



Abstract
Glioblastoma (GBM) is the most common and deadliest tumor of the central nervous system. GBM has poor prognosis and glioma stem cells (GSCs) are implicated in tumor initiation and therapy resistance. Estrogen receptor β (ERβ) is expressed in GBM and exhibit tumor suppressive function. However, the role of ERβ in GSCs and the therapeutic potential of ERβ agonists on GSCs remain largely unknown. Here, we examined whether ERβ modulates GSCs stemness and tested the utility of two ERβ selective agonists (LY500307 and Liquiritigenin) to reduce the stemness of GSCs. The efficacy of ERβ agonists was examined on GSCs isolated from established and patient derived GBMs. Our results suggested that knockout of ERβ increased the proportion of CD133+ and SSEA+ positive GSCs and overexpression of ERβ reduced the proportion of GSCs in GBM cells. Overexpression of ERβ or treatment with ERβ agonists significantly inhibited the GSCs cell viability, neurosphere formation, self-renewal ability, induced the apoptosis and reduced expression of stemness markers in GSCs. RNA sequencing analysis revealed that ERβ agonist modulate pathways related to stemness, differentiation and apoptosis. Mechanistic studies showed that ERβ overexpression or agonist treatment reduced glutamate receptor signaling pathway and induced apoptotic pathways. In orthotopic models, ERβ overexpression or ERβ agonists treatment significantly reduced the GSCs mediated tumor growth and improved the mice overall survival. Immunohistochemical studies demonstrated that ERβ overexpression decreased SOX2 and GRM3 expression and increased expression of GFAP in tumors. These results suggest that ERβ activation could be a promising therapeutic strategy to eradicate GSCs.

Glioma stem cells (GSCs) are implicated in glioblastoma (GBM) development, progression and chemo/radiation resistance, and their elimination is essential for the development of effective therapies. The present study shows that estrogen receptor β (ERβ) activation by agonists or upregulation of its expression, reduced viability, self-renewal, and increased apoptosis in GSCs. Mechanistically, ERβ activation reduced glutamate receptor signaling in GSCs, reduced the tumor progression and improved the survival in orthotopic murine models. The present study proposes that ERβ agonists may represent promising therapeutic agents for eliminating GSCs, and thereby improving the overall survival of GBM patients.
INTRODUCTION
Glioblastoma (GBM), the most aggressive tumor of the central nervous system exhibits dismal prognosis.1,2 The 1-year survival rate for GBM is 40.8% and 5 year survival rate is 6.8%.2 Standard therapy for GBM consists of surgery followed by external beam radiation, adjuvant chemotherapy with temozolomide, and tumor treatment fields.3 Although patients may initially respond, they inevitably relapse and eventually succumb to therapy resistant disease. GBMs are highly infiltrative and migratory in nature and exhibit extensive cellular heterogeneity and plasticity.4-6 Glioma stem cells (GSCs) are implicated in tumor initiation, progression, and therapy resistance.5,6 Furthermore, GSCs contribute to tumor angiogenesis, immune evasion and possess high DNA repair capacity.7-11 Identifying the molecular mechanisms that contribute to stemness of GSCs and therapeutics that efficiently eliminate GSCs are urgently needed to improve outcomes in GBM.
GBM often exhibit gender bias with the incidence of GBM higher in men compared with women2 and hormones including estrogen playing a protective role in GBM development.12-14 Estrogen mediates its effects via estrogen receptor α (ERα, ESR1) and estrogen receptor β (ERβ, ESR2).15 ERβ is considered as a tissue specific tumor suppressor.16-21 ERβ plays a vital role in brain development, and neuronal and glial migration. Furthermore, it is strongly expressed in the human and mouse brain tissues.22-24 During brain development ERβ expression is induced in embryoid bodies and neural precursor cells (NPCs).24 Yet, little is known regarding the role and significance of ERβ signaling in GSCs.
Several selective synthetic and natural ERβ agonists have been identified and are being investigated for therapeutic use. LY500307 is a synthetic, potent and selective ERβ agonist that has a 12-fold higher affinity for ERβ than ERα and exhibits 32-fold more functional potency.25 Liquiritigenin (Liq), a natural, potent ERβ agonist isolated from the plant Glycyrrhiza uralensis, exhibits a 20-fold higher affinity for ERβ than for ERα.26 Furthermore, both LY500307 and Liq reduce the GBM cell viability, survival, migration and in vivo tumor progression.27,28 The effect of ERβ agonists on GSCs functions and GSCs mediated tumor progression remain unknown.
In the present study, we investigated the effect of ERβ (ERβ isoform 1) on stemness of GSCs and test the utility of ERβ agonists in reducing GSCs mediated functions. Our results demonstrated that ERβ knockout increases GBM GSCs representation whereas overexpression results in loss of GSCs. Furthermore, ERβ agonists reduce cell viability, sphere formation, and self-renewal of GSCs via the attenuation of glutamate receptor signaling. Finally, ERβ overexpression and ERβ agonist treatment reduced the GSCs mediated tumor progression and improved the survival of tumor bearing mice.
