



Abstract
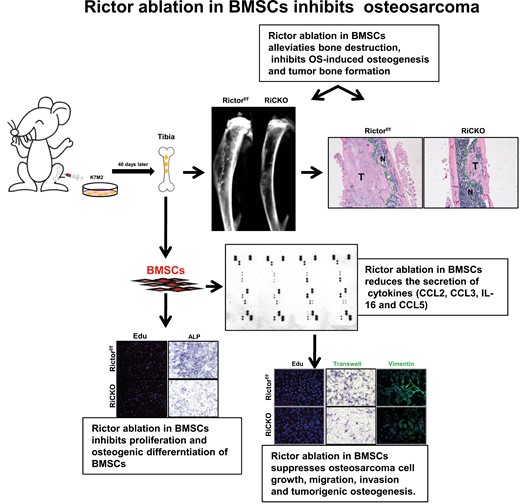
Bone marrow mesenchymal stem cells (BMSCs) are indispensable cells constituting the bone marrow microenvironment that are generally recognized as being involved in the development and progression of osteosarcoma (OS). To explore whether mTORC2 signaling inhibition in BMSCs suppressed OS growth and tumor-caused bone destruction, 3-month-old littermates genotyped Rictorflox/flox or Prx1-cre; Rictorflox/flox (with same gender) were injected with K7M2 cells in the proximal tibia. After 40 days, bone destruction was alleviated in Prx1-cre; Rictorflox/flox mice, as observed on X-ray and micro-CT. This was accompanied by decreased serum N-terminal propeptide of procollagen type I (PINP) levels and reduced tumor bone formation in vivo. Interactions between K7M2 and BMSCs were studied in vitro. Rictor-deficient BMSCs, which were cultured in tumor-conditioned medium (TCM), caused reduced bone proliferation and suppressed osteogenic differentiation. Moreover, compared with the control group, K7M2 cells cultured in BCM (culture medium extracted from Rictor-deficient BMSCs) displayed less proliferation, migration, and invasion, and attenuated osteogenic activity. Forty types of cytokines were then analyzed by mouse cytokine array and decreased levels CCL2/3/5 and interleukin-16 were detected in Rictor-deficient BMSCs. These results suggested that inhibition of mTORC2 (Rictor) signaling pathway in BMSCs exerted anti-OS effects through 2 mechanisms: (1) by suppressing the proliferation and osteogenic differentiation of BMSCs induced by OS to alleviate bone destruction; (2) by reducing the secretion of cytokines by BMSCs, which are closely related to OS cell growth, migration, invasion, and tumorigenic osteogenesis.
Introduction
Osteosarcoma (OS), also known as osteogenic sarcoma, is the most common primary malignant tumor of the bone. It is highly metastatic and has the highest incidence in children and adolescents.1 OS occurs mainly in the long bones, close to the growth plate in the bone metaphysis, and is characterized by the presence of osteoid matrix and neoplastic tissue, thereby disrupting the bone structure.2,3 With the introduction of neoadjuvant chemotherapy, the prognosis of patients with OS has improved considerably postsurgery.4 However, the 5-year survival rate has not improved significantly in the past 40 years mainly due to chemotherapy intolerance and recurrent metastases. High heterogeneity in OS tumor cells makes it difficult to define genomic-initiating biological processes driving genesis of OS and, hence, no therapeutic targets in OS have been delineated.5,6 Therefore, innovative therapeutic strategies for targeting OS tumor cells remain to be explored in research and clinical settings.
Bone marrow mesenchymal stem cells (BMSCs) are major components that constitute the bone marrow microenvironment and are considered to be essential and supportive for growth, dissemination, and osteoid structure formation during OS development.7,8 BMSCs are heavily recruited in the mesenchyme of progressive OS tissues and exert their functions partly through secretion of active substances, including growth factors, cytokines, chemokines, and matrix metalloproteinases (MMPs).9-13 BMSCs can also transform into cancer-associated fibroblasts (CAFs) with increased expression of MMPs, chemokines, vascular endothelial growth factor, which in turn promote OS cell growth, survival, migration, and invasiveness.14
Another implication of BMSCs is associated with tumor-caused bone regeneration and destruction. As osteoblastic progenitors, BMSCs are able to differentiate into to mature osteoblasts which directly regulate bone matrix synthesis. Cytokines or growth factors such as TGF-βs, fibroblast growth factors (FGFs), or wingless-type MMTV integration site family members (WNTs) secreted by OS cells activate transcriptional factors including RUNX2, SP7, COL1, etc. to regulate osteoblastic differentiation of BMSCs.7,15 Extracellular vesicles (EVs) derived from BMSCs involves in the OPG-RANKL-RANK signaling pathway to regulate the differentiation and function of osteoclasts.16 The crosstalk between BMSCs-osteoblasts and monocyte-macrophage-osteoclasts implies BMSCs exerts an indispensable role in bone reconstruction alone with OS cells. Therefore, the supportive roles of BMSCs in OS development suggest a supplementary therapeutic option of targeting BMSCs in addition to tumor cells.
