





Abstract
Acute kidney injury (AKI) is a complex syndrome with an abrupt decrease of kidney function, which is associated with high morbidity and mortality. Sepsis is the common cause of AKI. Mounting evidence has demonstrated that long non-coding RNAs (lncRNAs) play critical roles in the development and progression of sepsis-induced AKI. In this study, we aimed to illustrate the function and mechanism of lncRNA SNHG14 in lipopolysaccharide (LPS)-induced AKI. We found that SNHG14 was highly expressed in the plasma of sepsis patients with AKI. SNHG14 inhibited cell proliferation and autophagy and promoted cell apoptosis and inflammatory cytokine production in LPS-stimulated HK-2 cells. Functionally, SNHG14 acted as a competing endogenous RNA (ceRNA) to negatively regulate miR-495-3p expression in HK-2 cells. Furthermore, we identified that HIPK1 is a direct target of miR-495-3p in HK-2 cells. We also revealed that the SNHG14/miR-495-3p/HIPK1 interaction network regulated HK-2 cell proliferation, apoptosis, autophagy, and inflammatory cytokine production upon LPS stimulation. In addition, we demonstrated that the SNHG14/miR-495-3p/HIPK1 interaction network regulated the production of inflammatory cytokines (TNF-α, IL-6, and IL-1β) via modulating NF-κB/p65 signaling in LPS-challenged HK-2 cells. In conclusion, our findings suggested a novel therapeutic axis of SNHG14/miR-495-3p/HIPK1 to treat sepsis-induced AKI.
Introduction
Acute kidney injury (AKI) is a complex syndrome with an abrupt decrease of kidney function, which is associated with high morbidity and mortality [1,2]. Sepsis is the common cause of AKI [3,4]. Enormous efforts have been made to develop efficient therapeutic strategies for sepsis-induced AKI [5]. However, the mechanisms underlying sepsis-induced AKI are not fully understood. Thus, it is extremely important to investigate the pathogenesis of sepsis-induced AKI and develop potential novel therapies for AKI.
Mounting evidence has shown that renal tubular epithelial cell proliferation, apoptosis, and autophagy participate in the process of sepsis-induced AKI [6]. Growth factors such as vascular endothelial growth factor, insulin-like growth factor, and hepatocyte growth factor promote renal tubular epithelial cell proliferation and suppress cell apoptosis, leading to the recovery from AKI [7–9]. Studies also demonstrated that inflammation and metabolic response to inflammation are critical pathogenesis mechanisms of sepsis-induced AKI [10,11]. Therefore, novel therapeutic targets that regulate renal tubular epithelial cell proliferation, apoptosis, autophagy, and inflammatory cytokine production should be explored.
Long non-coding RNAs (lncRNAs) are a family of long nucleotides (>200 nt) without protein-encoding capability [12]. Accumulating studies have demonstrated that lncRNAs are involved in various physiological and pathological processes, including sepsis-induced AKI [13,14]. SNHG14 is a newly identified lncRNA located on chromosome 15q11.2 [15]. It has been demonstrated that SNHG14 promotes microglia activation via regulating miR-145-5p/PLA2G4A [16]. SNHG14 also exerts an oncogenic function in multiple malignancies, such as gastric cancer, breast cancer, and non-small cell lung cancer [17–19]. However, the function of SNHG14 in sepsis-induced AKI remains unclear.
In this study, we found that SNHG14 was highly expressed in the plasma of sepsis patients with AKI. In vitro experiments revealed that SNHG14 regulates human kidney proximal tubular HK-2 cell proliferation, apoptosis, autophagy, and inflammatory cytokine production via sponging miR-495-3p. HIPK1 has been identified as a direct target of miR-495-3p. Moreover, the regulatory axis of SNHG14/miR-495-3p/HIPK1 regulates inflammatory cytokines (TNF-α, IL-6, and IL-1β) via modulating NF-κB/p65 signaling in lipopolysaccharide (LPS)-challenged HK-2 cells. Taken together, our results indicate that the SNHG14/miR-495-3p/HIPK1 axis may be a novel therapeutic target to treat sepsis-induced AKI.
Materials and Methods
Patient samples
Blood samples were collected from patients with sepsis-induced AKI or healthy controls between March 2017 and December 2018 at the Second Affiliated Hospital of Xi’an Jiaotong University (Xi’an, China). The experiment protocols were reviewed and approved by the Ethics Committee of the Second Affiliated Hospital of Xi’an Jiaotong University. All participants signed the written informed consent forms.
Cell culture and treatment
HK-2 cells were purchased from Cell Bank, Chinese Academy of Sciences (Shanghai, China) and cultured in Ham’s F-12 K medium supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, USA), 50 μg/ml streptomycin, and 50 U/ml penicillin (Solarbio, Beijing, China) in a humidified incubator at 37°C with 5% CO2. Cells were treated with 5 μg/ml LPS (Sigma-Aldrich, St Louis, USA).