EXPERIMENTAL PROCEDURES
Cell culture, reagents, and generation of ERβ overexpression cell lines
Human GBM cell lines, U87, U251, and LN229 were obtained from the American Type Culture Collection (ATCC, Manassas, VA). GBM cells were maintained in DMEM supplemented with 10% fetal bovine serum (Sigma Chemical Co, St. Louis, Missouri). Neurobasal medium and B27 serum-free supplement and mGluR3 (GRM3) antibody were purchased from Invitrogen (Carlsbad, California). Nestin, SOX2, OCT-4, GFAP, and GAPDH antibodies were procured from Cell Signaling Technology (Beverly, Massachusetts). GADD45α and ATF-3 antibodies were procured from Santa Cruz Biotechnology (Dallas, Texas). GRIK5 and GRIK1 antibodies were purchased from Novus Biologicals (Littleton, Colorado). β-actin and all secondary antibodies were purchased from Sigma Chemical Co (St. Louis, Missouri). PE or APC conjugated CD133 antibodies were obtained from Miltenyi Biotec (Auburn, California). SSEA-1 antibody conjugated with APC or PE, OCT4 and SOX2 conjugated with Alexa Fluor 488, EGF, bFGF, LIF, and Annexin V/PI kit were purchased from BioLegend (San Diego, California). ERβ agonist LY500307 was purchased from APExBio Technology (Houston, Texas) and Liquiritigenin (Liq) was purchased from Biopurify Phytochemicals (Chengdu, China). The generation of U87 and U251 ERβ-KO and ERβ-FLAG cells were described earlier.29 Briefly, U87-ERβ-KO cells were generated using Santa Cruz CRISPR guide RNA plasmids (SC-400213). The gRNA sequences include (a) 5′-TGTATATGGAGCCGTGCTCC-3′; (b) 5′-TGTCTGCAGCGATTACGCAT-3′; (c) 5′-CGTTGCGCCAGCCTGTTAC-3′. U251-ERβ-KO cells were generated using Horizon CRISPR guide RNA plasmid (31033). The gRNA sequence is 5′-GATGGATTGACTGCAGTTGT-3′. ERβ-FLAG overexpression model cells were generated by infecting them with pLenti6/V5-D-FLAG ERβ and empty control vectors and stable clones were selected with blasticidine (5 μg/mL). Mission shRNA lentivirus vectors specific for ATF3 (TRCN0000329690), GADD45A (TRCN0000062350), GRM3 (TRCN0000357417), GRIK1 (TRCN0000423262), and GRIK5 (TRCN0000438409) were purchased from Sigma Chemical Co (St. Louis, Missouri).
Isolation of GSCs and flow cytometry
Patient-derived primary GBM cells were isolated from discarded specimens obtained from GBM patients undergoing surgery at UT Health San Antonio under an IRB approved biorepository and deidentified by the biorepository before provision. Patients provided informed consent for surgery and use of their tissues for research. Primary GBM lines GSC080409 (GSC08), GSC101310 (GSC10), GSC111010 (GSC11), and GSC040815 (GSC04) were cultured as neurospheres in neurobasal medium supplemented with B27 serum-free supplement, EGF (20 ng/mL), bFGF (20 ng/mL), LIF (10 ng/mL), and heparin (5 μg/mL) as described previously.30 Furthermore, GSCs were enriched using established stem cell marker CD133 from primary GBMs and U251 GBM cells by labeling with PE or APC-conjugated CD133 antibody and sorting by FACS. To induce differentiation, GSCs were cultured in 10% FBS for 7 days. To determine the percent CD133, SSEA-1, and OCT4 positive cells, ERβ-KO (U87 and U251) model cells and U87 and LN229 cells that stably express either empty vector or ERβ-FLAG vector were incubated with PE or APC conjugated CD133 or SSEA-1 or Alexa Fluor 488 conjugated OCT4 antibodies as per manufacturer's protocol and the percent of positive cell population was analyzed using flow cytometry.
Cell viability, Caspase3/7, and Annexin V assays
The effect of ERβ agonists LY500307 and Liq on the viability of GSCs was determined using CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, Wisconsin) according to the manufacturer's instructions. Briefly, GSCs were seeded in 96-well, flat, clear-bottom, opaque-wall microplates and treated with vehicle or LY500307 or Liq for 3 days and the total ATP content as an estimate of total number of viable cells was measured on an automatic Fluoroskan Luminometer. The effect of LY500307 and Liq on apoptosis was determined using Caspase3/7 glo assay as per the manufacturer's instructions. GSCs were treated with vehicle or LY500307 or Liq for 48 hours and the activity of caspase3/7 in terms of luminescence was determined using Promega GloMax Luminometer. Annexin V assay was performed as described previously.30 Briefly, GSC model cells were treated with either vehicle or LY500307 (10 μM) for 48 hours. Cells were then harvested and 100 μL of the cell suspension was incubated with Annexin V FITC and propidium iodide (PI) for 15 minutes at room temperature in the dark. Annexin V binding buffer (400 μL) was then added to each sample and stained cells were analyzed using flow cytometry. To determine the proliferation potential of ERβ model cells in real time, RealTime-Glo MT cell viability assay was performed as per the manufacturer's instructions. Briefly, ERβ model cells were seeded and assay reagent was added to cells during plating and the number of cells in culture was determined using Promega GloMax Luminometer.
Neurosphere formation and self-renewal assays
For neurosphere assays, single cell suspensions of GSCs were seeded in 24-well plates (100 cells/well) in triplicates and treated with vehicle or LY500307 (5 μM) or Liq (100 μM) for 7 days and the newly formed neurospheres were counted. The effect of ERβ agonists on self-renewal of GSCs was determined by extreme limiting dilution analysis (ELDA). Briefly, GSCs were seeded in decreasing numbers (50, 20, 10, 5, and 1 cells/well) in 96 well ultralow attachment plates and treated with vehicle or LY500307 (5 μM) or Liq (100 μM). After 14 days, the number of wells containing neurospheres per each plating density was recorded and stem cell frequency between control and treatment groups was calculated using ELDA software (http://bioinf.wehi.edu.au/software/elda/).
Western blotting
Whole cell lysates were prepared from GSCs using RIPA buffer containing protease and phosphatase inhibitors. Total proteins were mixed with SDS sample buffer and subjected to SDS-PAGE. Proteins were transferred onto nitrocellulose membranes and the blots were blocked with 5% non-fat dry milk powder solution for 1 hour at room temperature followed by incubation with primary antibodies overnight at 4°C. Blots were then incubated with secondary antibodies for 1 hour at room temperature and developed using the ECL kit (Thermo Fisher Scientific, Waltham, Massachusetts).
RNA sequencing and qRT-PCR
Cells were subjected to CD133 sorting and isolated GSCs were treated with either vehicle or LY500307 (5 μM) for 24 hours. Total RNA was isolated using RNeasy mini kit according to the manufacturer's instructions (Qiagen, Valencia, California). RNA sequencing and analysis were performed as described previously.31 Differential expression analysis was performed by DEseq, and significant genes with adjusted P value <.05 were chosen for analysis. The interpretation of biological significance of differentially expressed genes was determined with ingenuity pathway analysis (IPA) and gene ontology (GO). Gene set enrichment analysis (GSEA) was conducted on all expressed genes to study gene enrichment in specific pathway/function. Selected genes were validated using quantitative real time-PCR (qRT-PCR) using gene specific primers obtained from Harvard Primer Bank (http://pga.mgh.harvard.edu/primerbank/). Data were normalized to GAPDH or TBP1 and the difference in fold change was calculated using delta-delta-CT method.