Mammalian target of rapamycin (mTOR) is a highly conserved serine/threonine kinase that exists in 2 distinct multiprotein complexes in organisms: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2).17Rictor (mAVO3), SIN1, and mLST8 bind mTOR to comprise the rapamycin-insensitive mTORC2. mTORC2 phosphorylates a distinct set of downstream targets, including members of the AGC family of kinases such as Akt (at position Ser 473), serum- and glucocorticoid-induced protein kinase 1 (SGK1), and protein kinase C-α (PKC-α).
mTORC2 functions as an important regulator for the organization of the actin cytoskeleton, cell survival, and lipid metabolism.18 Because of a lack of specific pharmacological inhibitors, deciphering the biology of mTORC2 is circumscribed by deleting Rictor or by RNAi-mediated Rictor silencing. mTORC2 has been proven to maintain bone homeostasis by genetic rictor deletion in MSCs. Some studies reported that Rictor deletion inhibited differentiation of MSCs to osteoblasts, suppressed activities of the RANKL/RANK pathway, and finally maintained a fine balance between osteogenesis and osteolysis.19,20Rictor deficiency was also found to reduce the transition of BMSCs to CAFs coupled with decreased secretion of cytokines (IL-6, RANKL, and TGFβ) during bone metastasis of breast cancer.21It is hypothesized that targeted deletion of mTORC2 in BMSCs could prevent bone reconstruction and inhibit OS growth.
In the present study, we found that Rictor deletion in BMSCs alleviated bone destruction during OS progression. Rictor-deficient BMSCs displayed reduced proliferation and suppressed osteogenic differentiation in tumor-conditioned medium (TCM). We further confirmed the reduced secretion of the cytokines by Rictor-deficient BMSCs influenced the bioactivity of K7M2 cells. We revealed for the first time that targeting mTORC2 could act on BMSCs to prevent OS progression and bone destruction, thereby suggesting an effective adjunct therapy for OS.
Materials and Methods
Animals
Prx1-Cre mice and Rictorflox/flox (hereafter Rictorf/f) mice were kindly provided by Dr. Fanxin Long (Washington University, St Louis, MO, USA). Mice with the genotype of Prx1-Cre; Rictorf/f (hereafter RiCKO) were produced as previously described.20 The genotype of the mice was confirmed by PCR using mouse tail samples. Rictorf/f littermates were used as control animals. The use of animals in this study was approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (Approval NO.IACUC-1601205).
Cell Line and Cell Culture
The OS cell line K7M2 was cultured in α-MEM (HyClone, Logan, UT, USA), supplemented with 10% fetal bovine serum (v/v) (FBS, HyClone), 100 IU/mL penicillin, and 100 mg/mL streptomycin (HyClone) at 37 °C.
BMSCs were isolated from tibias and femora of 4-month-old mice, as described previously.20 Briefly, bone marrow cells were seeded into 100 mm tissue culture dishes in α-MEM (Gibco/Life Technologies, Carlsbad, CA, USA) containing 10% FBS and 1% penicillin/streptomycin. After 72 hours, the non-adherent cells were removed. On the seventh day, the cells were trypsinized for subsequent experiments.
Intratibial Implantation Models
Three-month-old sex-matched littermate pairs (Rictorf/f vs. RiCKO) were injected intratibially with K7M2 cells. As previously described, the K7M2 cell suspension was slowly injected into the left tibia using a 26G needle to generate OS.21 Mice were finally sacrificed and by cervical dislocation after inhaled anesthesia with ether at 40 days post injection. Total 17 successful sex-matched littermate pairs (Rictorf/f, n = 10; RiCKO, n = 7) with transplanted osteosarcoma were confirmed by skeletal radiography.
Skeletal Radiography and Micro-computed Tomography Analysis
The tibias with OS were dissected free of soft tissue. X-ray imaging was performed using a Faxitron model 805 (Faxitron Contact, Faxitron, Hennef, Germany) radiographic inspection system (22-kV voltage and 4-minute exposure time). Micro-CT was performed using a SkyScan 1072 scanner and analysis software (SkyScan, Antwerp, Belgium), with voxel size of 10.5 μm.
