



Abstract
Quiescence and self-renewal of human corneal epithelial progenitor/stem cells (LEPC) are regulated by the limbal niche, presumably through close interaction with limbal (stromal) niche cells (LNC). Paired box homeotic gene 6 (Pax6), a conserved transcription factor essential for eye development, is essential for proper differentiation of limbal and corneal epithelial stem cells. Pax6 haploinsufficiency causes limbal stem cell deficiency, which leads to subsequent corneal blindness. We previously reported that serial passage of nuclear Pax6+ LNC resulted in the gradual loss of nuclear Pax6+ and neural crest progenitor status, the latter of which was reverted upon recovery of Pax6. These findings suggest Pax6 plays a pivotal role in supporting the self-renewal of LEPC in limbal niche. Herein, we show that HC-HA/PTX3, a unique matrix purified from amniotic membrane (AM) and consists of heavy chain 1of inter-α-trypsin inhibitor covalently linked to hyaluronic acid and complexed with pentraxin 3, is capable of reverting senescent LNC to nuclear Pax6+ neural crest progenitors that support self-renewal of LEPC. Such reversion is causally linked to early cell aggregation mediated by activation of C-X-C chemokine receptor type 4 (CXCR4)-mediated signaling followed by activation of bone morphogenetic protein (BMP) signaling. Furthermore, CXCR4-mediated signaling, but not BMP signaling, controls recovery of the nuclear Pax6+ neural crest progenitors. These findings not only explain why AM helps in vivo and ex vivo expansion of human LEPC, but they also illuminate the potential role of HC-HA/PTX3 as a surrogate matrix niche that complements stem cell-based therapies in regenerative medicine.

The authors show that HC-HA/PTX3 purified from human amniotic membrane uniquely restores senescent limbal niche cells to nuclear Pax6+ neural crest progenitors to support self-renewal of limbal progenitor/stem cells. Such reprogramming is linked to early cell aggregation mediated by activation of C-X-C chemokine receptor type 4 (CXCR4) mediated signaling highlighted by transient nuclear translocation of CXCR4 followed by activation of bone morphogenetic protein signaling. This finding not only explains why amniotic membrane transplantation helps in vivo and ex vivo expansion of human limbal epithelial stem cells but also envisage that this new paradigm based on regenerative matrix HC-HA/PTX3 as a surrogate niche can set a new standard for regenerative medicine in and beyond ophthalmology.
INTRODUCTION
The corneal epithelium is composed of stratified, nonkeratinized, squamous epithelial cells that protect the ocular surface while maintaining corneal transparency and clear vision. The quiescence, self-renewal, and fate decision of corneal epithelial stem cells are regulated within the limbal niche in distinctive anatomical features known as the palisades of Vogt. Within this locale, limbal epithelial progenitor/stem cells (LEPC) reside in the basal epithelial layer1 in palisade epithelial ridges that extend deep into the stroma beyond the limbal basement membrane to form limbal epithelial crypts.2,3
We have successfully isolated limbal niche cells (LNC) by keeping them in close proximity with LEPC and have characterized them as a unique subset of mesenchymal cells that express embryonic stem cell (ESC) and neurovascular markers.4-6 To recapitulate the in vivo niche support of LNC, we established an in vitro reunion of isolated single LEPC and LNC in three-dimensional (3D) Matrigel (MG) and noted that restoration of their close contact led to sphere growth of LEPC through C-X-C chemokine receptor type 4 (CXCR4)/stromal derived factor (SDF-1) signaling similar to their in vivo pattern expression of CXCR4 by LEPC and SDF-1 by LNC.7 Such close contact is pivotal in preventing corneal cell fate decision and maintaining self-renewal of LEPC.7 This supporting niche function is mediated by the activation of canonical Wnt signaling and the inhibition of bone morphogenetic protein (BMP) signaling in LNC.8
Paired box homeotic gene 6 (Pax6) is an evolutionally conserved transcription factor that is essential for proper development of the eyes, central nervous system, craniofacial skeleton, olfactory epithelium, and pancreas (reviewed in Reference 9). The primary function of Pax6 in the eye is to mediate the commitment of the head ectoderm of the optic vesicle into the lens ectoderm and promote the formation of the lens vesicle.10 Homozygous Pax6-deficient mice embryos exhibit lack of eyes and nose and die soon after birth.11,12 Expression of Pax6 is dosage dependent, as mutation or missing allele leads to small eye mutants (Sey, Pax6+/−) in mice13 and aniridia in humans (reviewed in Reference 9), which is a prototypic genetic disease that causes corneal blindness due to limbal stem cell deficiency.14,15 Postnatal expression of Pax6 is restricted to corneal and limbal epithelial cells.16 Abnormal levels of Pax6 in human17 and mice16 corneal epithelium leads to abnormal differentiation. Interestingly, Pax6+/− heterozygous mice corneas also exhibit profound defects in the stroma and endothelium but less so than is observed in the epithelium.18,19 We have shown that LNC possess neurovascular phenotypes6 with multipotent capabilities for angiogenic, tri-lineage and neuroglial differentiation.5,6,20 The above neural crest progenitor status is lost through serial passage but can be regained by overexpression of Pax6 via adenovirus in late-passaged P10 LNC; recovery of neural crest progenitor status enables LNC to prevent corneal fate decision and maintain self-renewal of LEPC upon in vitro reunion.6 These salient features of LNC, including their possession of the neural crest progenitor phenotype highlighted by nuclear Pax6 staining, are summarized in a recent review.21
Since its reintroduction in 1995, transplantation of cryopreserved human amniotic membrane (AM) has been shown to promote wound healing following ocular surface reconstruction due to its anti-inflammatory22-24 and anti-scarring properties.25,26 Transplantation of AM also augments the success of in vivo27-29 and ex vivo30,31 expansion of limbal stem cells to treat corneal blindness caused by limbal stem cell deficiency. Through further exploration of AM's mechanism of action, we successfully purified HC-HA/PTX3 from water-soluble AM extract, and identified it as a unique matrix consisting of high molecular weight hyaluronic acid (HA) covalently linked with heavy chain 1 (HC1) from inter-α-trypsin inhibitor (“–” is used to denote the covalent linkage) and further complexed with pentraxin 3 (PTX3) (“/” is used to denote the noncovalent linkage).32-34 HC-HA/PTX3 has been shown to exert anti-inflammatory actions that extend from innate immune responses, through facilitating apoptosis of stimulated neutrophils and polarizing M2 macrophages, to adaptive immune responses, by suppressing the activation of Th1 and Th17 lymphocytes to downregulate alloreactive immune responses.35,36 In addition, HC-HA/PTX3 reverts human corneal fibroblasts and myofibroblasts to keratocytes by inducing cell aggregation mediated by CXCR4 signaling followed by activation of BMP signaling.37 Different from 3D MG, in vitro reunion of LEPC and LNC on immobilized HC-HA/PTX3 promotes cell aggregation and sphere growth highlighted by quiescence of LEPC by activating BMP and noncanonical Wnt (PCP) signaling in LNC.38 Herein, we discovered that HC-HA/PTX3 differs from 3D MG in reverting senescent (differentiated) late passaged LNC to the nuclear Pax6+ neural crest progenitor phenotype by promoting early cell aggregation through CXCR4-mediated signaling followed by activation of BMP signaling.