Immunocytochemistry and immunohistochemistry
GSCs that stably express empty vector or ERβ-FLAG vector were seeded in poly-d-lysine/Laminin coated chamber slides and after 48 hours cells were fixed with 4% paraformaldehyde followed by permeabilization with 0.2% Triton X-100. Cells were then blocked in 5% normal goat serum followed by incubation with OCT4 primary antibody followed by fluorochrome-conjugated secondary antibody (Alexa Fluor 488) or Alexa Fluor 488 conjugated SOX2 antibody. 4′,6-Diamidino-2-phenylindole (DAPI) was used to visualize the nuclei and images were captured using confocal microscopy. Immunohistochemical studies were performed as described previously.32 Briefly, tumor tissue sections were incubated overnight with SOX2 antibody followed by secondary antibody incubation for 30 minutes at room temperature. Immunoreactivity was detected by using the DAB substrate and counterstained with hematoxylin. For the immunofluorescence studies, tissue sections were blocked in 5% normal goat serum for 1 hour at room temperature. Sections were then incubated with GFAP and GRM3 primary antibodies overnight at 4°C followed by incubation with fluorochrome-conjugated secondary antibody (Alexa Fluor 488) for 1 hour at room temperature. DAPI was used to visualize the nuclei and images were captured using confocal microscopy.
In vivo orthotopic tumor model
All animal experiments were conducted after obtaining UT Health San Antonio IACUC approval. Male athymic nude mice of 8-10 weeks old were purchased from Charles River (Wilmington, Missouri). CD133 positive U251-GSCs were transduced with empty vector (EV) or ERβ-FLAG vectors and injected (2 × 105) orthotopically into the mouse right cerebrum using established protocol.30 Mouse survival was determined using Kaplan-Meier survival curves and log-rank test using GraphPad Prism 8 software (GraphPad Software, San Diego, California). For patient derived GSCs experiment, GFP-Luciferase labeled CD133 positive GSC10 (1 × 104) cells were injected orthotopically into the mouse right cerebrum and after tumor establishment mice were randomized to receive vehicle (30% Captisol) or LY500307 (5 mg/Kg body weight/day) or Liq (20 mg/Kg body weight/day) orally in 30% Captisol. Tumor growth in terms of luciferase intensity (p/s) was measured weekly using the Xenogen IVIS system. At the end of the experiment, the mice were euthanized, and brains were isolated and processed for histological studies.
Statistical analyses
Statistical differences between groups were assessed using unpaired Student's t-test and one-way ANOVA using GraphPad Prism 8 software. All the data represented in graphs are shown as means ± SE and a value of P < .05 was considered as statistically significant.
RESULTS
ERβ knockdown increases and overexpression reduces the stemness of GBM cells
Several studies, including ours, have shown that ERβ signaling reduces cell proliferation and induces apoptosis of various cancer cell types; however, little is known about the role of ERβ signaling in GSCs. As shown in Supporting Information Figure S1A-C knockout of ERβ increased the proliferation rates of GBM cells whereas overexpression of ERβ resulted in reduction in proliferation. To determine the significance of ERβ in GSCs, we have analyzed the proportion of CD133 and SSEA-1 (CD15) positive cells, which are widely used GSCs specific phenotypic markers in GBM cells that under express or overexpress ERβ. Flow cytometry analysis of U87-ERβ knockout and U251-ERβ knockout cells showed that knockout of ERβ significantly increased the CD133 positive GSCs population compared with WT cells (Figure 1A,B,G,H). Furthermore, overexpression of ERβ in U87 cells significantly reduced the CD133 and SSEA-1 positive GSCs compared with control cells (Figure 1C,D,I). Similarly, overexpression of ERβ in other GBM cell line LN229 also significantly reduced the proportion of CD133 and SSEA-1 positive population compared with control cells (Figure 1E,F,J). We also examined the proportion of OCT4 positive cells in U251 ERβ model cells. Flow cytometry analysis revealed that the knockout of ERβ increased the abundance of OCT4 positive cells, however, its overexpression reduced the proportion of OCT4 cells compared with controls (Supporting Information Figure S1D-F). These results suggested that ERβ has potential to modulate the stemness of GBM cells.