Quantitative real-time polymerase chain reaction
Total RNA was purified using Trizol (Invitrogen, Carlsbad, USA) and reverse-transcribed into cDNA using PrimerScript RT reagent kit (TaKaRa Bio, Shiga, Japan). SYBR Green master mix (AccuRef Scientific, Xi’an, China) was used to perform quantitative polymerase chain reaction (PCR) analysis on ABI7000 qPCR system (Applied Biosystems, Foster City, USA). GAPDH and U6 were used as the internal controls. Relative expression was calculated using the 2-ΔΔCT method. The primer sequences used for quantitative real-time polymerase chain reaction (RT-qPCR) are listed in Table 1.
Sequences of primers used in this study
| Gene | Forward primer (5ʹ→3ʹ) | Reverse primer (5ʹ→3ʹ) |
|---|---|---|
| miR-495-3p | GCGGAAACAAACATGGTGCA | GTTGGCTCTGGTGCAGGGTCCGAGGTATTCGCACCAGAGCCAACAAGAAG |
| U6 | TTGGTCTGATCTGGCACATATAC | AAAAATATGGAGCGCTTCACG |
| HIPK1 | TCCCGCCTAAGCAGTGAAAAT | GGCAGGTATGATTCTTGTGCTG |
| SNHG14 | GGGTGTTTACGTAGACCAGAACC | CTTCCAAAAGCCTTCTGCCTTAG |
| GAPDH | GAAGGTGAAGGTCGGAGTC | GAAGATGGTGATGGGATTTC |
| Gene | Forward primer (5ʹ→3ʹ) | Reverse primer (5ʹ→3ʹ) |
|---|---|---|
| miR-495-3p | GCGGAAACAAACATGGTGCA | GTTGGCTCTGGTGCAGGGTCCGAGGTATTCGCACCAGAGCCAACAAGAAG |
| U6 | TTGGTCTGATCTGGCACATATAC | AAAAATATGGAGCGCTTCACG |
| HIPK1 | TCCCGCCTAAGCAGTGAAAAT | GGCAGGTATGATTCTTGTGCTG |
| SNHG14 | GGGTGTTTACGTAGACCAGAACC | CTTCCAAAAGCCTTCTGCCTTAG |
| GAPDH | GAAGGTGAAGGTCGGAGTC | GAAGATGGTGATGGGATTTC |
Sequences of primers used in this study
| Gene | Forward primer (5ʹ→3ʹ) | Reverse primer (5ʹ→3ʹ) |
|---|---|---|
| miR-495-3p | GCGGAAACAAACATGGTGCA | GTTGGCTCTGGTGCAGGGTCCGAGGTATTCGCACCAGAGCCAACAAGAAG |
| U6 | TTGGTCTGATCTGGCACATATAC | AAAAATATGGAGCGCTTCACG |
| HIPK1 | TCCCGCCTAAGCAGTGAAAAT | GGCAGGTATGATTCTTGTGCTG |
| SNHG14 | GGGTGTTTACGTAGACCAGAACC | CTTCCAAAAGCCTTCTGCCTTAG |
| GAPDH | GAAGGTGAAGGTCGGAGTC | GAAGATGGTGATGGGATTTC |
| Gene | Forward primer (5ʹ→3ʹ) | Reverse primer (5ʹ→3ʹ) |
|---|---|---|
| miR-495-3p | GCGGAAACAAACATGGTGCA | GTTGGCTCTGGTGCAGGGTCCGAGGTATTCGCACCAGAGCCAACAAGAAG |
| U6 | TTGGTCTGATCTGGCACATATAC | AAAAATATGGAGCGCTTCACG |
| HIPK1 | TCCCGCCTAAGCAGTGAAAAT | GGCAGGTATGATTCTTGTGCTG |
| SNHG14 | GGGTGTTTACGTAGACCAGAACC | CTTCCAAAAGCCTTCTGCCTTAG |
| GAPDH | GAAGGTGAAGGTCGGAGTC | GAAGATGGTGATGGGATTTC |
Transfection
Empty vector pcDNA3.1, SNHG14 overexpression vector pcDNA3.1-SNHG14, knockdown control sh-NC, SNHG14 knockdown plasmid sh-SNHG14, HIPK1 knockdown plasmid sh-HIPK1, miR-NC, miR-495-3p mimics, inhibitor NC, miR-495-3p inhibitors were purchased from GenePharma (Shanghai, China). The HK-2 cells were separately transfected with sh-SNH14 (sense: 5ʹ-GCAAAUGAAAGCUACCAAU-3′, antisense: 5ʹ-AUUGGUAGCUUUCAUUUGC-3ʹ), sh-HIPK1 (sense: 5′-GAGUAGCUGUGUUGUGUAA-3′; antisense: 5′-UUACACAACACAGCUACUC-3′) miR-495-3p mimics (sense: 5′-AAACAAACAUGGUGCACUUCUU-3′, antisense: 5′-GAAGUGCACCAUGUUUGUUUUU-3′), miR-495-3p inhibitor (5′-AAGAAGUGCACCAUGUUUGUUU-3′), and miR-NC (5′-GACCUUCAUGUACCUGGCACCG-3′) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. The transfection efficiency was confirmed by RT-qPCR and/or western blot analysis.