RESULTS
Progressive loss of nuclear Pax6+ neural crest phenotype in senescent limbal niche cells by serial passage
Serial passage of LNC leads to decreased cell doubling time and the loss of the neural crest progenitor status that is characterized by nuclear Pax6 staining, the expression of ESC markers and LNC progenitor markers, such as SRY box transcription factor 2 (Sox2), p75 neurotrophin receptor (p75NTR), Musashi-1, Nestin, Msh homeobox 1 (Msx1), and Forkhead box D3 (FoxD3), and the capacity to undergo neuroglial differentiation.6 To determine whether the above phenotypic change was associated with differentiation-induced senescence, we repeated the experiment and found that the transcript expression of Pax6, Sox2, p75NTR, Musashi-1, and Nestin by P10 LNC was significantly reduced when compared with that of P2 LNC (Figure 1A, ##P < .01, n = 3). This phenotypic change was coupled with the loss of nuclear Pax6 staining and notable reduction of staining to such NC markers, such as p75NTR and Musashi-1, when compared with P4 LNC (Figure 1B). Histochemical staining of β-galactosidase (SA-β-gal) has been detected in senescent cells in cell culture and aged skin in vivo.39 Our results showed that serial passage of cultured LNC significantly increased β-galactosidase in P10 LNC when compare to P4 LNC (Figure 1C, **P < 0.01, n = 1000). Therefore, we concluded that the senescence of LNC by serial passage was associated with the loss of the nuclear Pax6+ neural crest progenitor phenotype.
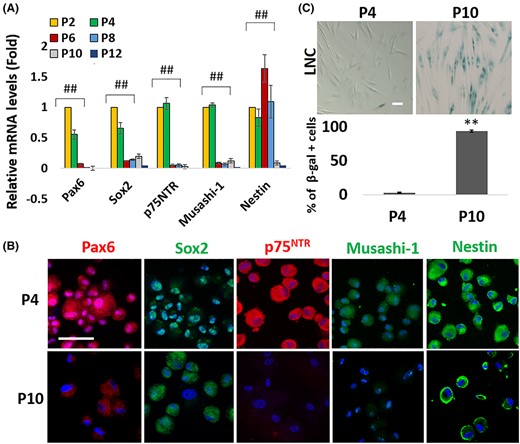
Progressive loss of nuclear Pax6 neural crest progenitor status in LNC after serial passage is accompanied by senescence
Immobilized HC-HA/PTX3 promotes cell aggregation and reverts senescent limbal niche cells to nuclear Pax6+ neural crest progenitors
P4 LNC expanded on coated MG in modified embryonic stem cell medium (MESCM) aggregate when reseeded on 3D MG5,40 or immobilized HC-HA/PTX3,38 the latter of which also helps to regain expression of ESC markers.5 Thus, we wondered whether P10 LNC might behave in the same manner and regain the nuclear Pax6+ neural crest progenitor status when reseeded on immobilized HC-HA/PTX3. After seeding of P10 LNC on coated MG, 3D MG, or immobilized HC-HA/PTX3 in MESCM for 48 hours, cell aggregation was similarly promoted between 3D MG and immobilized HC-HA/PTX3 (Figure 2A). HC-HA/PTX3, but not 3D MG, significantly reduced β-galactosidase-positive senescent cells (Figure 2B, **P < 0.01, n = 1000). However, transcript levels of Pax6, p75NTR, Musashi-1, Nestin, Msx-1, and FoxD3 were significantly upregulated on immobilized HC-HA/PTX3 when compared with coated MG (Figure 2C, **P < .01, n = 3) or 3D MG (Figure 2C, ##P < .01, n = 3). The immunofluorescence staining confirmed the reappearance of nuclear Pax6 staining (see inset). Furthermore, the significant difference was readily appreciated by the positive and strong staining of p75NTR on HC-HA/PTX3 and negative staining in 3D or coated MG and the stronger staining of Sox2 and Musashi-1 on HC-HA/PTX3 than 3D or coated MG without any difference in nestin (Figure 2D). To examine the specificity of cell aggregation promoted by HC-HA/PTX3, we examined several human fibroblasts and noted that immobilized HC-HA/PTX3, but not HA, induced cell aggregation in LNC and human corneal fibroblasts but not human skin fibroblasts and tenon fibroblasts, and the restoration of nuclear Pax6 staining was noted in LNC but not in human corneal, skin and tenon fibroblasts (Supplemental Data Figure S5). Furthermore, P10 LNC on HC-HA/PTX3 exhibited reduced cell size and expressed markers of oligodendrocyte progenitor cells, astrocytes and neurons when compared with their counterpart in 3D MG (Figure 2E). Quantitative analysis showed that HC-HA/PTX3 promoted significantly more cells expressing O4, glial fibrillary acidic protein (GFAP), β-III tubulin and neurofilament M than coated MG or 3D MG. (Figure 2E, **P < .01, *P < .1, n = 1000) Collectively, these results suggest that immobilized HC-HA/PTX3, but not 3D MG, uniquely reverts P10 LNC to neural crest progenitors highlighted by nuclear Pax6 staining and higher neuroglial differentiation potential.