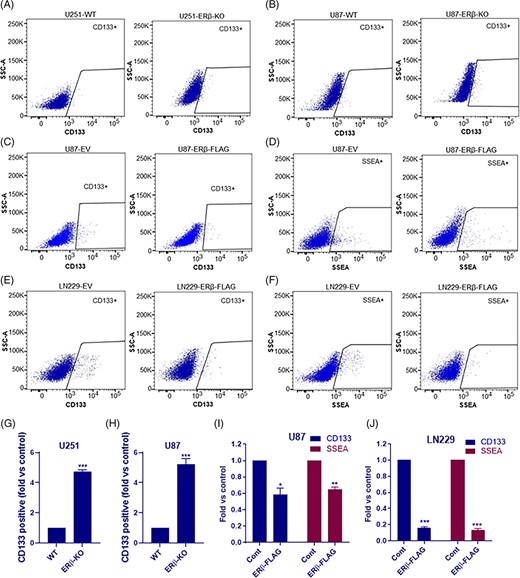
ERβ reduced the stemness of GBM cells. U251 (A, G) and U87 (B, H) WT or ERβ KO cells were stained with CD133 antibody and proportion of CD133-positive stem cells were determined using flow cytometry. U87-EV or U87-ERβ-FLAG cells were stained with CD133 (C, I) or SSEA-1 (D, I) antibodies and the proportion of CD133-positive and SSEA-1-positive stem cells were determined using flow cytometry. LN229-EV or LN229-ERβ-FLAG cells were stained with CD133 (E, J) or SSEA-1 (F, J) antibodies and the proportion of CD133-positive and SSEA-1-positive stem cells were determined using flow cytometry. Data are represented as mean ± SE. *P < .05; **P < .01; ***P < .001
ERβ agonist treatment reduces the cell viability, neurosphere formation, self-renewal, and induces the apoptosis of GSCs
We next examined the effect of the ERβ specific agonists LY500307 and Liq on the cell viability of GSCs. GSCs were isolated using established stem cell marker CD133 from primary patient derived GSC lines GSC101310 and GSC111011. CD133 positive-GSCs were treated with vehicle or ERβ agonists for 72 hours and the cell viability rates were determined. As shown in Figure 2A,B treatment with LY500307 and Liq significantly reduced the cell viability of GSCs. To determine whether ERβ agonists affect the neurosphere formation, GSCs were treated with LY500307 and Liq for 7 days and the number of newly formed sphered were counted. As shown in Figure 2C,D, LY500307 and Liq treatment significantly reduced the neurosphere formation ability of GSCs. To determine the effect of ERβ agonists on the self-renewal ability of GSCs, we have used limiting dilution assays. U251-GSCs that stably express EV or ERβ-FLAG vectors and CD133 positive GSC10 and GSC11 were seeded in decreasing numbers and treated with LY500307 or Liq. After 14 days, neurospheres per each plating density were determined and the stem cell frequency was analyzed using ELDA software. As shown in Figure 2E-G, overexpression of ERβ or treatment with ERβ agonists significantly reduced the self-renewal ability of GSCs compared with controls. To further examine the effect of ERβ agonists on apoptosis, GSC10 and GSC11 cells were treated with vehicle or LY500307 and apoptosis was measured by Caspase3/7 assay. As shown in Figure 2H, LY500307 treatment significantly induced apoptosis of GSCs. We then examined whether differentiation of GSCs promote alterations in ERβ expression. To induce the differentiation, GSCs were cultured in 10% FBS containing media for 7 days. As shown in Figure 2I, ERβ expression is significantly induced in differentiated cells compared with their undifferentiated counterparts.
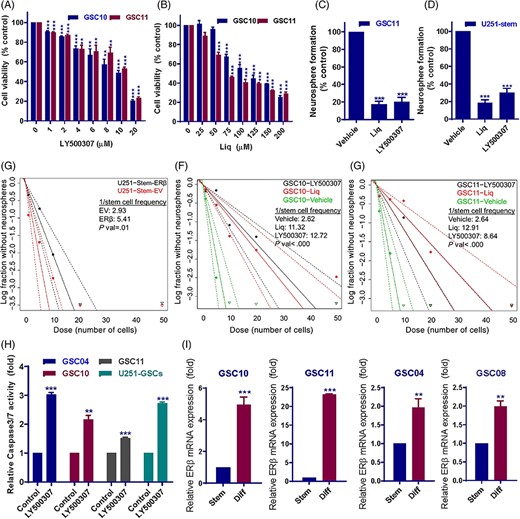
ERβ agonists reduced the cell viability, neurosphere formation, self-renewal, and promoted the apoptosis of GSCs. A and B, CD133 positive GSC10 and GSC11 cells were treated with vehicle or indicated concentrations of LY500307 or Liq for 72 hours and the cell viability was determined using CellTiter-Glo luminescence assay. C and D, CD133 positive GSC11 and U251-GSCs were seeded in 24 well plates (100/well) and treated with vehicle or LY500307 or Liq for 7 days and the number of neurospheres were counted. E, U251-GSCs that stably express EV or ERβ-FLAG vector were seeded in decreasing numbers (50, 20, 10, 5, and 1 cell/well) in 96 well plates and number of wells containing neurospheres per each plating density was recorded after 14 days. F and G, CD133-positive GSC10 and GSC11 cells were seeded in decreasing numbers (50, 20, 10, 5, and 1 cell/well) in 96 well plates and treated with vehicle, or LY500307 or Liq and number of wells containing neurospheres per each plating density was recorded after 14 days. Stem cell frequency between control and treatment groups was calculated using ELDA software. H, GSCs were treated with vehicle or LY500307 for 48 hours and the caspase3/7 activity was determined using caspsae3/7-Glo assay. I, GSCs were subjected to forced differentiation by culturing in the presence of 10% FBS for 7 days. ERβ expression was examined in stem and differentiated GSCs using qRT-PCR. Data are represented as mean ± SE. **P < .01; ***P < .001
ERβ overexpression or agonist treatment reduced the expression of stemness genes
To determine the role of ERβ on stemness of GSCs, we determined the expression of stem cell markers CD133, OCT4 and SOX2 in U87-ERβ-KO and ERβ overexpression cells. As shown in Figure 3A, qRT-PCR analysis showed that knockout of ERβ significantly increased the expression of stem cell markers while overexpression significantly reduced their expression. Next, we examined the expression of stemness markers in GSCs transduced with empty or ERβ expression vectors. As shown in Figure 3B-D, overexpression of ERβ significantly decreased the expression of stemness markers in GSCs derived from U251 and patient derived GSC lines. Furthermore, western blotting results showed that ERβ overexpression reduced the expression of stemness markers nestin and SOX2 (Figure 3E). Immunofluorescence analysis also revealed that ERβ-overexpressing GSC10 and GSC11 model cells have reduced expression of SOX2 and OCT4 when compared with control GSCs (Figure 3F,G). To further validate these findings, GSCs isolated from U251-GBM cells, GSC10 and GSC11 were treated with LY500307 and Liq for 5 days and analyzed for expression of stemness markers nestin and SOX2. Western blotting analysis showed that LY500307 and Liq treatment significantly reduced the expression of nestin and SOX2 in GSCs (Figure 3H-J). These results suggested that ERβ overexpression or agonist treatment reduces stemness of GSCs by downregulating the expression of stem cell markers.