MTT assay
MTT assay was performed to analyze HK-2 cell proliferation with different treatments. Briefly, a total of 1 × 104 HK-2 cells were seeded into each well of 96-well plates and cultured for 24 h and then treated with 5 μg/ml LPS for 8 h. Following this, 10 μl MTT solution (5 mg/ml; Sigma-Aldrich) was added to each well and incubated for 4 h at 37°C. Then, the absorbance of each well was measured at 570 nm with a microplate reader (Bio-Tek, Winooski, USA), followed by cell viability analysis.
Cell apoptosis
Apoptosis of HK-2 cells treated with LPS was analyzed by the Annexin V-FITC/PI double staining method. Briefly, cells were seeded in a six-well plate and cultured for 24 h, and then treated with 5 μg/ml LPS for 8 h. Then, cells were harvested and stained with Annexin-V/FITC and PI (BD Biosciences, Franklin Lakes, USA) according to the manufacturer’s protocol. Subsequently, the apoptotic cells were detected with a BD FACS Calibur cytometer (BD, San Diego, USA) for further analysis.
Western blot analysis
Total protein was extracted using RIPA buffer containing proteinase inhibitor cocktail (EXPrecision, Xi’an, China). Protein concentration was quantified using a BCA kit (AccuRef Scientific). An equal amount of proteins was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes (Millipore, Billerica, USA). The membranes were blocked with 5% non-fat milk for 1 h at room temperature. After being rinsed with Tris-buffered saline and Tween 20 (TBST), membranes were incubated with the primary antibodies at 4°C overnight. After being washed with TBST for three times (5 min each), membranes were incubated with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 1 h, followed by three times (10 min each) wash with PBST. Following this, protein bands on membranes were visualized using an enhanced chemiluminescence detection kit (Thermo Fisher Scientific, Waltham, USA). The primary antibodies used in this study were purchased from Abcam (Cambridge, UK) and listed as follows: anti-Bax antibody (1:1000, ab32503), anti-Bcl-2 antibody (1:1000, ab32124), anti-Cleaved caspase-3 antibody (1:1000, ab2302), anti-LC-3 antibody (1:1000, ab192890), anti-HIPK1 antibody (1:500, ab152109), anti-IκBα (E130) antibody (1:1000, ab32518), anti-p-IκBα (Ser36) [EPR6235] antibody (1:10,000, ab133462), anti-NF-κb p65 (E379) antibody (1:50,000, ab32536), and anti-p-p65 (Ser536) (EP2294Y) antibody (1:10,000, ab76302). Anti-GAPDH (AG0766) antibody (1:10,000, 60004-1-Ig) was from Proteintech (Shanghai, China). Secondary antibodies used were HRP-conjugated goat anti-mouse IgG (H + L) antibody (1:10,000, SA00001; Proteintech) and HRP-conjugated goat anti-rabbit IgG (H + L) antibody (1:10,000, SA00001-2, Proteintech).
Enzyme-linked immunosorbent assay
The levels of cytokines in HK-2 cells were determined by enzyme-linked immunosorbent assay (ELISA). Briefly, HK-2 cells were seeded into 24-well plates (2.0 × 105 cells/well) and transfected with sh-NC, sh-SNHG14, miR-495-3p mimics, or sh-HIPK1. LPS (5 μg/ml) was added to each well and incubated for 8 h. After that, the cell culture supernatant was collected, and the levels of TNF-α, IL-6, and IL-1β in the supernatant were determined using commercial ELISA kits (R&D Systems, Minneapolis, USA) according to the manufacturer’s protocol.
Prediction of the downstream targets of lncRNA and miRNA
To explore the potential mechanisms of action of lncRNAs, miRNAs that interact with the lncRNAs were predicted using the DIANA online tools (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=lncbasev2%2Findex-predicted). The underlying downstream targets of miRNAs were predicted using the ENCORI (http://starbase.sysu.edu.cn/agoClipRNA.php?source=mRNA), mi-RDB (http://mirdb.org/index.html) and TargetScan (http://www.targetscan.org/vert_72/) databases. Then, the overlapping targets of miRNAs among these three databases were screened using Venn analysis and used for subsequent investigations.