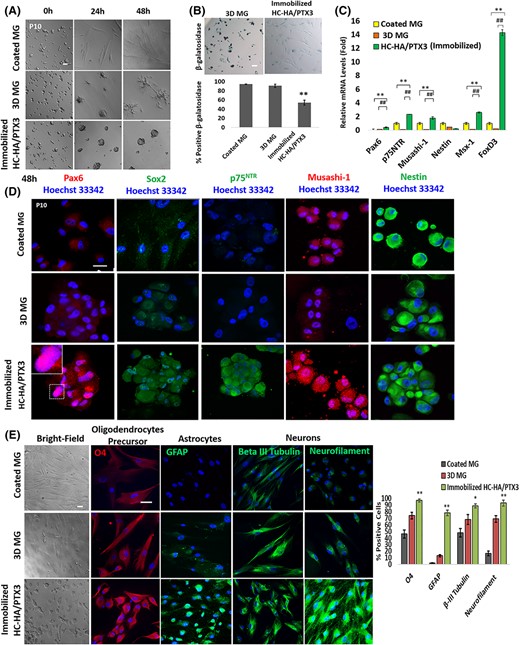
Immobilized HC-HA/PTX3, but not on 3D MG, reverts P10 LNC to nuclear Pax6+ neural crest progenitors
Soluble HC-HA/PTX3 also promotes cell aggregation and reverts to Pax6+ neural crest progenitors
When added on coated MG, soluble HC-HA/PTX3 also promoted cell aggregation in P10 LNC as early as 60 minutes (marked by a white arrow), but the aggregated cells spread to single spindle cells by 24 hours (Figure 3A). Quantitative PCR showed that both soluble and immobilized HC-HA/PTX3 promoted transcript expression of neural crest markers, Sox2, p75NTR, Musashi-1, and nestin, in P10 LNC at 48 hours (Figure 3B, **P < .01, n = 3). Furthermore, soluble HC-HA/PTX3 significantly upregulated transcript expression of p75NTR, nerve growth factor (NGF), and Musashi-1 at both 24 hours and 48 hours when compared with 3D MG (Figure 3C, ##P < .01, n = 3). Immunofluorescence staining also confirmed nuclear staining of Pax6 and Sox2 as well as cytoplasmic staining of p75NTR at 48 hours with soluble HC-HA/PTX3 (Figure 3D). The percentage of nuclear Pax6+ cells at 48 hours was significantly higher in soluble HC-HA/PTX3 than both 3D MG and coated MG (76 ± 11% vs 25 ± 5% and 18 ± 4%, respectively [Figure 3E, **P < .01]). These results support the comparability between immobilized HC-HA/PTX3 and soluble HC-HA/PTX3 in restoring the Pax6+ neural crest progenitor phenotype.
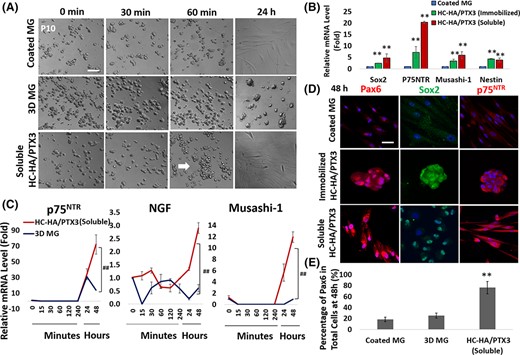
Soluble HC-HA/PTX3 also promotes early cell aggregation and nuclear Pax6+ NC progenitors in P10 LNC
Cell aggregation promoted by soluble HC-HA/PTX3 is facilitated by CXCR4 mediated signaling
In vitro reunion between “heterotypic” P4 LNC and LEPC in 3D MG is mediated by CXCR4/SDF-1 signaling and is pivotal in maintaining self-renewal of LEPC.7 To determine whether CXCR4/SDF-1 signaling is also involved in cell aggregation of “homotypic” P10 LNC by soluble HC-HA/PTX3, we perturbed CXCR4 by adding AMD3100, a small-molecule antagonist of CXCR4 receptor.41,42 The addition of AMD3100 to soluble HC-HA/PTX3 aborted cell aggregation of P10 LNC at 60 minutes and turned them into single cells (Figure 4A). The time course study showed that the CXCR4 transcript increased 4-fold as early as 15 minutes and reached a high peak at 60 minutes with a 500-fold increase when compared with their counterpart in 3D MG (Figure 4B, **P < .01 and **P < .01, n = 3). The addition of AMD3100 significantly downregulated the upregulation of CXCR4 transcript at 24 hours and completely aborted it at 48 hours (Figure 4B). In contrast, the SDF-1 transcript was not upregulated during the first 60 minutes in all cultures but demonstrated a 40-fold increase at 24 hours by 3D MG. Furthermore, the SDF-1 transcript demonstrated a 10-fold increase by soluble HC-HA/PTX3 at 24 hours, which was completely abolished by AMD3100 (Figure 4B, ##P < .01, n = 3). Immunofluorescence staining of CXCR4 showed membrane/cytoplasmic staining throughout the 60 minutes period in 3D MG. In contrast, CXCR4 staining was membrane/cytoplasmic at 0 and 5 minutes but nuclear at 5, 15 and 30 minutes and reverted to predominant membranous in cell aggregation at 60 minutes in soluble HC-HA/PTX3 (Figure 4C). The latter staining pattern reverted to that of 3D MG by AMD3100 (Figure 4C). In contrast, the immunofluorescence staining of SDF-1 was strongly membranous/cytoplasmic throughout the 60 minutes period in cells seeded on 3D MG but was reduced in soluble HC-HA/PTX3 and became negative after the addition of AMD3100 (Figure 4C). Western blot analysis of subcellular membranous, cytoplasmic, and nuclear fractions confirmed the cytolocalization of CXCR4 and Pax6 proteins. Both CXCR4 and Pax6 proteins decreased in the cytoplasmic fraction, but increased in the nuclear fraction, at 5, 15, and 30 minutes in soluble HC-HA/PTX3 (Figure 4D). After blocking CXCR4 by AMD3100, CXCR4 and Pax6 were only present in cytoplasmic fraction at all time points. (Figure 4D) AMD3100 not only prevented cell aggregation, but it also significantly downregulated Pax6, p75NTR, NGF, Musashi-1, Msx-1, and FoxD3 transcripts promoted by soluble HC-HA/PTX3 (Figure 4E, **P < .01, n = 3). This data collectively indicates that the cell aggregation promoted by soluble HC-HA/PTX3 was mediated by CXCR4 signaling, which was causatively linked to the recovery of nuclear Pax6+ neural crest progenitor phenotype in P10 LNC.