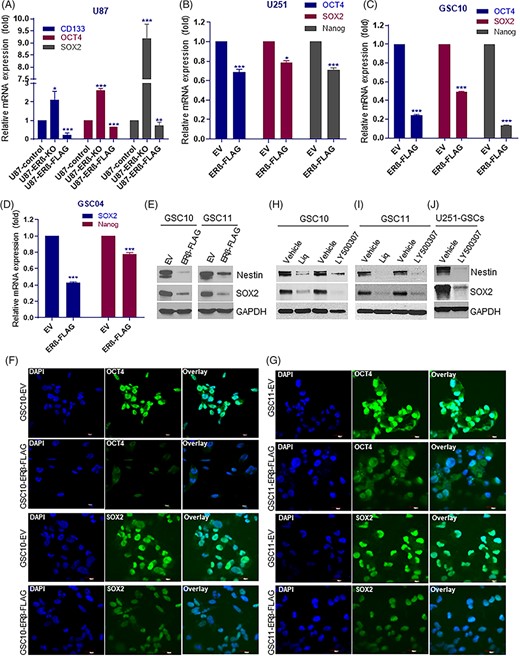
ERβ overexpression or agonist treatment reduced the expression of stemness markers. A, The expression levels of stem cell markers CD133, OCT4 and SOX2 in U87-ERβ knockout and overexpression model cells (A) and ERβ overexpression model cells of U251 (B) GSC10 (C) and GSC04 (D) cells was examined using qRT-PCR. E, CD133-positive GSC10 and GSC11 were transduced with either empty vector or ERβ-FLAG vector and the expression of nestin and SOX2 was determined using western blotting. CD133-positive GSC10 (F) and GSC11 (G) cells that stably express either empty vector or ERβ-FLAG vector were subjected to immunofluorescence staining of SOX2 and OCT4 and images were captured under confocal microscope. Scale bar = 20 μm. CD133-positive GSC10 (H), GSC11 (I) were treated with vehicle or LY500307 or Liq and the expression of nestin and SOX2 was determined using western blotting. J, U251-GSCs were treated with vehicle or LY500307 and the expression of nestin and SOX2 was determined using western blotting. Data are represented as mean ± SE. *P < .05; **P < .01; ***P < .001
Analysis of transcriptional changes altered by ERβ agonist in GSCs
To examine the global transcriptional changes in GSCs following ERβ agonist treatment, we performed RNA-sequencing on GSCs that were treated with either vehicle or LY500307. Differentially expressed genes with adjusted P value <.05 were chosen for analysis. Overall, 291 genes were differentially expressed in LY500307-treated GSCs: 113 genes were upregulated, and 178 genes were downregulated. A representative heat map is shown in Figure 4A. The sequenced data were deposited in the GEO database under accession number GSE162614. To examine the biological significance of the differentially expressed genes, pathway analysis was performed using IPA software and the top 10 canonical pathways altered by LY500307 are shown in Figure 4B. We found that the top pathway altered by LY500307 was the glutamate receptor pathway. Other pathways altered by LY500307 treatment include unfolded protein response, CREB signaling, death receptor signaling, PCP pathway, p53 signaling and NRF2-mediated oxidative stress response which are known to be involved in stemness and apoptosis. To understand the significance of differentially expressed genes, further analysis was performed using Gene Ontology. As shown in Figure 4C, differentially expressed genes were enriched in biological processes, such as regulation of programmed cell death, glutamate receptor signaling pathway, cell communication and response to unfolded protein. In terms of molecular function, the differentially expressed genes were mainly enriched in glutamate receptor activity, glutamate-gated and transmitter-gated ion channel activity (Figure 4D). Importantly, gene set enrichment analysis (GSEA) demonstrated that LY500307 regulated genes showed a negative correlation with the gene sets of glutamate receptor signaling pathway and positive correlation with unfolded protein response, p53 pathway, and apoptosis (Figure 4E).
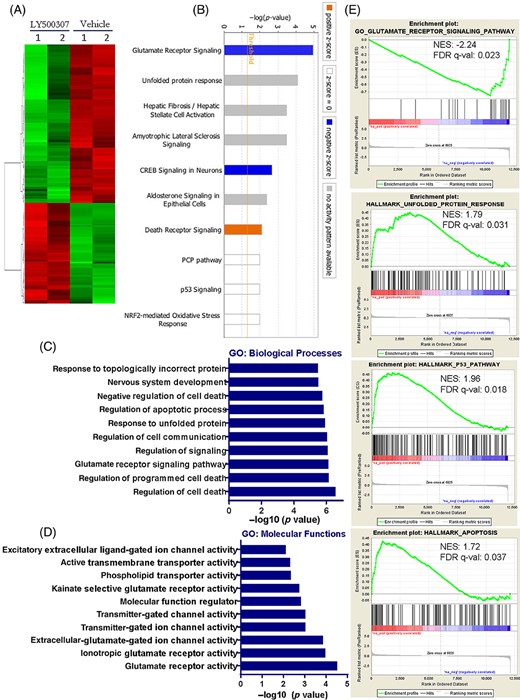
Global transcriptional changes regulated by ERβ agonist treatment. GSC11 cells were treated with vehicle or LY500307 (5 μM) and after 24 hours treatment total RNA was isolated and subjected to RNA-sequencing. A, Heat map of differentially expressed genes with adj-P < .05 between vehicle and LY500307 was shown. B, Differentially expressed genes were analyzed using ingenuity pathway analysis (IPA) program and the top 10 canonical pathways altered in LY500307 treated GSCs compared with vehicle were shown. C and D, Gene ontology (GO) analysis of differentially expressed genes in terms of either biological processes or molecular functions was examined using DAVID software. E, Gene set enrichment analysis (GSEA) testing correlation of LY500307-regulated genes with signatures of the glutamate receptor signaling pathway, unfolded protein response, p53 pathway, and apoptosis gene sets
ERβ activation attenuates glutamate receptor signaling in GSCs
The glutamate receptor signaling pathway plays an essential role in neurotransmission and normal cognitive functions and deregulation of glutamate receptor signaling is implicated in various neurodegenerative diseases.33,34 Importantly, recent studies demonstrated that the glutamate pathway is essential in neural stem cell maintenance and its inhibition promotes differentiation and apoptosis.35-38 Our RNA-seq analysis of LY500307-treated GSCs revealed that several genes of glutamate receptor signaling, such as GRM3, GRIK1, GRIK3, GRIA3, GRIA4, and GRIK5 were significantly downregulated in LY500307 treated group when compared with control (Figure 5A). We further validated these findings with other ERβ agonist, Liq. As shown in Figure 5B, Liq treatment significantly downregulated the expression of glutamate receptor signaling components in GSCs. Next, we examined whether overexpression of ERβ exhibited similar effects in GSCs. As shown in Figure 5C,D, overexpression of ERβ significantly reduced the expression of glutamate receptor signaling components in GSCs. We next validated these findings by examining the protein levels of glutamate receptor pathway components in ERβ overexpression GSCs models or after agonist treatment. As shown in Figure 5E, western blotting results showed that overexpression of ERβ markedly reduced the expression of GRM3, GRIK1 and GRIK5 in GSC10, GSC11 and GSC04 models. Furthermore, we also observed that treatment with ERβ agonists LY500307 and Liq resulted in decrease in GRM3, GRIK1 and GRIK5 expression in GSCs when compared with vehicle treated cells (Figure 5F).