Luciferase reporter assay
Wild-type (Wt) and mutant (Mut) sequences of lncRNA SNHG14 or 3′-UTR of HIPK1 were cloned into the pGL3 luciferase reporter vector (Promega, Madison, USA). HK-2 cells were co-transfected with reporter vectors, with negative control (NC) or miR-495-3p mimics. The relative luciferase activity was measured 48 h later using a dual-luciferase reporter kit (Promega).
Statistical analysis
Statistical analysis was conducted using GraphPad Prism (version 6; Prism, La Jolla, USA). The results are presented as the mean ± standard deviation (SD). Unpaired Student’s t-test was used for statistical analysis. A P-value of < 0.05 was considered to be statistically significant.
Results
SNHG14 is highly expressed in the plasma of sepsis patients with AKI
The expression of SNHG14 was analyzed in the plasma of sepsis patients with AKI. The results showed that sepsis patients had significantly higher expression of SNHG14 (Fig. 1A). To determine whether the dysregulation of SNHG14 was derived from kidney injury, HK-2 cells were treated with LPS to establish in vitro sepsis-induced AKI. In comparison with that in untreated cells, LPS treatment markedly upregulated the expression of SNHG14 in HK-2 cells (Fig. 1B).
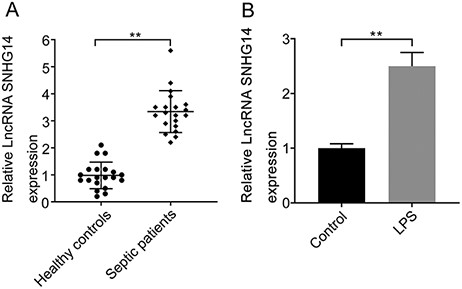
SNHG14 is highly expressed in the plasma of sepsis patients with AKI and LPS treatment induces SNHG14 expression (A) Analysis of the relative expression of SNHG14 in the serum of patients with sepsis-induced AKI and healthy controls measured by RT-qPCR (n = 20). (B) The relative expression levels of SNHG14 in LPS-induced HK-2 cells and untreated cells were measured by RT-qPCR. **P<0.01.
SNHG14 inhibits cell proliferation and autophagy but promotes cell apoptosis and inflammatory cytokine production in LPS-stimulated HK-2 cells
To investigate the function of SNHG14, pcDNA3.1-SNHG14 or sh-SNHG14 was transfected into HK-2 cells to overexpress or knock down the expression of SNHG14 (Fig. 2A). LPS treatment significantly inhibited cell viability (Fig. 2B). Overexpression of SNHG14 further decreased the viability of HK-2 cells treated with LPS, while knockdown of SNHG14 enhanced cell viability (Fig. 2B). In contrast, LPS stimulation led to a higher rate of cell apoptosis, which was further increased upon SNHG14 overexpression (Fig. 2C). Knockdown of SNHG14 suppressed the apoptosis of HK-2 cells (Fig. 2C). Consistent with this, LPS stimulation enhanced the expressions of the pro-apoptotic proteins Bax and cleaved-Caspase3 but inhibited Bcl-2 expression (Fig. 2D). Overexpression of SNHG14 further upregulated the expressions of Bax/cleaved-Caspase3 but downregulated the expression of Bcl-2, while knockdown of SNHG14 had the opposite effect (Fig. 2D). In addition, overexpression of SNHG14 enhanced the production of inflammatory cytokines (TNF-α, IL-6, and IL-1β) in HK-2 cells treated with LPS (Fig. 2E). Moreover, overexpression of SNHG14 inhibited cell autophagy, with a decreased LC3-II/LC3-I ratio and enhanced p62 expression (Fig. 2F). Meanwhile, knockdown of SNHG14 significantly enhanced LC3-II expression and inhibited p62 expression in HK-2 cells upon LPS stimulation (Fig. 2F).