Histological and Immunohistochemical Analysis
Paraffin-embedded tissues were cut into 5-μm thick sections for histological analysis. For total collagen staining, sections were exposed to 1% Sirius red (Direct red) in saturated picric acid for 1 hour, then washed with water thoroughly to remove nonspecific staining and counterstained with hematoxylin.
Bone sections were deparaffinized, treated with 0.01 M sodium citrate buffer by heating at sub-boiling temperature for 15 minutes and processed for immunohistochemical and immunofluorescence staining. The staining process was performed with antibodies against collagen-I (1:100 dilution, Abcam, Cambridge, UK), osteocalcin (1:100 dilution, Abcam). The positive area normalized to bone surface was determined using ImageJ (https://imagej.nih.gov/ij/).
Western Blot and qPCR
Proteins were extracted from cells or tibias with OS, and 30 µg samples were fractionated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), then transferred to nitrocellulose membranes. Membranes were blocked with 5% BSA in tris buffered saline with 0.05% Tween 20 (TBS-T) and incubated with primary antibodies against p-Akt (S473) and Rictor (1:1000 dilution, Cell Signaling Technology); actin, SP7, RUNX2, Col-I, Bax, proliferating cell nuclear antigen (PCNA), cleaved-caspase3, vimentin, α-SMA, mmp9 (1:1000 dilution, Abcam), CCL-2, and CCL-5 (1:1000 dilution, Proteintech).
Total RNA was extracted from cells using RNAeasy mini kit (Qiagen, Valencia, CA, USA). One microgram (1 μg) of total RNA was reverse-transcribed to cDNA using iScript Reverse Transcription Supermix (Bio-Rad, Hercules, CA, USA). qPCR was performed with SYBR green Supermix (Bio-Rad). Expression levels were first normalized to β-actin, and then to control samples. The experiment was repeated 3 times. The primers used were listed as follows:
Runx2 Forward 5ʹ-CCG TGGCCTTCAAGGTTGT-3ʹ
Reverse 5ʹ-TTCATAACAGCGGAGGCATTT-3ʹ
Sp7 Forward 5ʹ-CCCTTCTCAAGCACCAATGG-3ʹ
Reverse 5ʹ-AAG GGTGGGTAG TCATTT GCATA-3ʹ
Alpl Forward 5ʹ-TGACCTTCTC TCCT C CATCC-3ʹ
Reverse 5ʹ-CTTCCTGGGAGTCTCATCCT-3ʹ
CCL2 Forward 5ʹ-ATGCAGTTAATGCCCCACTC-3ʹ
Reverse 5ʹ-TTCCTTATTGGGGTCAGCAC-3ʹ
CCL5 Forward 5ʹ-CTTGACCTGTGGACGACTGC-3ʹ
Reverse 5ʹ-ATCCAGTGAGAAAAGCCCGT-3ʹ
Migration
For transwell migration assay, K7M2 cells were first incubated in the culture supernatant of Rictor-deficient BMSCs for 72 hours and then 1 × 104 cells were transferred to the upper chamber of 24-well 8 μmTranswell chambers (Millipore, Massachusetts, UA) with 100 μL serum-free medium for 24 hours. The non-migrated cells were removed from the upper chamber with a cotton swab. The cells that had migrated through the membrane were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. The cell number was counted from at least 5 random central fields.
Serum Biochemistry
N-terminal propeptide of procollagen type I (PINP) was evaluated by enzyme immunoassay (EIA) (Rat/Mouse PINP EIA; IDS; Fountain Hills, AZ, USA). The animals were fasted for 6 hours before serum collection.
ELISA
Culture supernatants were collected, and the levels of CCL-2 and CCL-5 were measured by ELISA, according to the manufacturer’s instructions (Catalog no: ab208979 and ab100739, Abcam).
Statistical Analysis
Data indicate mean ± SD of triplicates from at least 3 separate experiments. Student’s t test was used to determine statistical significance (P < .05). Statistical analyses were performed using GraphPad Prism 7.0 (Graphpad Software Inc., La Jolla, CA, USA).
Results
Rictor Ablation in BMSCs Attenuates Bone Reconstruction After K7M2 Injection In Vivo
To explore the effect of targeted inhibition of the mTORC2 (Rictor) signaling pathway in BMSCs on growth of OS in vivo, K7M2 cells were injected intratibially into RiCKO or Rictorf/f mice (as controls) to construct OS models (Fig. 1A). After 40 days, OS growth and bone reconstruction were compared between the 2 mice genotypes by X-ray and micro-CT. The images revealed that both genotypes of mice displayed reactive trabecular hyperplasia (Fig. 1B and 1C). However, Codman’s triangle and sunray sign, which signified periosteum invasion, appeared in control mice on X-ray (Fig. 1B). Meanwhile, micro-CT analysis showed that the volume, thickness, and number of bone trabeculae were reduced in the medullary cavity of RiCKO mice (Fig. 1D). These results indicated that inhibition of the Rictor signaling pathway in BMSCs could reduce osteogenic bone reconstruction induced by K7M2 cells.