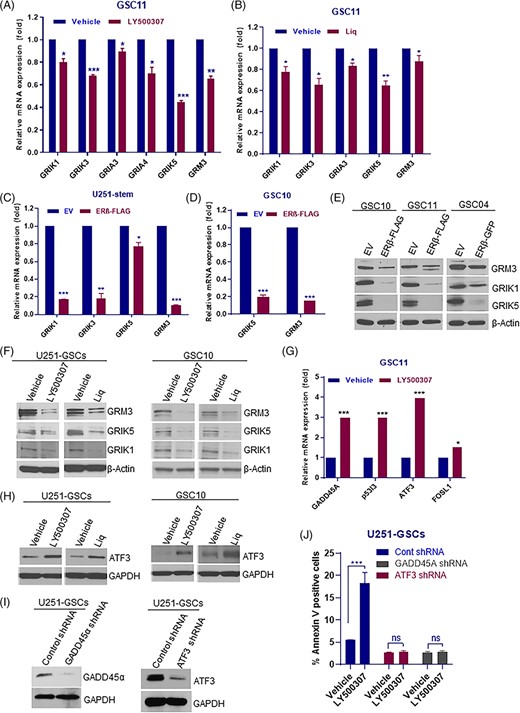
ERβ downregulated the glutamate receptor signaling in GSCs. A and B, GSC11 cells were treated with vehicle or LY500307 (5 μM) or Liq (100 μM) for 24 hours, and the expression of glutamate receptor pathway components were validated using RT-qPCR. C and D, U251-GSCs, and GSC10 cells were transduced with EV or ERβ-FLAG expression vector and the expression of glutamate receptor pathway components were analyzed using qRT-PCR. E, GSC10, GSC11, and GSC04 cells were transduced with EV or ERβ-FLAG expression vector and the expression levels of glutamate receptor pathway components were analyzed using western blotting. F, U251-GSCs and GSC10 cells were treated with vehicle or LY500307 (5 μM) or Liq (100 μM) for 24 hours, and the expression levels of glutamate receptor pathway components were determined using western blotting. G, GSC11 cells were treated with vehicle or LY500307 and the expression of apoptotic molecules was determined using qRT-PCR. H, U251-GSCs and GSC10 cells were treated with vehicle or LY500307 (5 μM) or Liq (100 μM) for 72 hours, and the expression of ATF3 was determined using western blotting. I, U251-GSCs were transduced with control shRNA or ATF3 shRNA or GADD45A shRNA and the knockdown of ATF3 and GADD45A was confirmed using western blotting. J, U251-GSCs control and ATF3-, GADD45A-knockdown cells were treated with vehicle or LY500307 for 48 hours, and the proportion of early apoptotic cell populations was determined using Annexin V assay. Data are represented as mean ± SE. *P < .05; **P < .01; ***P < .001
To determine the essential role of glutamate receptor pathway in stemness, GSCs were transduced with GRM3, GRIK1 and GRIK5 specific shRNAs and their self-renewal ability was determined using ELDA. As shown in Supporting Information Figure S2A-F knockdown of GRM3, GRIK1 and GRIK5 significantly reduced the self-renewal ability of GSCs when compared with control shRNA cells. To confirm the importance of glutamate receptor signaling pathway in stemness of GSCs, the expression of GRM3 and GRIK1 were examined in undifferentiated and differentiated GSCs. As shown Supporting Information Figure S3A,B, the expression of glutamate receptor pathway components was significantly reduced in differentiated cells than in undifferentiated GSCs. Altogether, these results suggest that ERβ activation significantly attenuates the glutamate receptor pathway leading to differentiation of GSCs.
Since, ERβ agonists also induce apoptosis, we further validated the expression of genes involved in apoptosis. As shown in Figure 5G, LY500307 treatment significantly increased the expression of apoptotic genes, such as, GADD45A, ATF3, and p53I3. Western blotting results also confirmed that ERβ agonists increased the expression of ATF3 in GSCs (Figure 5H). To validate the function link between these genes and ERβ mediated apoptosis, we knockdown ATF3 and GADD45A using lentiviral transduction in GSCs and treated them with ERβ agonist LY500307. Our results suggested that LY500307 significantly increased the apoptosis in GSCs that stably express control shRNA, however, LY500307 treatment did not induce apoptosis significantly in GSCs that stably express ATF3-shRNA or GADD45A-shRNA (Figure 5I,J). These results suggest that ERβ agonist mediated apoptosis involves the upregulation of ATF3 and GADD45A.
ERβ overexpression or agonist treatment suppressed the GSCs driven tumor progression and increased the survival of tumor bearing mice
Since our in vitro results demonstrated that ERβ activation decreases the stemness of GSCs, we further evaluated the effect of ERβ activation on GSCs driven tumor progression in vivo. To determine whether overexpression of ERβ suppresses the GSCs-mediated tumor progression and improves mice survival, CD133 positive U251-GSCs that were transduced with either empty vector or ERβ-FLAG vector were implanted intracranially in immunocompromised mice and survival was determined. As shown in Figure 6A, mice implanted with U251-GSCs expressing ERβ survived significantly longer compared with controls. To determine the therapeutic benefit of ERβ agonists on GSCs mediated tumor growth, CD133 positive GSCs separated from GSC101310 were transduced with GFP-luciferase expression vector and implanted into the mice brains. After tumors were established, mice were randomized to receive vehicle, LY500307 or Liq orally and tumor progression in terms of luciferase intensity was measured weekly using Xenogen in vivo imaging system. Compared with vehicle, LY500307 or Liq treatment significantly reduced the in vivo tumor growth of GSCs (Figure 6B).