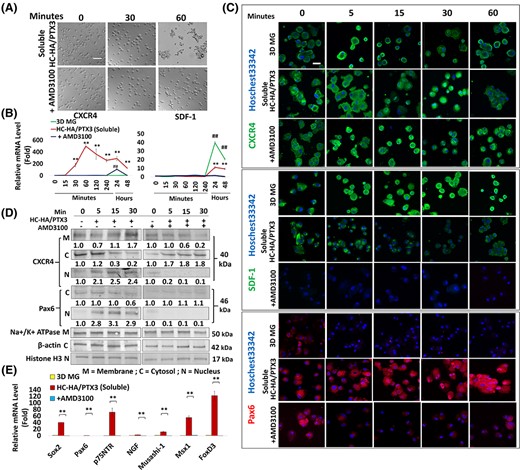
Cell aggregation and nuclear Pax6 expression promoted by soluble HC-HA/PTX3 is facilitated by CXCR4-mediated signaling
CXCR4-mediated signaling is required for activation of BMP signaling
Immobilized HC-HA/PTX3, but not 3D MG, upregulates BMP signaling in P4 LNC, which maintains limbal stem cell quiescence.38 Herein, we noted serial passage from P4 LNC to P10 LNC resulted in the significant downregulation of transcript expression of BMP6, but not BMP2 or BMP4, together with that of BMP receptor 1B (BMPR1B), BMP receptor 2 (BMPR2), and Activin A receptor 1 (ACVR1) (Figure 5A, **P < .01, n = 3). During serial passage, nuclear phosphor-Smad1/Smad5/Smad8 (pSmad1/5/8) staining was also markedly attenuated to nil (Figure 5B). To examine the role of BMP6, we tested exogenous BMPs and noted BMP6 and BMP2 + BMP4 + BMP6, but not BMP 2 or BMP4, induced nuclear pSmad1/5/8 staining in P4 LNC (Figure 5C). Compared with 3D MG, soluble HC-HA/PTX3 significantly upregulated the transcript expression of BMP2, BMP4, and BMP6 (Figure 5D, ##P < .01, n = 3). The upregulation of BMP4 and BMP6 was observed as early as 15 minutes and cyclic to a higher level toward 48 hours, while that of BMP2 gradually increased, with the highest peak observed at 48 hours (Figure 5B, **P < .01, n = 3). Addition of AMD3100 aborted expression of BMP2, BMP4, and BMP6 transcripts throughout the 48 hours period (Figure 5D, **P < .01, n = 3). Nuclear pSmad1/5 staining was promoted by soluble HC-HA/PTX3 in P10 LNC but aborted by AMD3100 (Figure 5E). Western blot confirmed that pSmad1/5 in the nuclear fraction increased as early as 5 minutes with soluble HC-HA/PTX3 but abolished by AMD3100 (Figure 5F). These findings collectively suggest that cell aggregation mediated by CXCR4 signaling by HC-HA/PTX3 in P10 LNC was causally linked to the activation of BMP signaling.
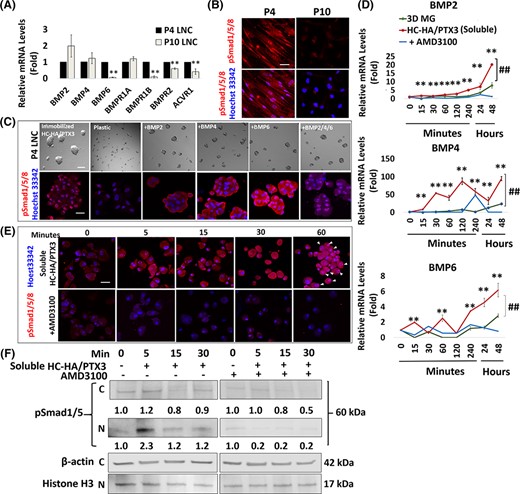
CXCR4-mediated signaling is required for activation of BMP signaling by soluble HC-HA/PTX3
BMP signaling is not involved in cell aggregation and nuclear Pax6 staining promoted by HC-HA/PTX3
To determine the involvement of BMP signaling in cell aggregation facilitated by CXCR4-mediated signaling, we perturbed BMP signaling by LDN-193189, a small molecule BMP inhibitor43 or short interfering RNAs (siRNA) of BMP receptors, that is, BMPR1A, BMPR1B, BMPR2, and ACVR1. LDN-193189 (data not shown) and siRNAs were confirmed to reduce the transcript expressions of BMP receptors (Figure 6A, **P < .01, n = 3) and prevent nuclear pSmad1/5/8 staining (Figure 6B). Our results showed that cell aggregation of P10 LNC by soluble HC-HA/PTX3 was not affected by either LDN-193189 or siRNAs to BMP receptors when compared with the control that was pretreated with scrambled RNA (scRNA) (Figure 6C). Furthermore, there was no significant difference in transcript expression of CXCR4 and SDF-1 throughout 48 hours when P10 LNC were pretreated with siRNAs to BMP receptors (Figure 6D, *P < .05, n = 3). Transient nuclear staining of CXCR4 and positive nuclear Pax6 staining were not affected by pretreated LDN-193189 or BMP receptors siRNA (Figure 6E). Collectively, this data supports the notion that inhibition of BMP signaling does not impact cell aggregation mediated by CXCR4-mediated signaling and nuclear Pax6 staining promoted by HC-HA/PTX3 in P10 LNC.
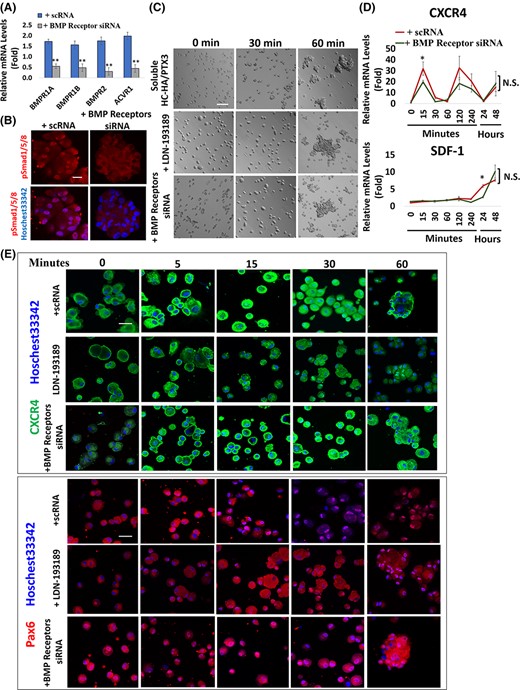
Inhibition of BMP signaling does not impact on cell aggregation and CXCR4-mediated signaling promoted by HC-HA/PTX3
HC-HA/PTX3 restores niche function in P10 LNC to promote self-renewal of the limbal epithelial stem/progenitor cells
We then determined whether HC-HA/PTX3 could restore the niche support function of P10 LNC and further support self-renewal of LEPC using the in vitro reunion assay.38 There was no appreciable difference in the sphere growth of LEPC after reunion with P10 LNC with or without immobilized or soluble HC-HA/PTX3 when compared with the positive control of P4 LNC (Figure 7A). However, compared with the negative control of P10 LNC, transcript expression of ΔNp63α was significantly upregulated while that of cytokeratin 12 (CK12) was significantly reduced in LEPC spheres to the same level as P4 LNC after reunion with P10 LNC pretreated with immobilized or soluble HC-HA/PTX3 (Figure 7B, **P < .01, n = 3). The cell counting of double immunostaining of ΔNp63α or CK12 within PCK+ cells corroborated this finding. That is, there were significantly higher p63α + cells among PCK+ cells, but less CK12+ cells, after reunion with P10 LNC that were pretreated with immobilized or soluble HC-HA/PTX3 when compared with that of untreated P10 LNC (Figure 7C, n > 500, **P < .01). After reunion, LEPC spheres were dissociated, and 1000 single cells were seeded on mitomycin treated 3T3 fibroblast feeder layers to assess the resultant clonal growth by rhodamine B staining at Day 10. As expected, a low number of colonies in LEPC alone has previously been reported.4-6,8,20,38,40 However, clonal growth of LEPC significantly increased when reunited with LNCs.5,8,38,40 Similarly, clonal growth of LEPC was promoted to the same extent as the positive control of P4 LNC upon reunion with P10 LNC pretreated with immobilized or soluble HC-HA/PTX3 when compared with untreated P10 LNC (Figure 7D). Therefore, we concluded that such a low clonal number did not result from cell death or donor age during LEPC isolation. Detailed counting of clonal types based on characterization of morphology (Figure 7E) and double immunostaining of p63α and cytokeratin 12 (Figure 7F) disclosed a significantly higher numbers of holoclones, signifying self-renewal, when LEPC were reunited with P10 LNC pretreated with immobilized or soluble HC-HA/PTX3 compared with untreated P10 LNC to the same extent as P4 LNC (Figure 7E, **P < .01, n = 4). Collectively, this data confirmed that HC-HA/PTX3 reverted P10 LNC to the nuclear Pax6+ neural crest phenotype and thus regained their niche support function to prevent corneal fate decision and maintain self-renewal of LEPC when tested in in vitro reunion assay in 3D MG.