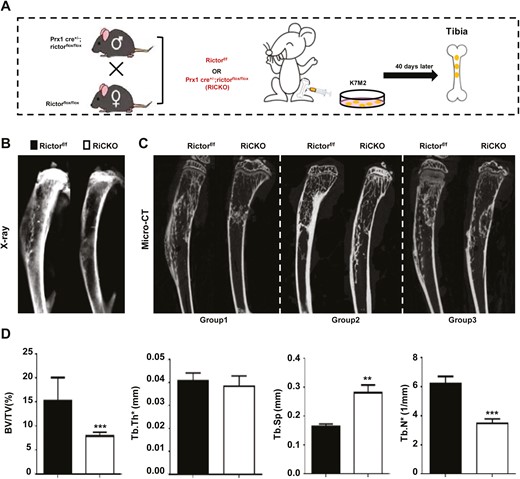
Rictor ablation in BMSCs attenuates osteoblastic lesions caused by osteosarcoma (OS) cell K7M2 injection in vivo. (A) Schematic diagram of OS mouse model preparation. Total 17 successful sex-matched littermate pairs (Rictorf/f, n = 10; RiCKO, n = 7) with transplanted osteosarcoma were obtained. Representative images of tibias with OS detected by (B) X-ray, and (C) micro-CT in longitudinal sections. The wireframes show the sites of OS. (D) Trabecular bone parameters in proximal tibia as seen on micro-CT. Data indicate mean ± SD of triplicates from individual mice **P < .01, ***P < .001.
Rictor Ablation in BMSCs Lessens Reactive Pathological Osteogenesis In Vivo
In accordance with the radiological data, the hematoxylin-eosin staining displayed a minor tumor area in RiCKO mice (Fig. 2A). Histochemical staining of total collagen and immunohistochemical staining of collagen I confirmed that less osteogenesis occurred in the RiCKO mice. Immunofluorescent staining revealed decreased expression of osteocalcin (OCN), a protein that indicates osteoblast activity, in the RiCKO mice. The serum level of PINP, a common marker of bone formation activity, was significantly lower in the RiCKO mice than in their control littermates (Fig. 2B). Consistent with the lower PINP levels, the expressions of several known osteoblast differentiation-related transcription factors, including Osterix (Sp7) and RUNX2 were significantly reduced in the RiCKO mice (Fig. 2C). In summary, these results indicated that Rictor ablation in BMSCs inhibited reactive pathological osteogenesis and osteoproliferation after injection of K7M2 cells and lessened bone reconstruction.
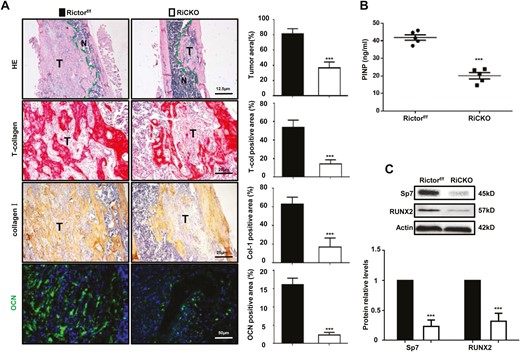
Rictor ablation in BMSCs lessens pathological osteogenesis in vivo. (A) Representative images and quantification of hematoxylin-eosin (HE), total collagen, collagen-I(Col-I), and osteocalcin (OCN) staining. T means tumor. (B) Serum PINP levels. (C) Western blot analyses and quantification of osteogenesis-related proteins extracted from tibial tissue with osteosarcomatous lesions. The protein expression levels are calculated as a ratio to the actin protein level and expressed relative to levels in group of Rictorf/f. ***P < .001.
Rictor Ablation Leads to Decreased Proliferation and Osteogenic Differentiation of BMSCs After Subculturing in TCM
It is generally considered that OS-induced proliferation and osteogenic differentiation of BMSCs are complementary mechanisms of bone reconstruction. To explore the influence of K7M2 cells on the proliferation and differentiation of BMSCs, K7M2 cell supernatant was collected as TCM for BMSCs culture (Fig. 3A). The genotypes of BMSCs isolated from the 2 genotypes of mice and Rictor-deficient BMSCs were further confirmed by detecting Rictor levels and p-Akt (S473), a direct readout of mTORC2 activity (Fig. 3B). After 72 hours, the proliferation and apoptosis of BMSCs were examined. Decreased EdU-positive nuclei and diminished cell proliferation of Rictor-deficient BMSCs were detected by EdU staining (Fig. 4A) and the MTT assay (Fig. 4B). Meanwhile, lower PCNA expression and increased expression of proapoptotic proteins Bax and cleaved-caspase3 in Rictor-deficient BMSCs were determined by western blot (Fig. 4C).
