University of Science and Technology. His research interests include pattern recognition, machine learning and bioinformatics.
Jiangning Song is an associate professor in the Monash Biomedicine Discovery Institute, Monash University. He is also affiliated with the Monash Data Futures Institute, Monash University. His research interests include bioinformatics, computational biology, machine learning, data mining and pattern recognition.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}