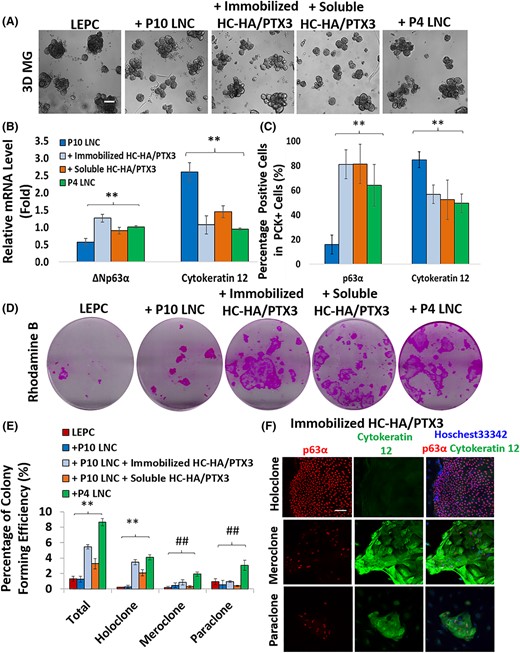
Restoration of niche support function in P10 LNC pretreated with HC-HA/PTX3 to maintain self-renewal of limbal epithelial progenitor/stem cells
DISCUSSION
The progressive loss of the neural crest progenitor phenotype and the niche supporting function of LNC due to serial passage6 was associated with senescence (Figure 1). When compared with coated and 3D MG, both immobilized and soluble HC-HA/PTX3 uniquely reverted such senescence by restoring the nuclear Pax6+ neural crest progenitor phenotype (Figure 2 and Figure 3) and ultimately their niche function of supporting LEPC self-renewal (Figure 7). This finding suggests that HC-HA/PTX3 is capable of reverting “senescent and differentiated” LNC to neural crest progenitors in vitro. Although our study has not provided direct evidence to support whether HC-HA/PTX3 also promotes reversal of senescent cells in vivo, a recent study by Gesteira et al44 did show that the murine limbal niche is rich in HA, and disruption of this HA matrix in knockout mice of HA synthetases or TSG-6, both of which are involved in the biosynthesis of HC-HA/PTX3, leads to limbal stem cell deficiency as evidenced by compromised corneal epithelial regeneration. Thus, we postulate that HC-HA/PTX3 can serve as a surrogate matrix for the limbal niche (also reviewed in Reference 21). This notion explains why HC-HA/PTX3-containing AM augments the success of in vivo27-29 and ex vivo,30,31,45 expansion of LEPC in the treatment of limbal stem cell deficiency. Because nuclear Pax6+ neural crest progenitors have the differentiation potential to give rise to neurovascular cells,6 HC-HA/PTX3 may also be considered as a surrogate niche matrix to support stem cells in other neurovascular niches of the body.46,47
The rejuvenation of senescent LNC by HC-HA/PTX3 involves a reprogramming (de-differentiation) process, which is highlighted by early “morphological” cell aggregation that uniquely occurs in P4 LNC38 and P10 LNC (Figure 2). Such aggregation also occurs as an early event in the reprogramming of human corneal fibroblasts and myofibroblasts into keratocytes,37 yet it does not occur in human skin and tenon fibroblasts (Supplemental data Figure S5). Although cell aggregation is similarly promoted by HC-HA/PTX3, restoration of nuclear Pax6 staining occurs in P10 LNC but not human corneal fibroblasts (Supplemental data Figure S5). Although similar cell aggregation can also be induced by 3D MG40 (also see Figure 2A), the signaling pathway that is induced by HC-HA/PTX3 is mediated by the activation of CXCR4-mediated signaling (Figure 4). The expression of CXCR4 is high in LNC subjacent to limbal basal epithelial stem/progenitors7 and declines during serial passage on coated MG (data not shown). Herein, we noted that HC-HA/PTX3 significantly upregulated the expression of the CXCR4 transcript and promoted membrane and nuclear translocation of the CXCR4 protein prior to cell aggregation (Figure 4). Suppression of CXCR4 with AMD3100 abolished cell aggregation and the ensuing phenotypic reversal by preventing transient nuclear translocation of CXCR4. This was accompanied with the downregulation of the CXCR4 transcript, a switch from the membranous and nuclear localization of the CXCR4 protein to the cytoplasmic localization, and the elimination of SDF-1 cytoplasmic staining without changing SDF-1 transcript expression (Figure 4). The latter finding resembles bone marrow stromal cells, which are mobilized into the blood circulation through the rapid release of intracellular SDF-1 following AMD3100 administration.48 The finding of nuclear CXCR4 in LNC after HC-HA/PTX3 is unique, because as of now, such a finding has only been found in several highly malignant cancer cells.49-52 Herein, we noted that nuclear translocation of CXCR4 in LNC occurred 15 and 30 minutes after the addition of HC-HA/PTX3, much faster than what has been reported for cancer cells treated with sustained SDF-1 stimulation.50 We, thus, envision a different mechanism by which HC-HA/PTX3 activates CXCR4-mediated signaling through the nuclear translocation of CXCR4. Because cell aggregation and nuclear translocation of CXCR4 also occur during the reprogramming of human corneal fibroblasts into keratocytes,37 future studies are needed to delineate the mechanism of how HC-HA/PTX3 promotes nuclear translocation of CXCR4 in LNC and human corneal fibroblasts.