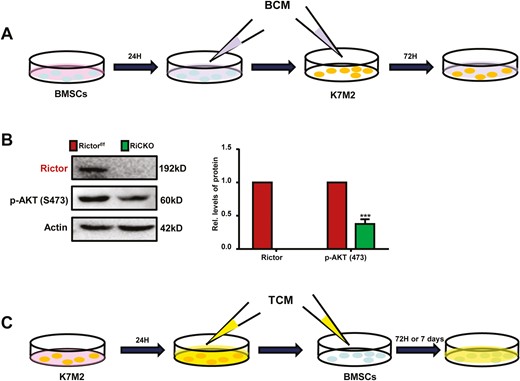
Schematic diagram of cell culture. (A) BMSCs isolated from Rictorf/f or RiCKO mice were grown in α-MEM supplemented with 10% FBS and 1% penicillin/streptomycin for24 hours, and then the supernatant was collected as BCM for subsequent culture of K7M2 cells. (B) Western blot analyses of mTORC2 pathway-related proteins. (C) Tumor-conditioned medium (TCM) was collected after growing K7M2 cells in α-MEM supplemented with 10% FBS and 1% penicillin/streptomycin for 24 hours and then filtered for subsequent culture of BMSCs.
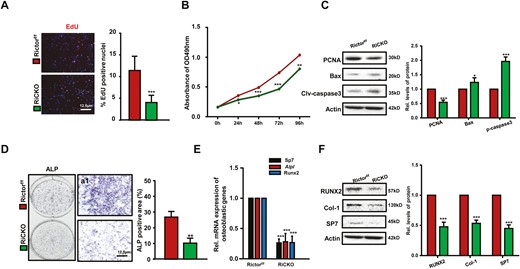
Loss of Rictor leads to decreased proliferation and osteogenic differentiation of BMSCs after subculturing in TCM. BMSCs isolated from Rictorf/f or RiCKO mice cells were cultured in TCM for 72 hours, the conditioned medium was changed every 24 hours, thrice. (A) Representative images of EdU staining and quantification of EdU-positive cells relative to total cells. (B) The growth and proliferation of BMSCs was investigated using MTT method. (C) Western blot analyses of PCNA, Bax, and cleaved-caspase3 expression in BMSCs cultured in TCM. (D) Representative ALP staining of osteoblasts differentiated from BMSCs induced by TCM and quantification of ALP-positive area relative to section area. (E) Quantitative Real-time PCR (q-PCR) analyses of osteogenic gene expression in BMSCs after culturing in TCM for 72 hours. (F) Western blot analyses and quantification of osteogenesis-related proteins extracted from BMSCs after culturing in TCM for 72 hours. *P <.05; **P < .01; ***P < .001.
To explore the influence of K7M2 cells on osteogenic differentiation of BMSCs, BMSCs were cultured in TCM for 1 week and then alkaline phosphatase (ALP) staining was performed. The results showed that positive area of ALP was diminished; the mRNA or protein levels of osteogenic differentiation-related transcription factors (Sp7 and RUNX2) and osteogenic activity-related proteins (ALP and Col-1) were significantly reduced in Rictor-deficient BMSCs (Fig. 4D-4F). These results suggested that targeted inhibition of the mTORC2 (Rictor) signaling pathway reduced the proliferation and osteogenic differentiation of BMSCs and was partly associated with alleviating K7M2-induced bone reconstruction.
Lower Bioactivity of K7M2 Cells Were Verified After Incubating in the Culture Supernatant From Rictor-Deficient BMSCs
To clarify the effects of Rictor ablation in BMSCs on K7M2 cells, the supernatants of Rictor-deficient BMSCs or control cells were collected as BCM RiCKO or BCM Rictorf/f for incubating K7M2 cells. Then, the proliferation, apoptosis, migration, and invasion abilities of K7M2 cells were analyzed after 72 hours (Fig. 3C). The results showed that compared with the control group, K7M2 cells cultured in BCM RiCKO displayed fewer EdU-positive cells, lower cell proliferation ability, decreased PCNA expression, and increased pro-apoptoti proteins Bax and cleaved-caspase3 (Fig. 5A-5C). In addition, the results of Transwell assay suggested that fewer cells migrated to the lower chamber in 24 hours ((Fig. 5D); furthermore, there was decreased expression of migration and invasion-related proteins, including vimentin, α-SMA, and mmp9 in K7M2 cells, which were cultured in BCM RiCKO (Fig. 5E and 5F). Next, the expressions of transcription factors that promote osteogenic differentiation (RUNX2, Sp7) or proteins related to osteogenic activity (Col-1) were detected in K7M2 cells, and the results confirmed reduced expression levels of RUNX2 and Col-1 proteins in K7M2 cells cultured in RiCKO BCM. These results indicated K7M2 cells in the culture supernatant of Rictor-deficient BMSCs showed lower bioactivity.