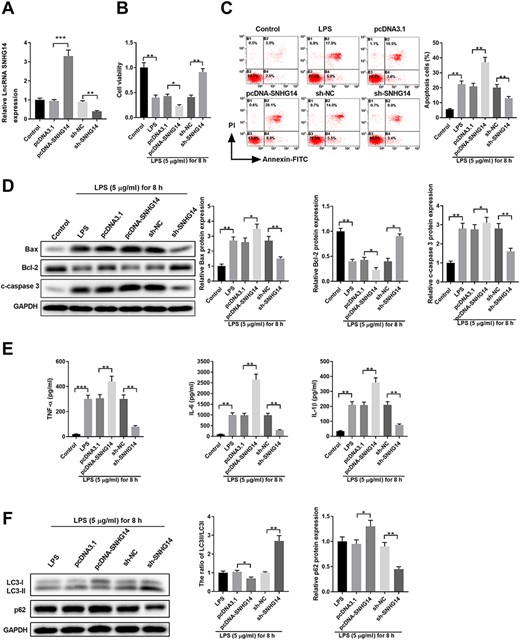
SNHG14 regulates cell viability, apoptosis, cytokines production, and autophagy in LPS-stimulated HK-2 cells (A) HK-2 cells were left untreated (control) or transfected with pcDNA3.1, pcDNA3.1-SNHG14, sh-NC, or sh-SNHG14. The relative expression of SNHG14 was examined by qPCR 48 h later. (B-F) HK-2 cells were left untreated (control), treated with 5 μg/ml LPS for 8 h (LPS), or transfected with pcDNA3.1, pcDNA3.1-SNHG14, sh-NC, or sh-SNHG14. After 48 h, cells were treated with 5 μg/ml LPS for 8 h. (B) Cell viability was analyzed by MTT assay. (C) Cell apoptosis was analyzed by Annexin-V-FITC/PI staining. (D) Protein expressions of Bax, Bcl-2, and cleaved-caspase 3 were analyzed by western blot analysis. GAPDH was used as the loading control. (E) The inflammatory cytokine production of TNF-α, IL-6, and IL-1β in cell culture supernatant was analyzed by ELISA. (F) Protein expressions of LC3-I, LC3-II, and p62 were analyzed by western blot analysis. GAPDH was used as the loading control. *P<0.05, **P<0.01, ***P<0.001.
SNHG14 functions as a ceRNA to negatively regulate miR-495-3p expression in HK-2 cells
For further investigation, the potential miRNA targets of SNHG14 were predicted using the DIANA tools, and a number of miRNAs were obtained. Then, the expressions of five miRNAs, i.e. miR-342-3p, miR-495-3p, miR-377-3p, miR-27a-3p, and miR-27b-3p that were rarely reported in AKI, were determined in SNHG14-silenced HK-2 cells. Our results showed that knockdown of SNHG14 significantly enhanced the expression of miR-495-3p (Fig. 3A). Further analysis revealed that miR-495-3p has complementary binding sequences targeting the 3ʹ-UTR of SNHG14 (Fig. 3B). Luciferase reporter assay further confirmed the interaction between miR-495-3p and the 3′-UTR of SNHG14 (Fig. 3C). In addition, transfection with miR-495-3p mimics suppressed the expression of SNHG14, while the inhibition of miR-495-3p enhanced SNHG14 expression in HK-2 cells (Fig. 3D). The expression of miR-495-3p was also analyzed in the plasma of sepsis patients with AKI. The results showed that sepsis patients had a significantly lower level of miR-495-3p (Fig. 3E).
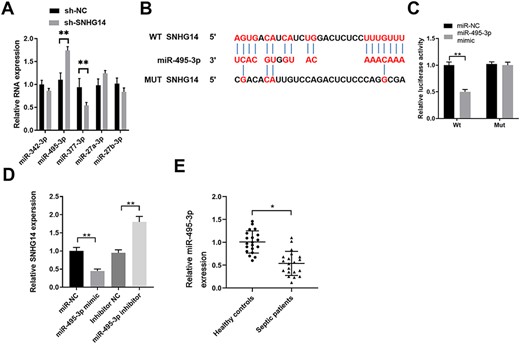
SNHG14 functions as a ceRNA to negatively regulate miR-495-3p expression in HK-2 cells (A) HK-2 cells were transfected with sh-NC or sh-SNHG14. The relative expressions of miR-342-3p, miR-495-3p, miR-377-3p, miR-27a-3p, and miR-27b-3p were analyzed by qPCR 48 h later. (B) Bioinformatics analysis predicted the putative binding sequences between miR-495-3p and WT or Mut 3ʹ-UTR of SNHG14. (C) HK-2 cells were co-transfected with miR-NC or miR-495-3p mimics, and luciferase reporter containing WT or Mut 3ʹ-UTR of SNHG14. The relative luciferase activity was analyzed 48 h later. (D) HK-2 cells were transfected with miR-NC, miR-495-3p mimics, inhibitor NC, or miR-495-3p inhibitor. SNHG14 expression was analyzed by qPCR 48 h later. (E) The relative expression of miR-495-3p in the serum of patients with sepsis-induced AKI or healthy controls was analyzed by qPCR (n = 20). *P<0.05, **P<0.01.
HIPK1 is a direct target of miR-495-3p in HK-2 cells
To further investigate the function of miR-495-3p in LPS-induced AKI, its potential targets were searched using different online prediction tools (ENCORI, miRDB, and TargetScan) (Fig. 4A). Overexpression of miR-495-3p significantly suppressed the expression of HIPK1 in HK-2 cells (Fig. 4B). miR-495-3p was found to have a putative binding sequence targeting the 3′-UTR of HIPK1 (Fig. 4C). The interaction between miR-495-3p and 3′-UTR of HIPK1 was further validated by luciferase reporter assay (Fig. 4D). Overexpression of miR-495-3p was also found to inhibit the protein expression of HIPK1 in HK-2 cells (Fig. 4E). Consistent with this, sepsis patients had a significantly higher expression level of HIPK1 (Fig. 4F). These results suggested that HIPK1 is a direct target of miR-495-3p in HK-2 cells.