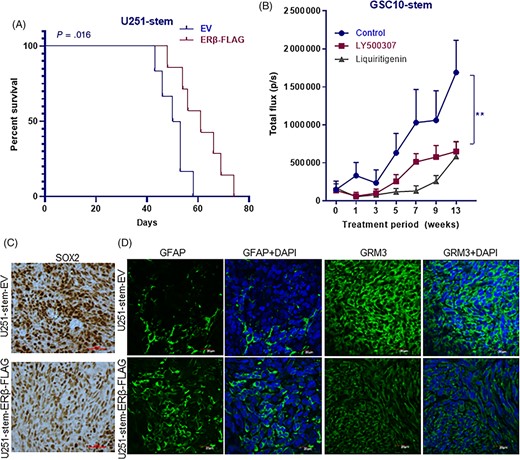
ERβ overexpression or ERβ agonist treatment reduced GSCs driven tumor progression. A, U251-GSCs were transduced with EV or ERβ-FLAG vector and implanted intracranially into the right cerebrum of nude mice and the survival was determined using Kaplan-Meier analysis (n = 6-7). B, GSC10 stably expressing GFP-luciferase were implanted orthotopically into the right cerebrum and after the tumors established, mice were treated with vehicle or LY500307 (5 mg/kg body weight/day) or Liq (20 mg/kg bodyweight) and the tumor growth in terms of luciferase intensity was monitored using Xenogen IVIS imaging every week (n = 6-7). C, Tumor sections collected from EV or ERβ-FLAG expressing U251-GSCs groups were processed and subjected to immunohistochemical staining for SOX2. D, Tumor sections collected from EV or ERβ-FLAG expressing U251-GSCs groups were processed and subjected to immunofluorescence staining for GFAP and GRM3. DAPI was used to visualize the nuclei. Data are represented as mean ± SE. **P < .01
To examine the effect of overexpression of ERβ on stemness and differentiation in vivo, tumor sections derived from empty vector and ERβ-FLAG expressing U251-GSCs tumors were analyzed immunohistochemically. As shown in Figure 6C, overexpression of ERβ significantly reduced the expression of stem cell marker SOX2 in tumors. Furthermore, the differentiation marker GFAP was significantly increased in ERβ overexpressing tumors compared with controls (Figure 6D). To validate the effect of ERβ-KO on stemness in vivo, we have determined SOX2 expression in the tumor sections collected from U251 and U251-ERβ-KO tumor bearing mice.29 Immunohistochemical analysis showed that ERβ-KO tumors had high expression of SOX2 compared with control tumors (Supporting Information Figure S3C). To examine whether ERβ overexpression reduces glutamate receptor pathway in vivo, the expression of GRM3 was examined immunohistochemically on tumor sections. As shown in Figure 6D, ERβ overexpressing tumors exhibited reduced expression of GRM3 compared with control tumors suggesting that the overexpression of ERβ attenuates glutamate receptor pathway in vivo.
DISCUSSION
The role of ERβ in stemness and differentiation of GSCs has not been previously elucidated. Furthermore, the potential of ERβ agonists as potential therapeutics via altering GSCs mediated functions has not been explored. In this study, we provided evidence that ERβ expression is increased in differentiated cells; and overexpression of ERβ or agonist treatment reduces the cell viability, sphere formation, and self-renewal of GSCs. Mechanistic studies demonstrated that ERβ agonist treatment or overexpression reduced the glutamate receptor pathway in GSCs. Furthermore, our results demonstrated that ERβ overexpression or agonist treatment has the potential to mitigate GSCs mediated tumor progression in vivo.
Recent studies demonstrated the involvement of ERβ in stem cells. In a prostate cancer study, activation of ERβ impairs prostatic regeneration by inducing apoptosis in stem/progenitor cells.39 Furthermore, agonist mediated activation of ERβ attenuates self-renewal of murine prostatic stem/progenitor cells.39 Another study has reported that ERβ-KO NPC exhibited higher proliferation rates and ERβ agonist treatment reduced their proliferation.24 Our studies support and are in agreement with these studies. Our results demonstrated that overexpression of ERβ reduces the proportion of GSCs, whereas ERβ knockout increases GSCs population. Furthermore, we also observed that ERβ expression induced in differentiated cells compared with undifferentiated GSCs; and overexpression of ERβ or agonist treatment reduced their viability, sphere formation, and self-renewal ability. Moreover, we observed a reduced expression of stemness markers nestin, SOX2, OCT4 and NANOG in GSCs after overexpression of ERβ or agonist treatment. Furthermore, in vivo data also supports the role of ERβ in reducing the stemness of GBM. We observed that reduced expression of stemness marker SOX2 and an increased expression of differentiation marker GFAP in ERβ overexpressing tumors compared with control tumors.
A recent study demonstrated that breast cancer stem cells (BSCs) express ERβ, not ERα; additionally, upregulation of ERβ in BSCs was associated with phenotypic stem cell markers.40 Furthermore, this study demonstrated that forced differentiation of BSCs was associated with a reduction in ERβ expression. Interestingly their study also showed that stimulation of ERβ by DPN treatment caused a decrease in mammosphere formation of normal mammary stem cells (MSCs), suggesting that ERβ is not proliferative in the MSCs population.40 This discrepancy could be in part due to the tissue specific effects of ERβ and the presence or absence of various coactivators or corepressors or differential interactome. ERβ exists in multiple isoforms which exhibit distinct roles in numerous cancers.41 The full length agonist activated ERβ1 is widely regarded as tumor suppressor; however, other isoforms in particular ERβ2 and ERβ5 are overexpressed in several cancers and associated with poor prognosis of breast, prostate and ovarian cancer patients.42-46 Interestingly, a recent study suggests that increased expression of the ERβ variants, β2 and β5, induce stem cell characteristics and chemotherapy resistance in prostate cancer through activation of hypoxic signaling.47 Since ERβ isoforms expression is tissue specific and mediates different functions and the levels of other isoforms dictate the function of tumor suppressive ERβ1, it is likely that it may have distinct roles in the cancer stem cell phenotype as well as in a tissue specific manner. Further counteracting oncogenic functions of other ERβ isoforms and ratio of levels of ERβ1 with other ERβ isoforms also has implications in the cancer stem cell phenotypes.