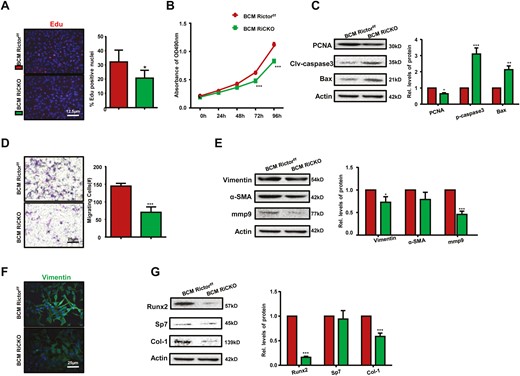
Lower capacities of proliferation, migration and osteogenesis in K7M2 cells are verified after incubating in the culture supernatant of Rictor-deficient BMSCs. K7M2 cells were incubating in the culture supernatant of BMSCs (BCM) for 72h, the medium was changed every 24 hours, thrice. (A) Representative images of EdU staining and quantification of EdU-positive cells relative to total cells. BCM Rictorf/f, K7M2 cells were cultured in BCM derived from Rictorf/f BMSCs; BCM RiCKO, K7M2 cells were cultured in BCM derived from Rictor-deficient BMSCs. (B) The growth and proliferation of K7M2 cells were investigated using the MTT method. (C) Western blot analyses and quantification of proliferative and apoptotic proteins extracted from K7M2 cells after culturing in BCM for 72 hours. The protein expression levels were calculated as a ratio to the Actin protein level and expressed relative to levels in the BCM Rictorf/f group. (D) Representative images and calculated number of migratory K7M2 cells. (E) Western blot analyses and quantification of migration or infiltration-associated proteins extracted from K7M2 after culture in BCM for 72 hours. (F) Representative images of vimentin staining of K7M2 after culture in BCM for 72 hours. (G) Western blot analyses and quantification of osteogenesis-related proteins extracted from K7M2. *P < .05; **P < .01; ***P < .001.
Rictor Ablation in BMSCs Reduces Secretion of Cytokines
To further explore whether Rictor ablation in BMSCs reduced cytokine secretion and thus inhibited the bioactivity of K7M2 cells, the expressions of more than 40 cytokines closely related to tumor proliferation, apoptosis, migration, invasion in BMSCs were examined using mouse cytokine array according to the manufacturer’s protocol (Catalog no. ARY006, Bio-techne). These cytokines included interleukins, chemokines, tumor necrosis factors, etc. The BMSCs genotypes were divided into 2 groups, one group was cultured in normal culture medium and the other group was cultured in TCM, and cell proteins were extracted after 72 hours for microarray detection. As shown in Fig. 6A, 8 kinds of cytokines were detected by the microarray, which were TIMP-1, M-CSF, CCL2, CD54, CCL3, IL-16, IL-1F3, and CXCL12; and 2 more cytokines CCL5 and IL-1α were newly found after culturing in TCM. Pixel analysis showed that of the above 10 cytokines, CCL2, CCL3, IL-16, and CCL5 were less expressed in Rictor-deficient BMSCs as compared to control cells (Fig. 6B).
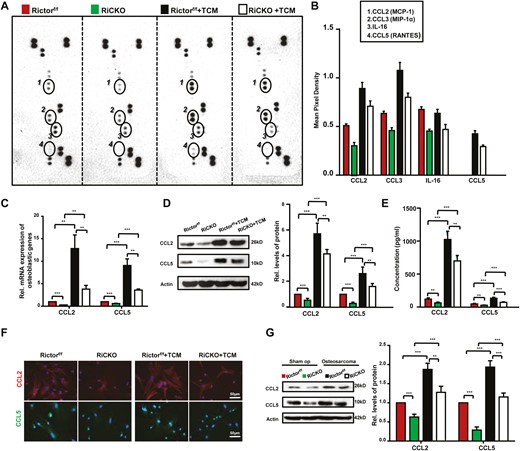
Rictor ablation in BMSCs downregulates the secretion of chemokines. Rictorf/f BMCSs and RiCKO BMSCs were treated with normal media or TCM for 72 hours. (A) Proteins extracted from BMSCs were used for array detection. (B) Circled spots are the proteins significantly differentially expressed among the different genotypes of BMSCs after densitometry analysis. Each cytokine/chemokine spot was normalized to positive and negative array controls. (C) Quantitative Real-time PCR (q-PCR) analyses and (D) Western blot of CCL2/5 expression in BMSCs. (E) ELISA analysis of CCL2/5 in culture supernatants from BMSCs. (F) Representative images show greater expressions of CCL2 and CCL5 in Rictorf/f BMCSs compared with RiCKO BMSCs. (G) Western blot analyses and quantification of CCL2/5 levels in tibial tissue from groups of sham operation and osteosarcoma. **P < .01; ***P < .001.