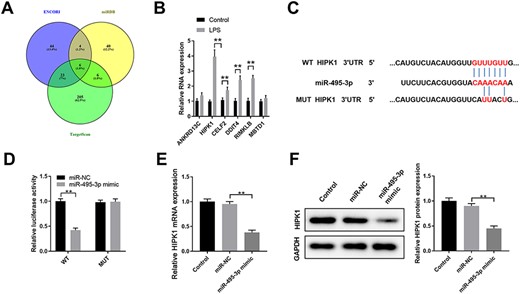
HIPK1 is a direct target of miR-495-3p in HK-2 cells (A) Bioinformatics analysis was performed using ENCORI, miRDB, and TargetScan to predict the potential targets of miR-495-3p. (B) HK-2 cells were transfected with miR-NC or miR-495-3p mimics. The relative mRNA expression levels of potential targets ANKRD13 C, HIPK1, CELF2, DDIT4, RIMKLB, and MBTD1 were analyzed by qPCR 48 h later. (C) The predicted binding sequences between miR-495-3p and WT or Mut 3ʹ-UTR of HIPK1 were shown. (D) HK-2 cells were co-transfected with miR-NC or miR-495-3p mimics, and luciferase reporter vector with WT or Mut 3ʹ-UTR of HIPK1. The relative luciferase activity was analyzed 48 h later. (E) HK-2 cells were left untreated (control), or transfected with miR-NC or miR-495-3p mimics. The protein expression of HIPK1 was analyzed by western blot analysis. (F) The relative expression of HIPK1 in the serum of patients with sepsis-induced AKI or healthy controls was analyzed by qPCR (n = 30). *P<0.05, **P<0.01.
SNHG14/miR-495-3p/HIPK1 interaction network regulates HK-2 cell proliferation, apoptosis, autophagy, and inflammatory cytokine production upon LPS stimulation
Next, we determined the regulatory interaction network of SNHG14/miR-495-3p/HIPK1 in HK-2 cells. Compared with the findings in control HK-2 cells, LPS treatment significantly suppressed cell proliferation and promoted cell apoptosis, with enhanced Bax/cleaved-caspase 3 expressions and decreased Bcl-2 expression (Fig. 5A–C). Intriguingly, knockdown of SNHG14, overexpression of miR-495-3p, and knockdown of HIPK1 exhibited similar effects upon LPS stimulation, with enhanced cell proliferation and inhibited cell apoptosis (Fig. 5A–C). Moreover, sh-SNHG14, miR-459-3p mimics, or sh-HIPK1 significantly decreased the expressions of inflammatory cytokines, such as TNF-α, IL-6, and IL-1β in HK-2 cells (Fig. 5D). Similarly, SNHG14 or HIPK1 knockdown and miR-495-3p overexpression promoted HK-2 cell autophagy, with an enhanced LC3-II/LC3-I ratio and decreased p62 expression compared with those in HK-2 cells transfected with sh-NC upon LPS treatment (Fig. 5E). These findings suggested that the SNHG14/miR-495-3p/HIPK1 interaction network modulates the HK-2 cell response to LPS stimulation.
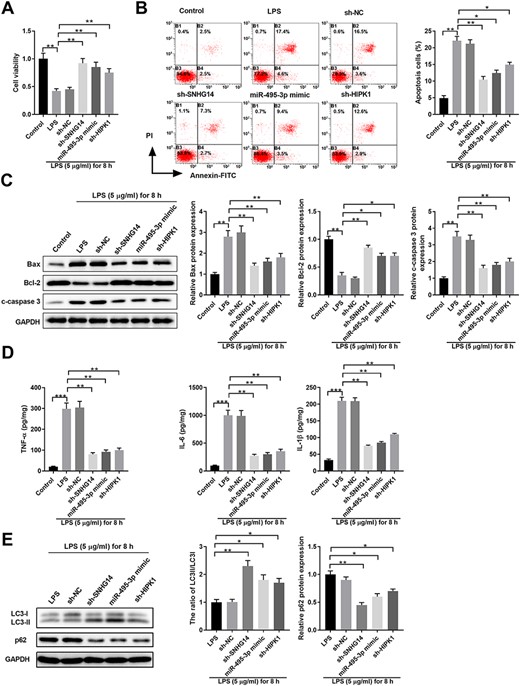
SNHG14/miR-495-3p/HIPK1 interaction network regulates HK-2 cell proliferation, apoptosis, autophagy, and inflammatory cytokine production upon LPS stimulation HK-2 cells were left untreated (control) or transfected with sh-NC, sh-SNHG14, miR-495-3p mimics, or sh-HIPK1, together with 5 μg/ml LPS treatment for 8 h. (A) Cell viability was analyzed by MTT assay. (B) Cell apoptosis was analyzed by Annexin-V-FITC/PI staining. (C) Protein expressions of Bax, Bcl-2, and Cleaved-Caspase 3 was analyzed by western blot analysis. GAPDH was used as the loading control. (D) The inflammatory cytokine production of TNF-α, IL-6, and IL-1β in cell culture supernatant was analyzed by ELISA. (E) Protein expressions of LC3-I, LC3-II, and p62 were analyzed by western blot analysis. GAPDH was used as the loading control. *P<0.05, **P<0.01, ***P<0.001.