HC-HA/PTX3, but not 3D MG, uniquely promotes BMP signaling in P4 LNC, which is pivotal for maintaining quiescence of LEPC.38 Senescence due to serial passage also resulted in significant downregulation of BMP6, but not BMP2 and BMP4, transcript (Figure 5A) and nuclear pSmad1/5/8 staining (Figure 5B). BMP6, but not BMP2 and BMP4, was responsible for promoting nuclear pSmad1/5/8 staining in P4 LNC seeded on plastic (Figure 5C). These results suggest that BMP6 may play a different role in maintaining the neural crest progenitor status of LNC, resembling the signature role of BMP6 in mesenchymal condensation of neural crest cells in the dermal papilla.53 Both immobilized (not shown) and soluble HC-HA/PTX3 also activated BMP signaling in P10 LNC before cell aggregation. Importantly, disruption of CXCR4-mediated signaling with AMD3100 abolished cell aggregation and the aforementioned BMP signaling (Figure 5), whereas disruption of BMP signaling by LDN-193189 or siRNAs to BMP receptors neither affected cell aggregation mediated by CXCR4-mediated signaling nor abolished nuclear Pax6 staining (Figure 6). Collectively, our results suggest that HC-HA/PTX3 promotes early cell aggregation by activating CXCR4-mediated signaling, which is also required to activate BMP signaling in P10 LNC. Furthermore, CXCR4-mediated signaling, but not BMP signaling, is pivotal in the reprogramming of P10 LNC. Although the reprogramming of human corneal fibroblasts into keratocytes by HC-HA/PTX3 also involves cell aggregation mediated by CXCR4-signaling followed by activation of BMP signaling, both CXCR4-signaling and BMP signaling are required for reprogramming of human corneal fibroblasts into keratocytes.37 Further studies are warranted to delineate the action mechanism of HC-HA/PTX3 in reprogramming differentiated cells into progenitor cells and will shed light upon its potential role as a surrogate niche matrix in regenerative medicine, particularly for stem cell-based therapies.
MATERIALS AND METHODS
Isolation and expansion of human limbal epithelial and niche cells
Human corneocscleral rims from six donors between 46- and 69-years of age were obtained from Florida Lion Eye Bank (Miami, Florida) in accordance with the declaration of Helsinki. Human corneolimbal rim and central cornea button were stored at 4°C in Optisol (Chiron Vision, Irvine, California) for less than 7 days. The tissue was rinsed three times with PBS (pH 7.4) containing 50 μg/mL gentamicin and 1.25 μg/mL amphotericin B. Under a dissecting microscope (Nikon SMZ800N, Irvine, California), the excess sclera, conjunctiva, iris, corneal endothelium and trabecular meshwork were removed up to the Schwalbe's line for the corneoscleral rim before being cut into superior, nasal, inferior, and temporal quadrants at 1 mm within and beyond the anatomic limbus. As previously reported,54 an intact epithelial sheet that included basal epithelial cells was obtained by subjecting each limbal quadrant to digestion with 10 mg/mL dispase at 4°C for 16 hours in the modified embryonic stem cell medium (MESCM), which was made of Dulbecco's Modified Eagle's Medium (DMEM)/F-12 nutrient mixture (F-12) (1:1) supplemented with 10% knockout serum, 10 ng/mL LIF, 4 ng/mL bFGF, 5 mg/mL insulin, 5 mg/mL transferrin, 5 ng/mL sodium selenite supplement (ITS), 50 μg/mL gentamicin, and 1.25 μg/mL amphotericin B. The remaining stroma was subjected to 2 mg/mL collagenase A at 37°C for 16 hours to generate floating clusters under the humidified 5% CO2 incubator.4,40 For experiments described herein, a minimum of three donors were processed using our standard protocol.40
For coated Matrigel, 5% Matrigel was prepared by diluting 50 μL of Matrigel into cold 950 μL MESCM and plating at 1000 μL per well in a 6-well plastic dish for 1 hour at 37°C before use. Single cells derived from limbal clusters after digestion with 0.25% trypsin and 1 mM EDTA (T/E) were seeded at 1 × 104/cm2 in the 6-well plate that was precoated with 5% MG in MESCM and cultured in humidified 5% CO2 with media change every 3 to 4 days for a total of 6 to 7 days. When cells reach at 80% to 90% confluence, cells were subcultured at the seeding density of 5 × 103 per cm2 for up to 13 passages. The extent of total expansion was measured by the number of cell doubling (NCD), which was calculated using the following formula: NCD = log10(y/x)/log 102, where “y” is the final density of the cells and “x” is the initial seeding density of the cells. For all experiments, at least three donors of P10 LNC were used, expanded, and pooled together.
Purification and characterization of HC-HA/PTX3
HC-HA/PTX3 was purified from cryopreserved human placentas provided by TissueTech, Inc (Miami, Florida) as previously reported32,34 with modification. In brief, AM retrieved from placenta was cryopulverilzed by FreezeMill (FreezerMill 6870, SPEX SamplePrep, Metuchen, New Jersey), extracted by PBS (pH 7.4) at 4°C for 1 hour, and then centrifuged at 48 000g at 4°C for 30 minutes to generate the supernatant, which was designated as AM extract. This extract was then fractionated by ultracentrifugation in a CsCl gradient at an initial density of 1.35 g/mL in 4 M GnHCl at 125 000g at 15°C for 48 hours (Optima L-80X, SW41 rotor, Beckman Coulter, Indianapolis, Indiana). A total of 12 fractions (1 mL/fraction) were collected from each ultracentrifuge tube. The weight of each fraction was measured to calculate the density, while HA content and protein content in each fraction were measured by the enzyme-linked immunosorbent HA Quantitative Test Kit (Corgenix, Broomfield, Colorado) and the BCA Protein Assay Kit (Life Technologies, Grand Island, New York), respectively. The fractions of 2 to 12, which contained most of HC-HA/PTX3, were pooled and further subjected to three consecutive runs of ultracentrifugation at 125 000g in CsCl/4 M guanidine HCl at a density of 1.40 g/mL for the second run and 1.42 g/mL for the third and fourth run, each run at 15°C for 48 hours. The fractions 3 to 9 after the fourth run were pooled and dialyzed against distilled water at 4°C for 48 hours for a total of 5 times, which were then lyophilized, stored at 80°C, and designated as HC-HA/PTX3. Before use, the biochemical composition of HC-HA/PTX3 was verified using agarose gel electrophoresis containing high molecular weight HA and Western blot with or without HAase digestion (1 U/μg HA) in the presence of protease inhibitors (Sigma-Aldrich, St. Louis, Missouri)32,34 to validate the presence of HC1 (ab70048, Abcam, Cambridge, Massachusetts) and PTX3 (ALX-804-464-C100, Enzo Life Sciences, Farmingdale, New York). Because of the negligible amount of protein therein, the amount of HC-HA/PTX3 used in the experiment was expressed using the optical density of HA amount with a SpectraMax M5 microplate reader (Molecular Device, San Jose, California).