CCL2 (also known as monocyte chemotactic protein-1, MCP-1) and CCL5 (also known as RANTES) were demonstrated to be associated with OS cell malignant phenotype, including chemotherapy resistance and tumor progression, migration, and invasion, so we emphatically reexamined CCL2 and CCL5.22,23 We analyzed mRNA expression and protein levels using qRT-PCR and western blot, and concentrations in supernatant using ELISA (Fig. 6C-6E). The results confirmed lower mRNA and protein expressions and reduced secretion of CCL2 and CCL5 of Rictor-deficient BMSCs. Comparison between the cells in normal medium and in TCM, we found strongly expressions of CCL2 and CCL5 after culturing in TCM. Intracellular location of CCL2 and CCL5 were visually determined using immunofluorescent staining (Fig. 6F). To confirm our in vitro data, western blot analysis were performed in tibia of RiCKO or Rictorf/f mice belong to sham operation groups (as control) and transplanted osteosarcoma groups. Compared with controls, expression of CCL2 or CCL5 showed a significant increase in the transplanted osteosarcoma groups (P < .001). Within the same group, expressions of CCL2/5 showed lower in RiCKO mice (Fig. 6G). Together, these results indicated that Rictor ablation in BMSCs reduced cytokines secretion, and the expressions of CCL2/5 were proven to be less in vivo and in vitro.
Next, to confirm the influences of lower CCL2/5 secreted by Rictor-deficient BMSCs on bioactivity of K7M2 cells, we cultured K7M2 cells in BCM RiCKO, BCM Rictorf/f (as control group) or BCM Rictorf/f with cenicriviroc (CVC, Selleck) for 72 hours. CVC is a kind of a dual CCR2/5 antagonist used to blockade CCR2/5 signaling axis in K7M2 cells. The efficacy of CVC was confirmed by detecting protein expressions of phosphorylated STAT3 and Akt, which were downstream of CCR2/5 (Fig. 7A). In the group of K7M2 cells cultured in BCM Rictorf/f, we found CVC weakened the migration of, the number of cells crossing the membrane was significantly reduced (Fig. 7B). We also found a significant reduction in the proteins concerned with capacities for proliferation, migration, and osteogenesis of K7M2 cells after treated with CVC (Fig. 7C-7E). This result indicated that decreased secretion of CCL2/5 from Rictor-deficient BMSCs contributed to lower bioactivity of K7M2 cells, thus alleviated OS progression and bone destruction.
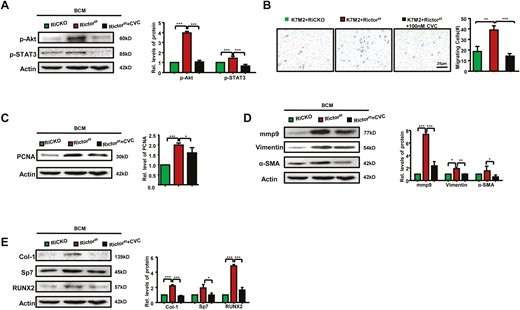
Bioactivity of K7M2 cells can be blocked by CVC. (A) Western blot analysis and quantification of phosphorylation of STAT3 (Tyr705) and Akt (ser473). (B) Representative images and calculated number of migratory K7M2 cells are shown. Western blot analyses and quantification of proteins concerned with proliferation (C), migration (D), and osteogenesis (E). *P < .05; **P < .01; ***P < .001.