SNHG14/miR-495-3p/HIPK1 interaction network regulates the production of inflammatory cytokines by modulating the NF-κB/p65 signal in LPS-challenged HK-2 cells
To further understand how the SNHG14/miR-495-ep/HIPK1 interaction network regulates HK-2 cell response upon LPS stimulation, we analyzed the signaling pathways. NF-κB signaling has been demonstrated to play critical roles in inflammation [20]. We examined the protein expressions of phosphorylated and total IκB, and phosphorylated and total p65 in control HK-2 cells or HK-2 cells with different transfections and subject to LPS stimulation (Fig. 6A). LPS stimulation significantly enhanced the phosphorylation of IκBα and p65. However, the knockdown of SNHG14, overexpression of miR-495-3p, and knockdown of HIPK1 markedly suppressed the activation of the NF-κB signaling pathway (Fig. 6A). In addition, blocking NF-κB signaling with JSH-23, an NF-κB inhibitor, significantly inhibited the phosphorylation of p65 in LPS-stimulated HK-2 cells, with or without sh-NC transfection (Fig. 6B). However, blocking NF-κB signaling only mildly suppressed the phosphorylation of p65 in HK-2 cells transfected with sh-SNHG14 (Fig. 6B), indicating that SNHG14 suppressed NF-κB signaling in HK-2 cells treated with LPS. Consistent with this, blocking NF-κB signaling suppressed the production of inflammatory cytokines (TNF-α, IL-6, and IL-1β) in HK-2 cells treated with LPS but not in LPS-stimulated HK-2 cells transfected with sh-SNHG14 (Fig. 6C). Taken together, these results indicated that the SNHG14/miR-495-3p/HIPK1 interaction network regulates the production of inflammatory cytokines by modulating the NF-κB/p65 signal in LPS-challenged HK-2 cells.
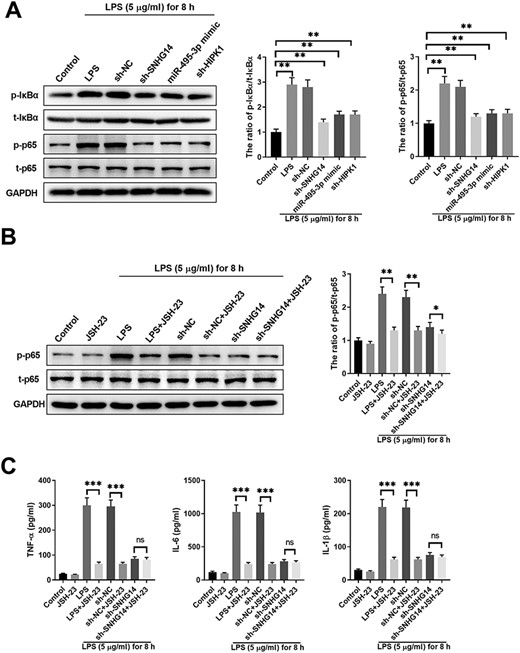
SNHG14/miR-495-3p/HIPK1 interaction network regulates production of inflammation cytokines via modulating NF-κB/p65 signal in LPS-challenged HK-2 cells (A) HK-2 cells were left untreated (control) or transfected with sh-NC, sh-SNHG14, miR-495-3p mimics, or sh-HIPK1, together with 5 μg/ml LPS treatment for 8 h. The expressions of NF-κB signaling pathway-related proteins, including p-IκBα, t-IκBα, p-p65, and t-p65 in LPS-induced cells after different treatments were detected by western blot analysis. (B) HK-2 cells were left untreated (control), treated with JSH-23, or transfected with sh-NC, sh-SNHG14, with or without JSH-23 and 5 μg/ml LPS treatment for 8 h. The expressions of p-p65 and t-p65 in LPS-induced cells with different treatments were detected by western blot analysis. (C) The expressions of TNF-α, IL-1β and IL-6 in LPS-stimulated cells with different treatments were detected by ELISA. *P<0.05, **P<0.01, ***P<0.001.