Cell culture and treatment
As reported,7,40 50% MG was prepared using an 8-well chamber slide by diluting 150 μL MG into 150 μL in cold MESCM per well followed by incubation for 1 hour at 37°C before use. For cell culture in 3D MG, cells expanded on coated MG at passage 10 were reseeded in 3D MG at the density of 5 × 104 cells/cm2 for 24 hours or 48 hours in MESCM. Aggregates for 3D MG were harvested by digestion with 10 mg/mL dispase II at 37°C for 2 hours before being prepared for cytospin.
P10 LNC were seeded at 1 × 105 cells/mL on immobilized and soluble HC-HA/PTX3 at 96-well for 24 or 48 hours in MESCM. The method of immobilizing HC-HA/PTX3 on Covalink-NH 96 wells has previously been reported32 and used in murine macrophage and CD4+ T cells,35,36 retinal pigment epithelial cells55 and limbal niche cells.38 In short, 100 μL of 20 μg/mL HC-HA/PTX3 was immobilized on Covalink-NH 96 wells by first sterilizing the Covalink-NH 96 wells in 70% alcohol for 30 minutes, and then the wells were washed with distilled water two times. HC-HA/PTX3 with the crosslinking reagents, Sulfo-NHS at 9.2 mg/mL and 1-ethyl-3(3-dimethylaminopropyl) carbodiimide (EDAC) at 6.2 mg/mL, were added to each well and incubated overnight at 4°C. After that, the un-crosslinked HC-HA/PTX3 and crosslinking reagents were removed, and the wells were washed twice with 2 M NaCl/50 mM MgSO4/PBS, followed by two washes of PBS.
Upon 80% confluence, P10 LNC cultured on coated MG were pretreated with 0.1% DMSO with or without 20 μg/mL AMD3100 or 100 nM LDN-193189 for 30 minutes before being trypsinized and seeded at 2 × 105/mL on coated MG in MESCM containing 20 μg/mL of AMD3100 or 100 nM LDN-193189 with 20 μg/mL soluble HC-HA/PTX3 in MESCM for another 48 hours. For the siRNA knockdown, 80% confluent P10 LNC on 6-well coated MG were subjected to transfection by mixing 200 μL of serum-free, antibiotic-free MESCM with 4 μL of HiPerFect siRNA transfection reagent (Final dilution, 1:300) and 6 μL of 20 μM of scRNA or siRNAs for BMPR1A, BMPR1B, BMPR2, and ACVR1 at the final concentration of 100 nM, drop-wise. This was followed by culturing in 1 mL of fresh MESCM at 37°C for 24 hours before soluble HC-HA/PTX3 was added at a final concentration of 20 μg/mL in MESCM. All material used are listed in Supplementary Table S1. Addition of 40 ng/mL BMP2,56 50 ng/mL BMP4,57 100 ng/mL BMP6,58 and BMP2/4/6 in MESCM was performed in 2 × 104 P4 LNC on 96-well plastic for 24 hours.
In vitro reunion assay in 3D MG
As reported,4 LEPC isolation from whole human corneolimbal rims were subjected to digestion with 10 mg/mL dispase in serum-containing supplemental hormonal epithelial medium (SHEM), which was made of an equal volume of HEPES-buffered DMEM and Ham's F-12 containing bicarbonate, 0.5% dimethyl sulfoxide, 2 ng/mL mouse-derived epidermal growth factor, 5 mg/mL insulin, 5 mg/mL transferrin, 5 ng/mL sodium selenite, 0.5 mg/mL hydrocortisone, 30 ng/mL cholera toxin A subunit, 5% fetal bovine serum (FBS), 50 mg/mL gentamicin, and 1.25 mg/mL amphotericin B. Three donors of entire intact limbal epithelial sheets were separated, pooled and obtained under dissecting stereo microscope. 5 × 104 cells/cm2 single LEPC were obtained from limbal epithelial sheets was reunion with 5 × 104 cells/cm2 LNC with or without pretreatment with soluble or immobilized HC-HA/PTX3 for 24 hours prior to the reunion in 3D MG in MESCM for 6 days.
Clonal assay for limbal epithelial progenitors
The feeder layer used for the colony forming assay was prepared by treating 80% subconfluent NIH-3T3 fibroblasts (ATCC, CRL-1658) with 6 mg/mL mitomycin C at 37°C for 2 hours in DMEM containing 10% fetal calf serum, and then mitomycin C-treated 3T3 fibroblasts were seeded at a density of 2 × 104/cm in SHEM. A total of 1000 single cells were obtained from reunion sphere growth from 3D MG and were seeded on mitomycin C-treated 3T3 fibroblast feeder layers for 10 days. The resultant clonal growth was fixed in 4% paraformaldehyde and assessed by 2% rhodamine B staining aqueous solution for marking clones for the measurement of colony-forming efficiency (CFE). The colony forming efficiency was calculated by dividing the percentage of the clone number by the total number of PCK/p63α positive cells. Clone morphology was subdivided into holoclones, meroclones, and paraclones based on the criteria established for skin keratinocytes.59 The epithelial morphology of holoclones, meroclones and paraclones were further characterized by immunostaining of p63α and cytokeratin 12.
Neuroglial differentiation
A total of 1 × 104/mL of P10 LNC was seeded on 50 μg/mL poly-l-ornithine and 20 μg/mL laminin-coated or Collagen Type IV coated cover glass in 48-well plate in NSCM supplement with 0.5% N2 and 1% B27 for 2 days. For neuronal differentiation,60 the medium was replaced with neuronal induction base medium containing DMEM/F12 (1:3) with 0.5% N2 and 1% B27 in additional to 10 ng/mL FGF2 and 20 ng/mL of BDNF (medium A) for 3 days and replaced with base medium in addition to 6.7 ng/mL FGF2 and 30 ng/mL of BDNF for another 3 days. Cell then replaced to base medium in addition to 2.5 ng/mL FGF2, 30 ng/mL BDNF, and 200 mM ascorbic acid for another 8 days. For oligodendrocyte precursor differentiation,60 medium then replaced with base medium containing DMEM/F12 (1:1) with 1% N2 in addition to 10 ng/mL FGF2, 10 ng/mL PDGF, and 10 μM forskolin for 4 days and then medium was replaced by the base medium in addition to 10 ng/mL FGF2, 30 ng/mL 3,3,5-triiodothyronine, and 200 μM ascorbic acid for another 7 days. For astrocyte differentiation (Thermo Fisher Scientific, Santa Clara, California), medium was replaced by DMEM containing 1% FBS, 1% N2, and 2 mM GlutaMax for 10 days. Induction media were changed every 3 to 4 days. Media and supplements are listed in Supplementary Table S2.