Glutamate is the major excitatory neurotransmitter molecule in the brain, and plays a crucial role in various neural functions including learning and memory, long-term potentiation, and synaptic plasticity.34,48 Glutamate exerts its signaling primarily through binding to glutamate-activated receptors. Glutamate receptors (GluRs) are classified into ionotropic glutamate receptors (iGluRs), which are ligand-gated cation channels, and the metabotropic glutamate receptors (mGluRs) which are G protein-coupled receptors.34,48 iGluRs family includes NMDA, AMPA, and Kainate receptor subfamilies and mGluRs are grouped into group I, II, and III subfamilies based on their sequence homology, signaling mechanisms and pharmacological properties.34,49 It has been shown that mGluRs have a significant effect on the proliferation, differentiation and survival of NSCs.35 Our RNA-sequencing analysis using IPA and GSEA demonstrated that the top pathway modulated by ERβ agonist treatment is glutamate receptor signaling. Further validation studies confirmed many of the genes in the glutamate receptor pathway were down regulated following treatment with both ERβ agonists LY500307 and Liq in multiple GSC lines. Our results also demonstrated similar findings using ERβ overexpressing GSC models.
Aberrant mGluR signaling is involved in the progression of various cancer types, including malignant brain tumors and mGlu3 receptors were expressed in GBM tissues, GSCs, GBM cell lines.50-52 Pharmacological activation of mGlu3 receptors enhances the proliferation and survival of GSCs, and that, in contrast, inhibition of mGlu3 induces GSCs differentiation into astrocytes.53,54 In addition to mGluRs, GBM cells also express iGluRs subunits and iGluR antagonists decrease GBM cell proliferation.48,55,56 GBM cells release high levels of glutamate, which regulates the proliferation and survival of NPCs.36 Moreover, tumors expressing low levels of mGluR3 correlated with a better survival rate in GBM patients.50,57 mGluR3 antagonists promoted astroglial differentiation and enhanced the cytotoxic activity of TMZ.50,51,57 Altogether these studies implicated glutamate receptor signaling in GBM progression and stemness. Our results suggest that attenuation of glutamate receptor pathway by ERβ overexpression or agonist treatment contribute to the reduction in stemness and induction of differentiation of GSCs. We also observed a decrease in glutamate receptors GRM3 and GRIK1 during the differentiation of GSCs. Importantly, ERβ agonist treatment resulted in a reduction in the levels of both metabotropic glutamate receptors (GRM3) and ionotropic glutamate receptors (GRIK1, GRIK3, GRIK5, GRIA3, and GRIA4). Furthermore, we observed a reduction in GRM3 expression in ERβ overexpressing tumors compared with control tumors. Our results also confirmed that knockdown of glutamate receptor pathway components such as GRM3, GRIK1, and GRIK5 resulted in decrease of self-renewal ability of GSCs suggesting that suppression of the glutamate receptor activity is sufficient to modulate stem-like phenotype of GSCs. RNA-seq data also showed alteration of genes in death receptor signaling and p53 signaling. GSEA showed that p53 pathway and apoptosis pathways were positively correlated with LY500307 modulated gene signatures. Furthermore, we found that LY500307 regulated genes showed a positive correlation with unfolded protein response which also shown to be involved in differentiation and apoptosis of GSCs.30 Functional validation results also confirmed that knockdown of proapoptotic molecules such as ATF3 and GADD45A compromised the ERβ agonist mediated induction of apoptosis in GSCs.
CONCLUSION
In summary, our study demonstrated that ERβ plays an essential role in GSCs and activation of ERβ reduced the stemness of GSCs in vitro and in in vivo by decreasing glutamate receptor signaling. These findings suggest that activation of ERβ may curtail GSC stemness and ERβ agonist may represent promising therapeutics for the treatment of GBM.
ACKNOWLEDGMENTS
We thank Jessica Perry (Ob/Gyn, UTHSA) for proofreading this manuscript. This study was supported by NIH-CA178499 (R.K.V., A.J.B.), Mays Cancer Center Support Grant P30CA054174-17, American Brain Tumor Association Discovery Grant, Max and Minnie Tomerlin Voelcker Fund Young Investigator Grant (G.R.S.) and NIH South Texas Medical Scientist Training Program (T32GM113898; S.A.). Data generated in the Genome Sequencing Facility was NIH Shared Instrument Grant 1S10OD021805-01 (S10 grant) and CPRIT Core Facility Award (RP160732). Flow cytometry data was generated in the UT Health San Antonio Flow Cytometry Shared Resource Facility, which is supported by UT Health San Antonio, the Mays Cancer Center P30 Cancer Center Support Grant (NIH-NCI P30 CA054174) and the National Center for Advancing Translational Sciences, NIH, through grant UL1 TR002645.
CONFLICT OF INTEREST
A.J.B. declared patent ownership with RNL186/NanoTX; advisory role with Vascular Biogenics, Alamab, Plus Therapeutics; research funding with Immunomedics and stock ownership with NanoTx, Plus Therapeutics. The other authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
G.R.S.: designed the experiments and interpreted the results, conducted the experiments, wrote the manuscript; U.P.P., P.P.V., M.Z., S.A., S.V.: conducted the experiments; R.R.T.: designed the experiments and interpreted the results; A.J.B., R.K.V.: designed the experiments and interpreted the results, wrote the manuscript.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
Author notes
Funding information National Cancer Institute, Grant/Award Number: CA178499; National Center for Advancing Translational Sciences, NIH, Grant/Award Number: UL1 TR002645; CPRIT Core Facility Award; NIH Shared Instrument Grant, Grant/Award Number: 1S10OD021805-01; NIH South Texas Medical Scientist Training Program, Grant/Award Number: T32GM113898; Max and Minnie Tomerlin Voelcker Fund Young Investigator Grant; American Brain Tumor Association Discovery Grant; Mays Cancer Center Support Grant, Grant/Award Number: P30CA054174-17
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}