Discussion
Advances in our understanding of the pathogenesis of sepsis-induced AKI provide insight into new therapeutic strategies for sepsis patients with AKI. However, available treatments are still not sufficiently effective [3,21]. In this study, we identified that the regulatory network of SNHG14/miR-495-3p/HIPK1 plays a critical role in sepsis-induced AKI (Fig. 7). SNHG14 positively regulated the expression of HIPK1 via sponging miR-495-3p. By using an in vitro model of LPS-stimulated HK-2 cells, we demonstrated that SNHG14/miR-495-3p/HIPK1 regulates HK-2 cell proliferation, apoptosis, autophagy, and inflammatory cytokine production. Furthermore, we demonstrated that SNHG14/miR-495-3p/HIPK1 modulated NF-κB/p65 signaling and regulated the production of TNF-α, IL-1β, and IL-6 in LPS-stimulated HK-2 cells. These findings might provide a new therapeutic strategy for treating sepsis-induced AKI.
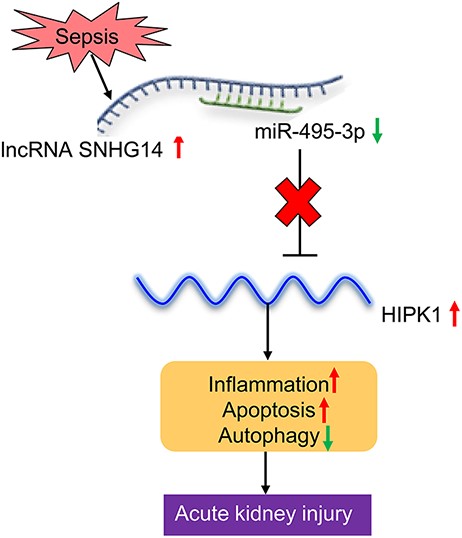
Schematic overview of the SNHG14/miR-495-3p/HIPK1 interaction network in aggravating acute kidney injury
The function of SNHG14 as an oncogene has been extensively studied in various tumors [19,22–25]. However, SNHG14 was reported to suppress cell proliferation and promote cell apoptosis via sponging miR-92a-3p in glioma [26]. Zhu et al. [27] found that SNHG14 was highly expressed in an acute lung injury mouse model, and its downregulation protected against LPS-induced acute lung injury through sponging miR-34c-3p. In another study, SNHG14 was found to promote the inflammatory response induced by cerebral ischemia/reperfusion injury by modulating miR-136-5p/ROCK1 [28]. Consistent with this, we found high expression of SNHG14 in sepsis-induced AKI patients, while SNHG14 was shown to promote the production of inflammatory cytokines in LPS-challenged HK-2 cells. Intriguingly, while SNHG14 functions as an oncogene and promotes cell proliferation in different tumors, we demonstrated that its overexpression suppressed HK-2 cell proliferation and enhanced cell apoptosis in LPS-stimulated HK-2 cells.
Multiple miRNAs have been predicted to interact with SNHG14, and overexpression of SNHG14 has been shown to significantly suppress the expression of miR-495-3p. In gastric cancer, SNHG10 and SNHG20 have also been found to competitively bind to miR-495-3p [29,30]. The lncRNA NEAT1 has been demonstrated to modify the miR-495-3p/STAT3 and miR-211/PI3K/AKT axes in sepsis-associated inflammation [31]. In this study, we demonstrated that SNHG14 functions as a ceRNA that can compete with miR-495-3p to regulate the expression of HIPK1. HIPK1 was identified as a p53-binding protein and shown to play a critical role in tumorigenesis [32]. In acute lung injury, the downregulation of HIPK1 was shown to exhibit a protective function and attenuate inflammation and oxidative stress, with enhanced autophagy [33]. Consistent with this, we found that knockdown of HIPK1 in HK-2 cells promoted cell proliferation, while suppressing cell apoptosis and the production of inflammatory cytokines.
A major limitation of this study is that the regulatory axis SNHG14/miR-495-3p/HIPK1 was only tested using an in vitro model of LPS-stimulated HK-2 cells. More efforts are needed to explore the function of the SNHG14/miR-495-3p/HIPK1 interaction network in in vivo models and sepsis patients with AKI. In addition, it remains a challenging issue to understand the complicated regulatory network between miRNA-495-3p and HIPK1 [34].
In conclusion, we demonstrated that SNHG14 aggravates sepsis-induced AKI through modulating the miR-495-3p/HIPK1 axis in LPS-challenged HK-2 cells. The SNHG14/miR-495-3p/HIPK1 interaction network might provide a novel therapeutic target for the clinical treatment of sepsis-induced AKI.
Funding
This work was supported by the grants from the Key Research and Development Projects of Shaanxi Province (No. 2020SF-121), Research Fund of the Second Affiliated Hospital of Xi’an Jiaotong University [YJ(QN)201711], and Shaanxi Administration of Traditional Chinese Medicine Research Program (JCMS037).
Conflict of Interest
The authors declare that they have no conflict of interest.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}