Subcellular fractionation and Western blotting
Membranous, cytoplasmic or nuclear fraction of cells was prepared from NE-PER Membrane Plus and MEM-PER Nuclear and Cytoplasmic Extraction Reagents Kit (Pierce, Rockford, Illinois) per manufacturer's instruction. Briefly, 3 × 106 P10 LNC after treatment were washed once on cold PBS and centrifuged at 500g for 5 minutes. The cell pellet was suspended in 100 μL of the cytoplasmic extraction reagent I containing protease inhibitor by vortexing. The suspension was incubated on ice for 10 minutes, followed by the addition of 6 μL of a second cytoplasmic extraction reagent II, vortexed for 5 seconds, incubated on ice for 1 minute and centrifuged for 5 minutes at 16 000g. The supernatant fraction (cytoplasmic extract) was transferred to a prechilled tube. The insoluble pellet fraction, which contains crude nuclei, was resuspended in 50 μL of nuclear extraction reagent by vortexing for 15 seconds three times and incubated on ice for 10 minutes each, then centrifuged for 10 minutes at 16 000g. The resulting supernatant, constituting the nuclear extract, was used for the subsequent experiments. For the membranous fraction, 150 μL was obtained from Solubilization Buffer fraction. Protein concentration was quantitated using the BCA protein assay kit (Pierce). Equal amounts of protein were loaded in each lane and separated on 4% to 15% gradient Mini-PROTEAN TGX Precast gels under denaturing and reducing conditions. The protein extracts were then transferred to a Trans-Blot Turbo Mini PVDF transfer pack by Trans-Blot Turbo Transfer System (Bio-Rad Laboratories, California). The membranes were then sequentially blocked with 5% (w/v) fat-free milk in TBST [50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% (v/v) Tween-20] for 1 hour followed by incubation with the specific primary antibodies against Pax6, CXCR4 or phosphor-Smad1/5 for 12 hours at 4°C. Antibody against Na+/K+ ATPase, β-actin or Histone H3 was used as the loading control for membrane, cytoplasmic or nucleus fraction, respectively. The respective horseradish peroxidase (HRP)-conjugated secondary antibody was incubated for 1 hour at room temperature. Immunoreactive protein bands were detected with Western Lighting Chemiluminescence (PerkinElmer, Waltham, Massachusetts), and images were captured by GE ImageQuant LAS 4010 (GE Healthcare Biosciences, Pittsburgh, Pennsylvania). Western blot images were quantified using Image Quant TL 8.1 software and normalized by the control protein band. The average of three measurements was reported.
Quantitative real-time PCR
Total RNAs were extracted from different passaged of LNC by RNeasy Mini Kit (Quiagen, Valencia, California) according to manufacturer's guideline, and 1 to 2 ug of RNA extract was reverse transcribed to cDNA using Applied Biosystem High Capacity Reverse Transcription Kit (Thermo Fisher Scientific, Santa Clara, California) and primers listed in Supplementary Table S3. The resultant cDNA was amplified by specific TaqMan gene expression assay mix and universal PCR master mix in QuantStudio 5 Real Time PCR System (Thermo Fisher Scientific, Santa Clara, California) with Real-Time PCR amplification profile. This consisted of 10 minutes of initial activation at 95°C, followed by 40 cycles of 15 seconds denaturation at 95°C, and 1 minute annealing and extension at 60°C. The threshold was set at 10 times the SD above the mean baseline emission value for the first 15 cycles. Threshold cycle number (Ct) was calculated with QuantStudio Design and Analysis v.1.4.3 (Thermo Fisher Scientific, Santa Clara, California). The relative gene expression data were analyzed using the comparative CT method (ΔΔCT). Fold change of in one sample was measured relative to the others. The results were normalized using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an internal control. All assays were performed in triplicate.
Immunofluorescence staining
Single cells of LNC at different passages were harvested with 0.05% trypsin and 1 mM EDTA at 37°C for 10 minutes and prepared for cytospin using Cytofuge (StatSpin, Inc, Norwood, Massachusetts) at 1000g for 8 minutes. Cells were fixed with 4% formaldehyde, pH 7.0, for 15 minutes at room temperature, permeabilized with 0.2% Triton X-100 in PBS for 15 minutes and blocked with 2% bovine serum albumin (BSA) for 1 hour before incubated with primary antibodies for 16 hours at 4°C. After 3 washes with PBS, the corresponding Alexa Fluor-conjugated secondary IgG (all 1:100 dilution) were incubated for 60 minutes. After three washes with PBS, the second primary antibodies were incubated for 60 minutes and followed with the corresponding Alex Fluor-conjugated secondary IgG. The nucleus was counterstained with Hoechst 33342 before being analyzed with Zeiss LSM 700 confocal microscope (Carl Zeiss, Thornwood, New York). Corresponding mouse and rabbit sera were used as negative controls for primary monoclonal and polyclonal antibodies, respectively. All experiments for immunofluorescence staining were repeated at least three times. Detailed information about primary and secondary antibodies used for immunofluorescence staining is listed in Supplementary Table S4.
Histochemical staining of β-galactosidase
Cells were stained for senescence-associated β -galactosidase (SA-β-gal) using a SA-β-gal Staining Kit (Cell signaling, #9860S, Danvers, Massachusetts) according to the manufacturer's instructions. Briefly, P4 LNC, P10 LNC or P10 LNC pretreated with 3D MG or Immobilized HC-HA/PTX3 for 48 hours were seeded at 1 × 104/cm2 in the 12-well plate precoated with 5% MG in MESCM for 48 hours. Cells were washed twice with PBS followed by fixative solution for 10 minutes at RT, followed by incubation of 0.5 mL pH 6 of β-galactosidase staining solution at 37°C overnight. The solution was removed, washed twice with 1× PBS before overlayed with 70% glycerol. Blue color development of β-galactosidase was observed and captured under the Zeiss Axio Observer Z1 inverted microscope (Carl Zeiss, Thornwood, New York).
Statistical analysis
All summary data was reported as mean ± SD. Significance was calculated for each group using the 2-tailed Student's t-test by Microsoft Excel (Microsoft, Redmond, Washington), and P < .05 was considered statistically significant. For observed statistical analyses between three donors, Bonferroni correction were employed, such that P-values are multiplied by 3.
ACKNOWLEDGMENT
The work was supported by a grant (RO1EY06819 to S.C.G.T.) from the National Eye Institute, National Institutes of Health, Bethesda, MD, USA.
CONFLICT OF INTEREST
S.Y.C. declared employment and patent ownership with TissueTech. The other authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
S.Y.C.: conception and design, data analysis, provision of study material, collection and/or assembly of data, data analysis and interpretation, manuscript writing; Y.T.Z.: conception and design, collection and/or assembly of data, data analysis and interpretation; Y.Z.: provision of study material, collection and/or assembly of data; D.H.: collection and/or assembly of data; S.C.G.T.: conception and design, financial support, data analysis and interpretation, manuscript writing, final approval of manuscript.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
Author notes
Funding information National Eye Institute, National Institutes of Health, Bethesda, MD, Grant/Award Number: RO1 EY06819
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}