Discussion
In the context of OS, bone homeostasis is destroyed by frequent osteolysis and osteogenesis, which stimulates the formation of characteristic osteoid matrix and causes bone reconstructure in patients.24-26 BMSCs are the progenitors of osteoblasts and are able to differentiate into mature osteoblasts under influence of some specific transcription factors, mainly RUNX2 and Osterix (also known as SP7).27-29 By deleting Rictor in the BMSCs with Prx1-Cre, we and others found impaired osteoblast differentiation in vitro and markedly lower mRNA level of Alpl in the Rictor-deficient cells, along with several other osteoblast markers, including Sp7, Ibsp, and Bglap.19,20 In addition, the Rictor-deficient cells expressed significantly less RANKL, even though other regulators of osteoclastogenesis, such as Opg and M-CSF, were relatively normal.21 These results indicate that mTORC2 signaling in BMSCs promotes osteoblast differentiation and concurrent osteoclastogenesis. In this study, we found Rictor ablation in BMSCs lessened bone reconstruction in vivo, the mRNA or protein levels of osteogenic differentiation-related transcription factors (Sp7 and RUNX2), and osteogenic activity-related proteins (ALP and Col-1) were significantly reduced in TCM-cultured Rictor-deficient BMSCs, which contributed to alleviate K7M2-induced bone reconstruction.
It has been suggested that BMSCs are important in the pathogenesis of OS; malignant proliferation and differentiation and osteogenesis following proto-oncogene activation in BMSCs are important pathogenic mechanisms of OS.30 In vivo studies revealed that overexpression of cMYC with loss of Ink4a/Arf can induce transformation of BMSCs to OS cells.31,32 Knockdown of P53 or double knockdown of Rb and P53 in BMSCs can induce formation of smooth muscle sarcoma, but knockdown of P53 and Rb after differentiation of BMSCs to osteoblasts can induce development of OS. In addition, BMP-2 and calcium ions in the bone marrow microenvironment affect the differentiation of P53−/−/Rb−/− BMSCs toward osteoblasts and also affect the osteogenic phenotype of OS.33 After the development of OS, BMSCs secrete chemokines CCL2, CCL5, CXCL8, IL, etc., which bind to the surface receptors of cancer cells and augment the proliferation, migration, and invasion abilities of cancer cells.34-39 Meanwhile, under the influence of BMSCs, OS cells also acquire stem cell potential, with increased expression of drug efflux pump proteins (Pgp, ABCG2, MDR-1, and MRP), activation of acetaldehyde dehydrogenase, and enhanced DNA damage repair ability, thereby becoming refractory to chemotherapeutic drugs which then have diminished clinical efficacy.40 In the present study, we found that Rictor deletion in BMSCs reduced the secretion of cytokines, which were closely related to bioactivity of OS cell.
Emerging evidence has shown that aberrant activation of mTOR is associated with the growth and metastatic behavior of OS. Inhibitors of mTOR can demonstrate anti-tumor effects in OS by suppressing cell growth and proliferation.41 However, mTOR-targeted monotherapy has shown modest anti-tumor activity in clinical trials, in part because inhibition of mTOR dually blocks mTORC1/2 activation in OS cells and then triggers autophagy, a pro-survival response contributing to drug resistance in OS treatment.42 Moreover, negative feedback loops are also induced by mTORC1 inhibition. There is significant interest in developing mTORC2-specific inhibitors that will keep the activities of mTORC1 intact, circumventing autophagy, feedback loops, and toxicities (including lesions within the oral mucosa, rash, and immune suppression) all of which are related to mTORC1 inhibition.
mTORC2 is emerging as a promising therapeutic target as its activity is essential for the transformation and viability of various types of cancer cells.43,44 Genomic aberrations in Rictor and mSin1 have been identified in several tumor types. Previous studies reported that targeted inhibition of mTORC2 prevented OS cell migration and promoted cell apoptosis in vitro. Specific inhibition of mTORC2 can promote cisplatin-induced apoptosis of OS cells.45 However, research into the function and regulation of mTORC2 in OS just gets started, and the comprehensive role of mTORC2 in OS treatment needs further exploration. To the best of our knowledge, this is the first study to confirm that mTORC2 inhibition in BMSCs restrains OS progression and bone reconstruction. It suggests that targeting mTORC2 not only works on restraining OS cells as previously reported but also influences BMSCs in tumor microenvironment, ultimately acquires synergistic anti-tumor effects.
Funding
This work is supported by the National Natural Science Foundation of China, Grant Number: 81200431, 82202932 and 81730066; Jiangsu Health Commission, Grant Number: M2021021; Youth Foundation of Jiangsu Natural Science Foundation, Grant Number: BK20220735.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author Contributions
Collection of data, data analysis, and interpretation: J.L. and D.D. Collection of data: J.Z. Administrative support: W.R. Provision of study material and financial support: D.M. Conception and design, assembly of data, and manuscript writing: W.S.
Data Availability
The data underlying this article are available in the article and in supplementary material.
References
Author notes
Jinhong Lu and Dongfang Dai. Contributed equally.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}